Abstract
Lung cancer is the leading cause of cancer death worldwide due to its high incidence and mortality. As the most common lung cancer, non-small cell lung cancer (NSCLC) is a terrible threat to human health. Despite improvements in diagnosis and combined treatments including surgical resection, radiotherapy and chemotherapy, the overall survival for NSCLC patients still remains poor. DNA damage is considered to be the primary cause of lung cancer development and is normally recognized and repaired by the intrinsic DNA damage response machinery. The role of DNA repair pathways in radio(chemo)therapy-resistant cancers has become an area of significant interest in the clinical setting. Meanwhile, some studies have proved that genetic and epigenetic factors can alter the DNA damage response and repair, which results in changes of the radiation and chemotherapy curative effect in NSCLC. In this review, we focus on the effect of genetic polymorphisms and epigenetic factors such as miRNA regulation and lncRNA regulation participating in DNA damage repair in response to radio(chemo)therapy in NSCLC. These may provide novel information on the radio(chemo)therapy of NSCLC based on the individual DNA damage response.
1. Introduction
Non-small cell lung cancer (NSCLC) accounts for about 85% of lung cancer cases. NSCLC is the leading cause of cancer death worldwide due to its high incidence and mortality for both men and women, and it has become a serious health problem [1]. It is widely accepted that lung cancer development is the result of many factors, such as genetic, environmental, food and lifestyle factors (particularly smoking related), among which DNA damage is considered to be the primary cause of lung cancer [2]. The systemic treatments for lung cancer consist of classical surgery, standard radiotherapy and chemotherapy, and monotherapy, and a combination therapy of these approaches is recommended in clinical practice. One main impediment for treating NSCLC is that most patients are diagnosed at an advanced stage, consequently missing the most opportune window for surgical intervention. So radiotherapy and chemotherapy become important therapeutic approaches for unresectable NSCLC [3]. Radiotherapy and platinum-based chemotherapy play key roles in the treatment of NSCLC by damaging DNA and inducing tumor cell death. However, regardless of these conventional therapies, the five-year survival rate of NSCLC has still not improved significantly, remaining at less than 15% [4]. In addition, clinical resistance (intrinsic or acquired) to radio(chemo)therapy is considered as another impediment in the treatment of human NSCLC, which can be attributed to many factors. One such factor is the DNA repair capacity of damaged cells in mediating resistance to radiation and platinum-based chemotherapy [5,6]. Thus, the roles of DNA repair pathways in radio(chemo)therapy-resistant cancers have attracted widespread interest in clinic.
An increasing number of studies have shown that large inter-individual variability exists in the capacity of DNA damage response and DNA repair, and sensitivity to radiation and chemotherapy varies from person to person, thus generating different therapeutic effects despite uniform treatment protocols [7,8]. Some studies have shown that genetic factors play a leading role in the inter-individual variation in radio(chemo)therapy sensitivity [9,10,11]. The expression level of DNA repair genes and the activity of the encoded proteins determine whether or not radio/chemo-resistance will occur by determining the capacity of DNA repair. In NSCLC, a deficiency in DNA repair could be a result of a mutation in one DNA repair gene, but much more frequently reduced DNA repair genes or the absent expression of DNA repair genes could be attributed to epigenetic alterations that reduce or silence corresponding gene expression. With the evolution of DNA sequencing and bioinformatics, genetic variations and epigenetic modification, such as miRNA and lncRNA regulation, have emerged as a novel research field in DNA repair pathways which influence radio(chemo)therapeutic outcome. In order to gain insights into the underlying mechanism of radio(chemo)therapy resistance in NSCLC, this review focuses on the various DNA repair genes from two aspects: genetic and epigenetic levels, with a particular emphasis on their role in mediating radio(chemo)therapy resistance in NSCLC. Understanding the DNA repair mechanism has huge implications for potentiating the anti-tumor effect of radiotherapy and genotoxic chemotherapy, helping to achieve personalized therapy for NSCLC (Figure 1).
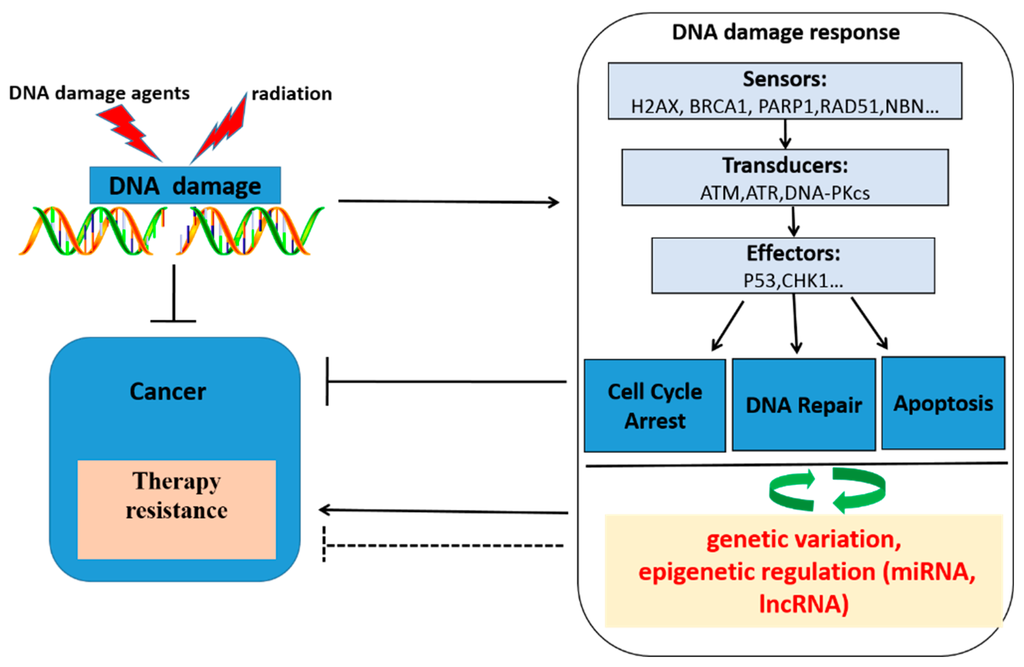
Figure 1.
Schematic overview of DNA damaging agents, radiotherapy, DNA damage, the cellular response upon DNA damage, genetic variation, epigenetic regulation and cancer therapy response. Genotoxic agents and radiation induce DNA damage, which activates the DNA damage response. The DNA damage response consists of three steps processes: At the first time, DNA damage was recognized by DNA damage association proteins, then transducing damage signals to the cell, lastly, cell cycle arrest, DNA repair and apoptosis biological process were activated by DNA damage. Thus, cancer cells will be killed. DNA damage response became a therapeutic target. However, tumors can develop therapy resistance by activating or inhibiting various processes within the DNA damage response by a bidirectional regulatory pathway exists between DNA damage response and miRNAs, lncRNA, genetic variation in DNA repair genes, which will influence DNA damage response, then regulation radio(chemo)therapy response.
2. Initiation of the DNA Damage Response and DNA Repair Pathway
DNA is vulnerable to damage that originates from endogenous metabolites, such as macrophages and neutrophils produced ROS (reactive oxygen species), RNS (reactive nitrogen species) [12] and exogenous agents including smoking, chemical carcinogens, radiation [13], and genotoxic cancer therapeutics [14]. For instance, platinum compounds such as cisplatin can form the platinum-DNA (Pt-DNA) adducts in the DNA which introduces covalent links between bases of the same or different DNA strands (intra-strand crosslinks or inter-strand crosslinks) [15]. Additionally, radiation may induce single-strand or double-strand DNA breaks (SSBs and DSBs) by inducing the oxidation of DNA bases causing lesions in the DNA. These lesions give rise to gene mutations and chromosomal damage, which are causal events in oncogenic transformation, malignant progression and radio(chemo)therapy resistance [16,17]. DSBs have the most harmful effect on cell survival among all DNA lesions, and are strong activators of apoptosis [18]; cell death could be induced by the persistence of DSBs if not repaired. To preserve genomic stability and survival, cells have developed DNA damage response (DDR) to handle DNA lesions [19]. When the genome gets damaged by radiation or chemotherapy, cells have to initiate an efficient response to faithfully repair the lesion and maintain genome integrity [20].
DNA repair is orchestrated by a series of pathways, mainly including nucleotide excision repair (NER), base excision repair (BER), DNA mismatch repair (MMR) and single-strand break repair (SSBs) [21]. Among these DNA repair pathways, NER repairs damaged DNA commonly caused by chemotherapeutics such as platinum drugs, which has been proven to be associated with chemotherapy resistance in NSCLC [22]. The repair of DSBs is carried out by two major repair pathways: non-homologous DNA end joining (NHEJ) and homologous recombination (HR). NHEJ acts on the G0/G1 cell cycle phase, and HR plays a role in the late S/G2 phase [19]. Blocking these repair pathways will likely result in increased radiotherapy and chemotherapy sensitivity.
In order to ensure that cells respond to DSBs quickly and accurately, three steps are supposed to be necessary: Firstly, the damage must be detected as soon as possible. Three interconnected sensor systems have been described that have the ability to detect a single DSB within minutes after its formation [23]. These sensors in the DDR are the PI3K-related kinases (PIKKs): ataxia telangiectasia and Rad3-related (ATR), ataxia telangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PK). ATR plays a key role in recognition of SSBs induced by cisplatin or IR, while ATM is mainly involved in the recognition of DSBs. Secondly, damage signals are transduced to the cell. Lastly, cells react to decide to either repair damaged DNA or to activate cell cycle checkpoints or induce apoptosis. Once DSBs are induced by cisplatin or IR, as a transducer, ATM/ATR targets many dual-function proteins that are key nodes in promoting survival or cell death. Then these proteins signal downstream checkpoint activation to prevent cell cycle progression that provides time for the cell to repair the damage and the recruitment of DNA repair proteins to facilitate DSB repair via the stimulation of NHEJ or HR, depending on the cell cycle phase [24]. When the damage to DNA is greater than the repair capacity, the remaining DNA damage will block replication and transcription, and activate DDR signals downstream cell death pathways. The repair capacity of the cell, the status of p53 and key DDR proteins including ATR, ATM and DNA-PK, the effectiveness of activating DNA repair genes, and the execution of downstream cell death pathways are determinants of a cell surviving death. The expression levels of these genes including XPA, ERCC3/XPB, XPC, ERCC2/XPD, DDB1/XPE, ERCC4/XPF, ERCC5/XPG, ERCC1, XRCC family, ATM, NBN, BRCA1, RAD51, P53, and EIF3A are important for the repair capacity and the response to radiotherapy and chemotherapy [25,26]. Although mutations of DNA repair genes and epigenetic alterations involved in DNA repair process are occasionally occurring events in cancer, they will change the expression of DNA repair genes, alter DNA repair capacity, and hence affect the response to radio(chemo)therapy in NSCLC.
3. Association of Genetic Polymorphisms in DNA Repair Genes with Radio(chemo)therapy Response in Non-Small Cell Lung Cancer (NSCLC)
Genetic variations in DNA repair genes are thought to modulate DNA repair capacity and be related to the sensitivity of radio(chemo)therapy in NSCLC. Thus, certain single nucleotide polymorphisms (SNPs) may be used to predict the sensitivity or clinical outcomes of radio(chemo)therapy in patients, which may play an important role in the individualized treatment of NSCLC. Through a number of case-control studies, SNPs at candidate loci were genotyped across patients with or without radio(chemo)therapy resistance and toxicity, and many genetic markers have been investigated in human NSCLC [27,28,29,30,31]. In the following text, we briefly recapitulate our knowledge of these SNPs in DNA repair genes, and how they play roles in radio(chemo)therapy response (Table 1).

Table 1.
Evidence for correlation of genetic variants with radio(chemo)therapy response in Non-Small Cell Lung Cancer (NSCLC).
3.1. X-ray Cross-Complementing (XRCC) Family Genes
The XRCC (X-ray cross-complementing) genes are discovered mainly through their roles in protecting mammalian cells from damage caused by ionizing radiations and anti-tumor chemotherapy agents. In the XRCC family, XRCC1, XRCC2, XRCC3, XRCC4 and XRCC5 are studied frequently in cancers. Among these genes, XRCC1 is the most frequently researched gene. It is a limiting factor in the BER pathway. XRCC1 is over-expressed in NSCLC [32], and the expression levels of XRCC1 have shown a significant correlation with cisplatin chemo-resistance in NSCLC cell lines [33]. Changes in the amino acids will affect the normal function of XRCC1 protein and cause altered DNA repair activity [34,35,36,37]. The Arg194Trp and Arg399Gln genetic polymorphisms are the most extensively studied SNPs of the XRCC1 gene. Evidence from lung cancer patients showed that XRCC1 (rs25487, Arg399Gln) might influence radiation and platinum-based chemotherapy wherein patients with the ancestral allele (G) were found to be more radiosensitive and have a higher overall survival [38], while the XRCC1 399Arg/Arg genotype carriers had a higher response rate than that of the Gln genotype carriers (OR = 4.81, 95% CI = 1.778–13.013, p = 0.002) [39,40] and a poor overall survival in a short-term period (HR = 1.718, p = 0.003; HR = 1.691, p = 0.003, respectively) in a treatment with platinum-based chemotherapy for NSCLC patients [41]. As for the XRCC1 Arg194Trp polymorphism, it was found to be significantly associated with better response rates to platinum-based chemotherapy in advanced NSCLC [36,42,43].
XRCC2 and XRCC3 genes encode a member of the RecA/Rad51-related protein family that is involved in the repair of DSBs by the HR pathway [44]. Previous findings demonstrated that XRCC2 (rs3218536, Arg188His) was correlated with overall survival (OS) in NSCLC patients treated with radiotherapy [45]. A meta-analysis result showed that the XRCC3 Thr241Met polymorphism had an impact on the response to platinum-based chemotherapy in patients with advanced NSCLC and XRCC3 carriers of the variant 241Met allele were significantly associated with better response [46]. In addition, one study showed that male patients with the TT genotype of XRCC4 rs6869366 (−1394G>T) and female patients with the AG/AA genotypes of XRCC5 rs3835 (2408G>A) were at increased risk of severe radiation-induced pneumonitis in NSCLC [47]. The use of XRCC family genes’ polymorphisms as predictors of clinical outcomes in personalized radio(chemo)therapy treatment requires further verification from large, well-designed pharmacogenetics studies.
3.2. Excision Repair Cross-Complementing (ERCC) Family Genes
ERCC (excision repair cross-complementing group) family genes mainly include ERCC1, ERCC2 (also named XPD), ERCC3 (also named XPB), ERCC4 (also named XPF) and ERCC5 (also named XPG), and they participate in DNA repair and DNA recombination. ERCC1 expression has proved to be related to the clinical benefit of platinum-based chemotherapy [6,48,49]. High ERCC1 expression is associated with a significantly worse OS in platinum-treated NSCLC patients [50]. Genetic polymorphisms of ERCC1 may alter the repair function, possibly by changing its expression level. Previous studies have demonstrated that the G/G genotype of ERCC1 rs11615 is associated with a better survival [51] and higher sensitivity to cisplatin in advanced NSCLC patients [52].
In addition, ERCC2 rs13181 has been studied regarding its effect on platinum-based chemotherapy sensitivity. A meta-analysis of clinical studies has shown that ERCC2 rs13181 with A/C and C/C genotypes is associated with low sensitivity in Asian populations and with high sensitivity in Caucasian NSCLC patients that were treated with platinum drugs [53]. Thus, ERCC2 Lys751Gln (A>C) may act as a predictor in NSCLC treated with platinum-based chemotherapy according to different ethnicities. More recently, another study has reported that patients with the ERCC3 rs3738948 G/G or G/A genotype show a better response, and those with the ERCC5 rs2296147 TT genotype and the T allele show a significantly reduced risk of developing progressive NSCLC when receiving platinum-based chemotherapy, while advanced NSCLC patients carrying the ERCC5 rs2094258 G/G and the G allele show a significantly decreased risk of developing a progressive disease [54,59]. There is little evidence that SNPs in other ERCC family genes have potentials to be used as predictors of radiotherapy or chemotherapy and more research is needed.
3.3. Ataxia Telangiectasia Mutated (ATM)
ATM (Ataxia telangiectasia mutated) is a serine/threonine protein kinase, a key DDR kinase and tumor suppressor, that is recruited and activated by DSBs induced by ionizing radiation or genotoxicity drugs [60]. It plays a central role in the recognition and signaling of DNA damage by regulating several key proteins (such as p53, CHK1, BRCA1, NBN, H2AX, ARF) that initiate activation of the DNA damage checkpoint, leading to cell cycle arrest, DNA repair or apoptosis [60,61]. Recently, a functional interplay between ATM and the alternative reading frame (ARF) tumor suppressor protein in response to oncogenic insults has been shown. ATM can suppress ARF protein levels and activity in a transcription-independent manner [62,63]. Loss of ATM will render tumors resistant to the cell death pathway.
Accompanied by a gain of therapeutic resistance, ATM is frequently mutated in tumors [64]. Some independent studies have shown the ATM (rs189037 (G>A)) A allele as a risk allele for radiation pneumonitis in NSCLC patients upon radiotherapy [55,56,57]. Additionally, further studies demonstrated that rs189037 might affect ATM expression by reducing transcriptional activity and interfering nuclear protein binding in vitro and in vivo [56]. In addition, haplotype analysis showed that patients carrying ATM rs228590 TT/CT or rs189037 AG/GG genotypes or rs228590T/rs189037G/rs1801516G (G-T-G) haplotypes had a lower risk of severe radiation pneumonitis when receiving definitive radio(chemo)therapy [57,58].
4. Noncoding RNAs and Radio(chemo)therapy Response in NSCLC
In the past, the most common approach to identifying genetic markers of drug response focused on SNPs [65]. However, along with the rapid development of sequencing technology and bioinformatics, epigenetic alterations have been increasingly identified and recognized as a frequent event in cancers. Non-coding RNAs (ncRNAs) are now widely thought to be critical in the biological processes of cancer incidence and radio(chemo)therapy sensitivity. Additionally, as a new epigenetic regulation, it affects the sensitivity of radio(chemo)therapy by regulating the DDR pathway through modulating the expression or location of key genes, such as cell cycle control genes, DNA repair genes and apoptosis genes. Thus, ncRNAs have great potential to be used as biomarkers for personalized therapy in NSCLC. Next, we will put emphasis on microRNAs and lncRNAs, summarize how they play roles in DDR to alter sensitivity to radio(chemo)therapy in NSCLC, and discuss the potential clinical applications of ncRNAs as predictors or therapeutic targets for NSCLC (Figure 2).
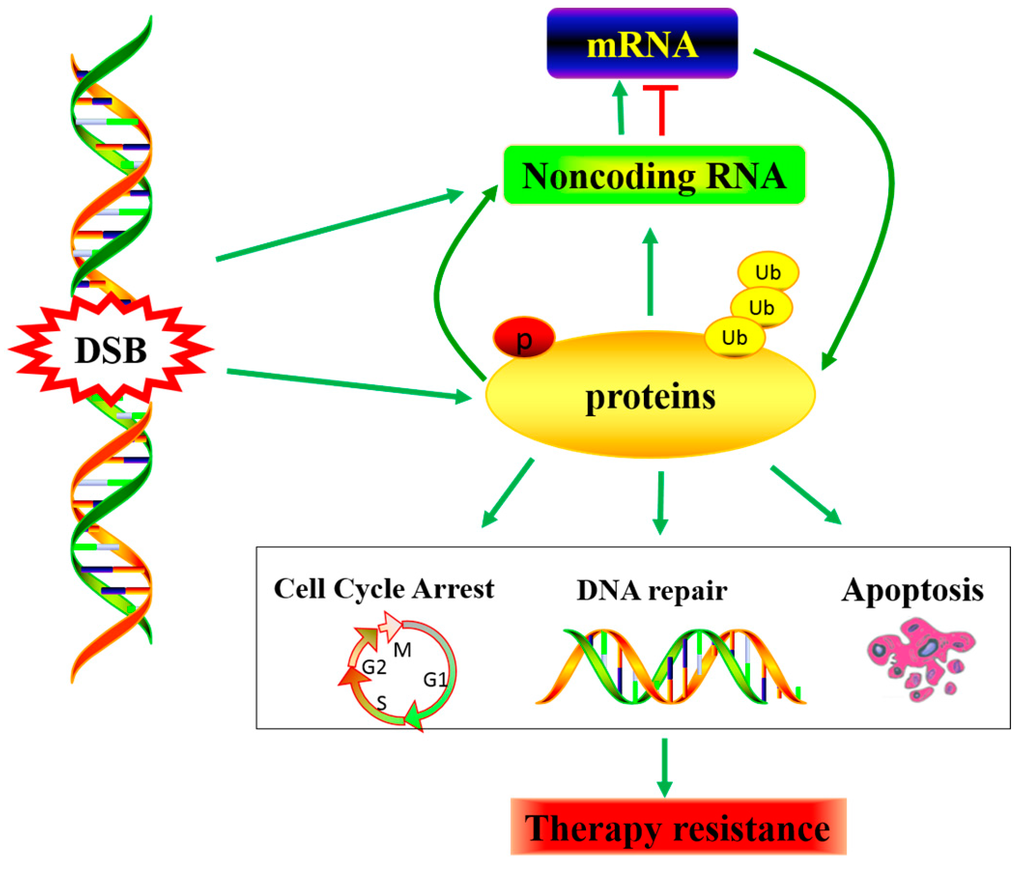
Figure 2.
DNA damage-induced non-coding RNA dys-regulating, Noncoding RNA as a connectivity node between the rapid DNA damage response mediated by protein modifications and the late response mediated by transcriptional regulation. Consequently, affecting the radiation and chemotherapy effect.
4.1. MicroRNAs and Radio(chemo)therapy Response in NSCLC
MicroRNAs (miRNAs) are a family of small non-coding RNAs that range in size from 19 to 25 nucleotides and negatively regulate the expression of their target genes at the post-transcriptional level, leading to a translational repression or mRNA degradation [66]. MicroRNAs are involved in radio(chemo)therapy resistance through DNA repair mechanisms [67,68]. Additionally, mRNAs are required for almost every aspect of cellular responses to DNA damage, including sensing DNA damage, transducing damage signals, repairing damaged DNA, activating cell cycle checkpoints, and inducing apoptosis [68].
With the development of gene chip technology and high throughput technology, gene expression profiles have been widely used to detect differentially expressed genes in DNA damage response. Accumulating evidence suggests that altered expression levels of candidate miRNAs may be involved in the acquisition of tumor cell resistance to radiotherapy and conventional chemotherapy through impacting the efficacy of DNA repair. For example, one study showed that 14 miRNAs were found dysregulated in doxorubicin-resistant A549 cells compared to A549 cells. Of these 14 miRNAs, four (has-mir-1973, 494, 4286 and 29b-3p) showed a 2.99- to 4.44-fold increase in their expression. Similarly, alteration of miRNA profiles was detected in A549 cells upon ionizing radiation [69]. These data suggest that miRNA expression is affected by DNA damage, and they may be involved in the regulation of DNA damage repair. This holds implications for using miRNA expression as resistance markers/targets to improve responses to chemotherapeutic drugs and radiotherapy [70].
DDR regulates miRNA expression via multiple mechanisms. Interestingly, recent studies have shown that a bidirectional regulatory pathway exists between miRNAs and DDR. In response to DNA damage, miRNAs can directly regulate cellular processes involved in DDR by altering their target genes, which provides a feedback regulatory loop for miRNA-mediated DDR [71]. For instance, the tumor suppressor p53 is a key factor that is induced by DNA damage. Various miRNAs are involved in the p53 network [72]. Among these miRNAs, the miR-34 family directly and post-transcriptionally targets p53, and they are activated by ionizing radiation (IR) or capsaicin-induced oxidative DNA damage [73,74]. Further functional analyses showed that the p53/miR-34a regulatory axis might be critical in sensitizing drug-resistant NSCLC cells [74]. MiR-34a can directly bind to the 3′ untranslated region of RAD51 and regulates homologous recombination, and inhibits DSB repair in NSCLC cells after DNA damage [75]. A recent study showed that miR-15b/16-2 can promote p53 phosphorylation and promote the DNA damage response through inhibiting Wip1 (PPM1D) expression following radiation in lung epithelial cells. Additionally, in miR-15b/16-2-overexpressing cells the ATM/Chek1/p53 pathway was activated by IR [76].
At the same time, some miRNAs can regulate the formation of cancer stem cells and the acquisition of the epithelial-mesenchymal transition (EMT), which is critically associated with drug resistance [77]. For instance, a recent study reported that miR-138 could reduce the DNA damage repair capacity of small cell lung cancer (SCLC) cells by targeting H2AX which plays an important role in DNA repair by increasing the local concentration of repair factors near the lesion [78]. Another typical example is CDC6, which is frequently overexpressed in lung cancer and leads to genomic instability and EMT [79]. CDC6 over-expression can give rise to a shortened 3′ UTR which lacks miRNA binding sites, thus increasing its mRNA stability [80]. Moreover, some miRNAs could target drug sensitivity-related genes. For example, miR-630 emerged as a novel modulator of the DNA damage response to cisplatin by targeting ATM kinase and the ATM substrates histone H2AX and p53. Therefore, it induces cell cycle arrest, counteracts early events of the response to DNA damage and results in the greatly diminished sensitivity of cisplatin in A549 NSCLC cells [81]. In addition, miR-138 plays a role in causing resistance to cisplatin in NSCLC by negatively regulating the ERCC1 gene which is involved in the NER pathway [82]. Furthermore, miR-513a-3p can sensitize A549 NSCLC cells to cisplatin by targeting GSTP1 (Glutathione S-Transferase P1), which has been reported to contribute to cisplatin resistance in many studies [83]. These results suggest that miRNAs play important roles in DDR, which may affect the sensitivity to chemotherapy or radiotherapy. Therefore, miRNA-based therapeutics provide an attractive anti-tumor approach for developing new and more effective individualized therapeutic strategies and for predicting the response to different anti-cancer drugs which target DDR and DNA repair. The differentially expressed miRNAs in DDR may become a novel strategy to overcome chemo(radio)therapy resistance for the treatment of NSCLC.
4.2. Long Non-Coding RNAs (LncRNAs) and Radio(chemo)therapy Response in NSCLC
Long non-coding RNAs (lncRNAs) are simply defined as ncRNAs of length greater than 200 nucleotides that are not translated into proteins [84]. However, a further large-scale sequencing study provides evidence that many transcripts thought to be lncRNAs may, in fact, be translated into proteins [85]. Although tremendous advances have been made to elucidate the role of lncRNAs, their functions are largely unknown and difficult to determine. For instance, their effect on genome integrity maintenance is not well understood. LncRNAs can interact with DNA, RNA, and proteins. Recently studies have shown that lncRNAs control the transcription of genes relevant to DNA damage response by four different regulatory models, including signal, decoy, guide, and scaffold [86,87]. It is well known that DNA damage response and repair ability is closely related to the sensitivity to radio(chemo)therapy. Thus, lncRNAs that regulate DDR may become biomarkers for DNA-damaging anti-cancer treatments including radiation and chemotherapy in NSCLC.
In the effort to understand the contribution of lncRNAs to the DNA damage repair pathway, microarray becomes a useful tool. For instance, in order to identify those non-coding transcripts expressed in a p53-dependent manner, Huarte et al. [88] designed an experiment using gene chip technology to prove the contribution of lncRNAs in a mouse model in which p53 was activated by DNA damage. They found that lincRNA-p21 (Trp53cor1) was involved in DNA damage and cell cycle control. LincRNA-p21 serves as a repressor in p53-dependent transcriptional responses, on the contrary, and inhibiting lincRNA-p21 affects the expression of hundreds of gene targets which are normally repressed by p53 [88], and lincRNA-p21 is required to trigger p53-dependent apoptosis following DNA damage by binding to heterogeneous nuclear ribonucleoprotein K (hnRNP-K); then lincRNA p21 is recruited to the promoters of genes and represses their expression in a p53-dependent manner [89]. LincRNA-p21 knockout mice present decreased expression of cdkn1a in cis-regulation to promote target gene expression and to enforce the G1/S checkpoint causing increased proliferation [90]. In addition, Sharma et al. [91] identify that expression of the DNA damage-induced lncRNA (DDSR1) is induced in an ATM-NF-κB pathway—dependent manner by several DNA double-strand break (DSB)-inducing agents. Additionally, DDSR1 can increase DNA repair capacity by homologous recombination (HR) through interacting with BRCA1 and hnRNPUL1 [91]. Thus, these studies suggest that lncRNAs play critical roles in the DDR pathway by regulating gene expression or modulating repair capacity.
Recently, numerous lncRNAs have been shown to correlate with drug resistance in lung cancer in a p53-dependent or independent manner. For instance, Lnc_bc060912 were proved to be regulated by p53 [92], and to suppress cell apoptosis via interacting with the two DNA damage repair proteins PARP1 and NPM1 [92], while MEG3 can regulate cisplatin resistance through controlling the expression of p53 and Bcl-xl in lung adenocarcinoma cells [93]. In addition, the long non-coding RNA HOTAIR contributes to cisplatin resistance in human lung adenocarcinoma cells via interaction with EZH2 which leads to chromosome modifications and the down-regulation of p21 (WAF1/CIP1) expression [94]. In addition, cisplatin resistance in NSCLC has also been associated with other lncRNAs such as AK126698 [95]. Additionally, more lncRNAs involved in DNA damage repair and drug resistance or radio-resistance of lung cancer cells are likely to be discovered in the further studies. Thus, lncRNAs may become biomarkers for improving the sensitivity and specificity of lung cancer treatment in the near future.
5. Conclusions and Perspectives
DNA repair is a vital target to improve NSCLC therapy and to reduce the resistance of tumor cells to chemotherapy or radiotherapy. We have discussed the factors affecting the DNA repair capacity from genetic and epigenetic aspects including single nucleotide polymorphisms and miRNA and lncRNA regulation in the DNA repair pathway. The involvement of these factors in DNA damage repair—induced chemo-resistance is an emerging area and many key questions need to be investigated further: (1) Many positive results have been reported in genetic polymorphism studies but they are always inconsistent. Thus, evidence from large-scale clinical trials is needed, and further biological mechanism research is necessary to understand how and to what degree a genotype will affect a phenotype; (2) Current research regarding the roles of miRNA and lncRNA regulation in the DNA repair pathway is still limited, and thus further study to test their contribution to DNA damage repair is highly needed. Regulation of the DNA repair pathway by ncRNAs offers opportunities to develop potential predictors for radio (chemo) sensitivity and therapeutic targets for NSCLC.
Acknowledgments
This work was supported by the National High Technology Research and Development Program of China (863 Program) (2012AA02A517), National Natural Science Foundation of China (81373490, 81573508, 81573463), and Hunan Provincial Science and Technology Plan of China (2015TP1043).
Author Contributions
Zhao-Qian Liu and Ling Li conceived of and wrote the paper. Tao Zhu, Yuan-Feng Gao, Wei Zheng, Chen-Jing Wang, Ling Xiao, Ma-Sha Huang, Ji-Ye Yin, Hong-Hao Zhou provided a significant contribution to the revision of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DSB | Double-Strand DNA Break |
| DDR | DNA Damage Response |
| NER | Nucleotide Excision Repair |
| NHEJ | Non-Homologous DNA End Joining |
| HR | Homologous Recombination |
| ATR | Ataxia Telangiectasia and Rad3-Related ATR Serine/Threonine Kinase |
| ATM | Ataxia Telangiectasia Mutated |
| DNA-PK | DNA-Dependent Protein Kinase |
| LncRNA | Long Non-Coding RNA |
| miRNA | MicroRNA |
| mRNA | Messenger RNA |
| EIF3A | Eukaryotic Translation Initiation Factor 3 Subunit A |
| RAD51 | RAD51 Recombinase |
| CHK1 | Checkpoint Kinase 1 |
| H2AX | Histone H2A |
| PARP1 | Poly [ADP-Ribose] Polymerase 1 |
| NPM1 | Nucleophosmin (Nucleolar Phosphoprotein B23, Numatrin) |
| EZH2 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit |
| BRCA1 | Breast Cancer 1 |
| NBN | Nibrin |
| SNPs | Single Nucleotide Polymorphisms |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B. DNA damage responses: Mechanisms and roles in human disease: 2007 G.H.A. clowes memorial award lecture. Mol. Cancer Res. 2008, 6, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Akerley, W.; Bazhenova, L.A.; Borghaei, H.; Camidge, D.R.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Dilling, T.J.; et al. NCCN guidelines insights: Non-small cell lung cancer, version 4.2016. J. Natl. Compr. Cancer Netw. 2016, 14, 255–264. [Google Scholar]
- Spira, A.; Ettinger, D.S. Multidisciplinary management of lung cancer. N. Engl. J. Med. 2004, 350, 379–392. [Google Scholar] [PubMed]
- Willers, H.; Held, K.D. Introduction to clinical radiation biology. Hematol. Oncol. Clin. N. Am. 2006, 20, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Cobo, M.; Isla, D.; Massuti, B.; Montes, A.; Sanchez, J.M.; Provencio, M.; Vinolas, N.; Paz-Ares, L.; Lopez-Vivanco, G.; Munoz, M.A.; et al. Customizing cisplatin based on quantitative excision repair cross-complementing 1 mRNA expression: A phase III trial in non-small-cell lung cancer. J. Clin. Oncol. 2007, 25, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, S.M.; Overgaard, J. Patient-to-patient variability in the expression of radiation-induced normal tissue injury. Semin. Radiat. Oncol. 1994, 4, 68–80. [Google Scholar] [CrossRef]
- Burdett, S.; Pignon, J.P.; Tierney, J.; Tribodet, H.; Stewart, L.; le Pechoux, C.; Auperin, A.; le Chevalier, T.; Stephens, R.J.; Arriagada, R.; et al. Adjuvant chemotherapy for resected early-stage non-small cell lung cancer. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Andreassen, C.N.; Alsner, J.; Overgaard, J. Does variability in normal tissue reactions after radiotherapy have a genetic basis—Where and how to look for it? Radiother. Oncol. 2002, 64, 131–140. [Google Scholar] [CrossRef]
- Barnett, G.C.; West, C.M.; Dunning, A.M.; Elliott, R.M.; Coles, C.E.; Pharoah, P.D.; Burnet, N.G. Normal tissue reactions to radiotherapy: Towards tailoring treatment dose by genotype. Nat. Rev. Cancer 2009, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [PubMed]
- Smela, M.E.; Hamm, M.L.; Henderson, P.T.; Harris, C.M.; Harris, T.M.; Essigmann, J.M. The aflatoxin B formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2002, 99, 6655–6660. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Ravanat, J.L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively generated complex DNA damage: Tandem and clustered lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.C.; Miller, J.A. Mechanisms of chemical carcinogenesis. Cancer Am. Cancer Soc. 1981, 47, 1055–1064. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Lips, J.; Kaina, B. DNA double-strand breaks trigger apoptosis in p53-deficient fibroblasts. Carcinogenesis 2001, 22, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Ferretti, C.; Dogliotti, E. The response to DNA damage during differentiation: Pathways and consequences. Mutat. Res. 2013, 743–744, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Taron, M.; Barnadas, A.; Scagliotti, G.; Sarries, C.; Roig, B. Nucleotide excision repair pathways involved in Cisplatin resistance in non-small-cell lung cancer. Cancer Control 2003, 10, 297–305. [Google Scholar] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Kaina, B. Transcriptional regulation of human DNA repair genes following genotoxic stress: Trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013, 41, 8403–8420. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.Y.; Shen, J.; Dong, Z.Z.; Huang, Q.; Zhong, M.Z.; Feng, D.Y.; Zhou, H.H.; Zhang, J.T.; Liu, Z.Q. Effect of eIF3a on response of lung cancer patients to platinum-based chemotherapy by regulating DNA repair. Clin. Cancer Res. 2011, 17, 4600–4609. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, L.; Wang, Y.; Yin, J.; Li, X.; Wang, Z.; Li, H.; Zou, T.; Qian, C.; Li, C.; et al. Effect of transporter and DNA repair gene polymorphisms to lung cancer chemotherapy toxicity. Tumor Biol. 2015, 37, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.Y.; Wang, W.; Ten, H.R.; Chen, J.; Bi, N.; Sadek, R.; Zhang, H.; Lawrence, T.S.; Kong, F.M. Use a survival model to correlate single-nucleotide polymorphisms of DNA repair genes with radiation dose-response in patients with non-small cell lung cancer. Radiother. Oncol. 2015, 117, 77–82. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.; Hoeijmakers, J.H. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Gurubhagavatula, S.; Liu, G.; Park, S.; Neuberg, D.S.; Wain, J.C.; Lynch, T.J.; Su, L.; Christiani, D.C. Excision repair cross-complementation group 1 polymorphism predicts overall survival in advanced non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin. Cancer Res. 2004, 10, 4939–4943. [Google Scholar] [CrossRef] [PubMed]
- Chang-Claude, J.; Popanda, O.; Tan, X.L.; Kropp, S.; Helmbold, I.; von Fournier, D.; Haase, W.; Sautter-Bihl, M.L.; Wenz, F.; Schmezer, P.; et al. Association between polymorphisms in the DNA repair genes, XRCC1, APE1, and XPD and acute side effects of radiotherapy in breast cancer patients. Clin. Cancer Res. 2005, 11, 4802–4809. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.H.; Jang, B.G.; Kim, D.W.; Chung, D.H.; Kim, Y.T.; Jheon, S.; Sung, S.W.; Kim, J.H. The prognostic significance of ERCC1, BRCA1, XRCC1, and betaIII-tubulin expression in patients with non-small cell lung cancer treated by platinum- and taxane-based neoadjuvant chemotherapy and surgical resection. Lung Cancer 2010, 68, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.A.; Crawford, E.L.; Warner, K.A.; Elkhairi, F.; Khuder, S.A.; Willey, J.C. ABCC5, ERCC2, XPA and XRCC1 transcript abundance levels correlate with cisplatin chemoresistance in non-small cell lung cancer cell lines. Mol. Cancer 2005, 4, 18. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bosken, C.H.; Wei, Q.; Amos, C.I.; Spitz, M.R. An analysis of DNA repair as a determinant of survival in patients with non-small-cell lung cancer. J. Natl. Cancer Inst. 2002, 94, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.E.; Yin, M.; Dong, Q.; Stewart, D.J.; Merriman, K.W.; Amos, C.I.; Spitz, M.R.; Wei, Q. DNA repair capacity in peripheral lymphocytes predicts survival of patients with non-small-cell lung cancer treated with first-line platinum-based chemotherapy. J. Clin. Oncol. 2011, 29, 4121–4128. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, J.; Zhou, Y.; Ying, J.; Zou, H.; Guo, S.; Wang, L.; Zhao, N.; Hu, J.; Lu, D.; et al. Predictive value of XRCC1 gene polymorphisms on platinum-based chemotherapy in advanced non-small cell lung cancer patients: A systematic review and meta-analysis. Clin. Cancer Res. 2012, 18, 3972–3981. [Google Scholar] [CrossRef] [PubMed]
- Lunn, R.M.; Langlois, R.G.; Hsieh, L.L.; Thompson, C.L.; Bell, D.A. XRCC1 polymorphisms: Effects on aflatoxin B1-DNA adducts and glycophorin A variant frequency. Cancer Res. 1999, 59, 2557–2561. [Google Scholar] [PubMed]
- Kelsey, C.R.; Jackson, I.L.; Langdon, S.; Owzar, K.; Hubbs, J.; Vujaskovic, Z.; Das, S.; Marks, L.B. Analysis of single nucleotide polymorphisms and radiation sensitivity of the lung assessed with an objective radiologic endpoin. Clin. Lung Cancer 2013, 14, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Su, T.; Zhao, L.; Tan, X.; Chang, W.; Zhang, H.; Cao, G. Associations of polymorphisms in DNA repair genes and MDR1 gene with chemotherapy response and survival of non-small cell lung cancer. PLoS ONE 2014, 9, e99843. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, Q.W.; Shi, G.M.; Wang, L.R. XRCC1 Arg399Gln and clinical outcome of platinum-based treatment for advanced non-small cell lung cancer: A meta-analysis in 17 studies. J. Zhejiang Univ. Sci. B 2012, 13, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Li, Z.; Zhang, S.; Xiong, Y.; Cun, Y.; Qian, C.; Li, M.; Ren, T.; Xia, L.; Cheng, Y.; et al. Association of DNA base excision repair genes (OGG1, APE1 and XRCC1) polymorphisms with outcome to platinum-based chemotherapy in advanced nonsmall-cell lung cancer patients. Int. J. Cancer 2014, 135, 2687–2296. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Li, J.; Hu, R.; Jiao, Y.; Han, Y.; Weng, Q. Predictive assessment in pharmacogenetics of XRCC1 gene on clinical outcomes of advanced lung cancer patients treated with platinum-based chemotherapy. Sci. Rep. 2015, 5, 16482. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, F.; Sun, N.; Shukui, Q.; Baoan, C.; Jifeng, F.; Lu, C.; Zuhong, L.; Hongyan, C.; YuanDong, C.; et al. Polymorphisms in XRCC1 and XPG and response to platinum-based chemotherapy in advanced non-small cell lung cancer patients. Lung Cancer 2009, 65, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Brenneman, M.; Meyne, J.; Oshimura, M.; Goodwin, E.H.; Chen, D.J. The XRCC2 and XRCC3 repair genes are required for chromosome stability in mammalian cells. Mutat. Res. 1999, 434, 75–88. [Google Scholar] [CrossRef]
- Yin, M.; Liao, Z.; Huang, Y.J.; Liu, Z.; Yuan, X.; Gomez, D.; Wang, L.E.; Wei, Q. Polymorphisms of homologous recombination genes and clinical outcomes of non-small cell lung cancer patients treated with definitive radiotherapy. PLoS ONE 2011, 6, e20055. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Xu, L.; Yang, X.; Ding, X.; Hu, J.; Jiang, F.; Xu, L.; Yin, R. XRCC3 Thr241Met is associated with response to platinum-based chemotherapy but not survival in advanced non-small cell lung cancer. PLoS ONE 2013, 8, e77005. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Liao, Z.; Liu, Z.; Wang, L.E.; O’Reilly, M.; Gomez, D.; Li, M.; Komaki, R.; Wei, Q. Genetic variants of the nonhomologous end joining gene LIG4 and severe radiation pneumonitis in nonsmall cell lung cancer patients treated with definitive radiotherapy. Cancer Am. Cancer Soc. 2012, 118, 528–535. [Google Scholar]
- Arriagada, R.; Bergman, B.; Dunant, A.; le Chevalier, T.; Pignon, J.P.; Vansteenkiste, J. Cisplatin-based adjuvant chemotherapy in patients with completely resected non-small-cell lung cancer. N. Engl. J. Med. 2004, 350, 351–360. [Google Scholar] [PubMed]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; Andre, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shi, W.; Wu, Z.; Zhang, G.; Yang, B.O.; Jiao, S. Prognostic significance of the mRNA expression of ERCC1, RRM1, TUBB3 and TYMS genes in patients with non-small cell lung cancer. Exp. Ther. Med. 2015, 10, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Hu, Z.; Shu, Y.; Pan, S.; Chen, W.; Wang, Y.; Hu, L.; Jiang, Y.; Dai, J.; Ma, H.; et al. Potentially functional polymorphisms in DNA repair genes and non-small-cell lung cancer survival: A pathway-based analysis. Mol. Carcinog. 2012, 51, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.Z.; Zhan, P.; Shi, M.Q.; Shi, Y.; Qian, Q.; Yu, L.K.; Song, Y. Predictive value of ERCC1 and XPD polymorphism in patients with advanced non-small cell lung cancer receiving platinum-based chemotherapy: A systematic review and meta-analysis. Med. Oncol. 2011, 28, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Zhang, C.; Yang, X.; Zhu, H.; Yang, B.; Cai, J.; Cheng, H.; Ma, J.; Lu, J.; Zhan, L.; et al. Polymorphisms in XPD gene could predict clinical outcome of platinum-based chemotherapy for non-small cell lung cancer patients: A meta-analysis of 24 studies. PLoS ONE 2013, 8, e79864. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, I.; Salazar, J.; Majem, M.; Pallares, C.; Del, R.E.; Paez, D.; Baiget, M.; Barnadas, A. Pharmacogenetics of the DNA repair pathways in advanced non-small cell lung cancer patients treated with platinum-based chemotherapy. Cancer Lett. 2014, 353, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, L.; Bi, N.; Ji, W.; Tan, W.; Zhao, L.; Yu, D.; Wu, C.; Wang, L.; Lin, D. Association of P53 and ATM polymorphisms with risk of radiation-induced pneumonitis in lung cancer patients treated with radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, M.; Bi, N.; Fang, M.; Sun, T.; Ji, W.; Tan, W.; Zhao, L.; Yu, D.; Lin, D.; et al. ATM polymorphisms are associated with risk of radiation-induced pneumonitis. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Liao, Z.; Liu, Z.; Xu, T.; Wang, Q.; Liu, H.; Komaki, R.; Gomez, D.; Wang, L.E.; Wei, Q. ATM polymorphisms predict severe radiation pneumonitis in patients with non-small cell lung cancer treated with definitive radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Ren, Y.; Li, X.; Zhang, X.; Zhou, B. Effect of single nucleotide polymorphism Rs189037 in ATM gene on risk of lung cancer in Chinese: A case-control study. PLoS ONE 2014, 9, e115845. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.Z.; Zhao, Y.Q. XPG polymorphisms are associated with prognosis of advanced non-small cell lung cancer treated with platinum-based doublet chemotherapy. Genet. Mol. Res. 2015, 14, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Birrell, G.; Chen, P.; Kozlov, S.; Scott, S.; Gueven, N. ATM signaling and genomic stability in response to DNA damage. Mutat. Res. 2005, 569, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Bartkova, J.; Kotsinas, A.; Pateras, I.S.; Liontos, M.; Velimezi, G.; Kosar, M.; Liloglou, T.; Trougakos, I.P.; Dyrskjot, L.; et al. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ. 2013, 20, 1485–1497. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, T.; Stewart, G.S.; Byrd, P.; Fegan, C.; Moss, P.A.; Taylor, A.M. ATM mutations in sporadic lymphoid tumours. Leuk. Lymphoma 2002, 43, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Gatti, R.A. MicroRNAs: New players in the DNA damage response. J. Mol. Cell. Biol. 2011, 3, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lu, X.; Cao, Y. MicroRNA and signal transduction pathways in tumor radiation response. Cell Signal. 2013, 25, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Cha, H.J.; Lee, E.M.; Lee, S.J.; Seo, S.K.; Jin, H.O.; Park, I.C.; Jin, Y.W.; An, S. Alteration of miRNA profiles by ionizing radiation in A549 human non-small cell lung cancer cells. Int. J. Oncol. 2009, 35, 81–86. [Google Scholar] [PubMed]
- El-Awady, R.A.; Hersi, F.; Al-Tunaiji, H.; Saleh, E.M.; Abdel-Wahab, A.H.; Al, H.A.; Suhail, M.; El-Serafi, A.; Al-Tel, T. Epigenetics and miRNA as predictive markers and targets for lung cancer chemotherapy. Cancer Biol. Ther. 2015, 16, 1056–1070. [Google Scholar] [CrossRef] [PubMed]
- Okada, N.; Lin, C.P.; Ribeiro, M.C.; Biton, A.; Lai, G.; He, X.; Bu, P.; Vogel, H.; Jablons, D.M.; Keller, A.C.; et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014, 28, 438–450. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Mert, U.; Ozgur, E.; Tiryakioglu, D.; Dalay, N.; Gezer, U. Induction of p53-inducible microRNA miR-34 by γ radiation and bleomycin are different. Front. Genet. 2012, 3, 220. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Mazumdar, M.; Mukherjee, S.; Bhattacharjee, P.; Adhikary, A.; Manna, A.; Chakraborty, S.; Khan, P.; Sen, A.; Das, T. Restoration of p53/miR-34a regulatory axis decreases survival advantage and ensures Bax-dependent apoptosis of non-small cell lung carcinoma cells. FEBS Lett. 2014, 588, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Valdecanas, D.; Niknam, S.; Peltier, H.J.; Diao, L.; Giri, U.; Komaki, R.; Calin, G.A.; Gomez, D.R.; Chang, J.Y.; et al. In vivo delivery of miR-34a sensitizes lung tumors to radiation through RAD51 regulation. Mol. Ther. Nucleic Acids 2015, 4, e270. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Lovat, F.; Romano, G.; Calore, F.; Acunzo, M.; Bell, E.H.; Nana-Sinkam, P. miR-15b/16–2 regulates factors that promote p53 phosphorylation and augments the DNA damage response following radiation in the lung. J. Biol. Chem. 2014, 289, 26406–26416. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, F.H.; Li, Y.; Wang, Z.; Kong, D.; Ali, S. Implication of microRNAs in drug resistance for designing novel cancer therapy. Drug Resist. Updat. 2010, 13, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Luo, J.; Liu, Z.; Zhou, R.; Luo, H. MicroRNA-138 regulates DNA damage response in small cell lung cancer cells by directly targeting H2AX. Cancer Investig. 2015, 33, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Sideridou, M.; Zakopoulou, R.; Evangelou, K.; Liontos, M.; Kotsinas, A.; Rampakakis, E.; Gagos, S.; Kahata, K.; Grabusic, K.; Gkouskou, K.; et al. Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. J. Cell Biol. 2011, 195, 1123–1140. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, T.G.; Komseli, E.S.; Papaioannou, M.; Vougas, K.; Polyzos, A.; Myrianthopoulos, V.; Mikros, E.; Trougakos, I.P.; Thanos, D.; Branzei, D.; et al. Exploring and exploiting the systemic effects of deregulated replication licensing. Semin. Cancer Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Morselli, E.; Vitale, I.; Kepp, O.; Senovilla, L.; Criollo, A.; Servant, N.; Paccard, C.; Hupe, P.; Robert, T.; et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010, 70, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhong, M.; Liu, W.; Li, J.; Huang, J.; Zheng, L. Alterations of microRNAs in cisplatin-resistant human non-small cell lung cancer cells (A549/DDP). Exp. Lung Res. 2011, 37, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, J.; Xing, R.; Tie, Y.; Fu, H.; Zheng, X.; Yu, B. miR-513a-3p sensitizes human lung adenocarcinoma cells to chemotherapy by targeting GSTP1. Lung Cancer 2012, 77, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Perkel, J.M. Visiting “noncodarnia”. Biotechniques 2013, 54, 303–304. [Google Scholar]
- Smith, J.E.; Alvarez-Dominguez, J.R.; Kline, N.; Huynh, N.J.; Geisler, S.; Hu, W.; Coller, J.; Baker, K.E. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep. 2014, 7, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Meng, X.M.; Huang, C.; Wu, B.M.; Zhang, L.; Lv, X.W.; Li, J. Long noncoding RNAs: Novel insights into hepatocelluar carcinoma. Cancer Lett. 2014, 344, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.R.; Messenger, Z.J.; Tam, H.W.; Phillips, S.L.; Recio, L.; Smart, R.C. Long noncoding RNA lincRNA-p21 is the major mediator of UVB-induced and p53-dependent apoptosis in keratinocytes. Cell Death Dis. 2015, 6, e1700. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Zamudio, J.R.; Jong, R.M.; Soukup, D.; Resnick, R.; Sarma, K.; Ward, A.J.; Raj, A.; Lee, J.T.; Sharp, P.A.; et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol. Cell 2014, 54, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Khurana, S.; Kubben, N.; Abdelmohsen, K.; Oberdoerffer, P.; Gorospe, M.; Misteli, T. A BRCA1-interacting lncRNA regulates homologous recombination. EMBO Rep. 2015, 16, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Sun, Y.; Wei, G.; Luo, J.; Yang, X.; Liu, W.; Guo, M.; Chen, R. Functional characterization of long noncoding RNA Lnc_bc060912 in human lung carcinoma cells. Biochemistry 2015, 54, 2895–2902. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wan, L.; Lu, K.; Sun, M.; Pan, X.; Zhang, P.; Lu, B.; Liu, G.; Wang, Z. The long noncoding RNA MEG3 contributes to Cisplatin resistance of human lung adenocarcinoma. PLoS ONE 2015, 10, e0114586. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, M.; Lu, K.; Liu, J.; Zhang, M.; Wu, W.; De, W.; Wang, Z.; Wang, R. The long noncoding RNA HOTAIR contributes to cisplatin resistance of human lung adenocarcinoma cells via downregualtion of p21(WAF1/CIP1) expression. PLoS ONE 2013, 8, e77293. [Google Scholar]
- Yang, Y.; Li, H.; Hou, S.; Hu, B.; Liu, J.; Wang, J. The noncoding RNA expression profile and the effect of lncRNA AK126698 on cisplatin resistance in non-small-cell lung cancer cell. PLoS ONE 2013, 8, e65309. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).