Old and New Biological Therapies for Psoriasis

Abstract
:1. Introduction
2. Definition of Biological Therapy
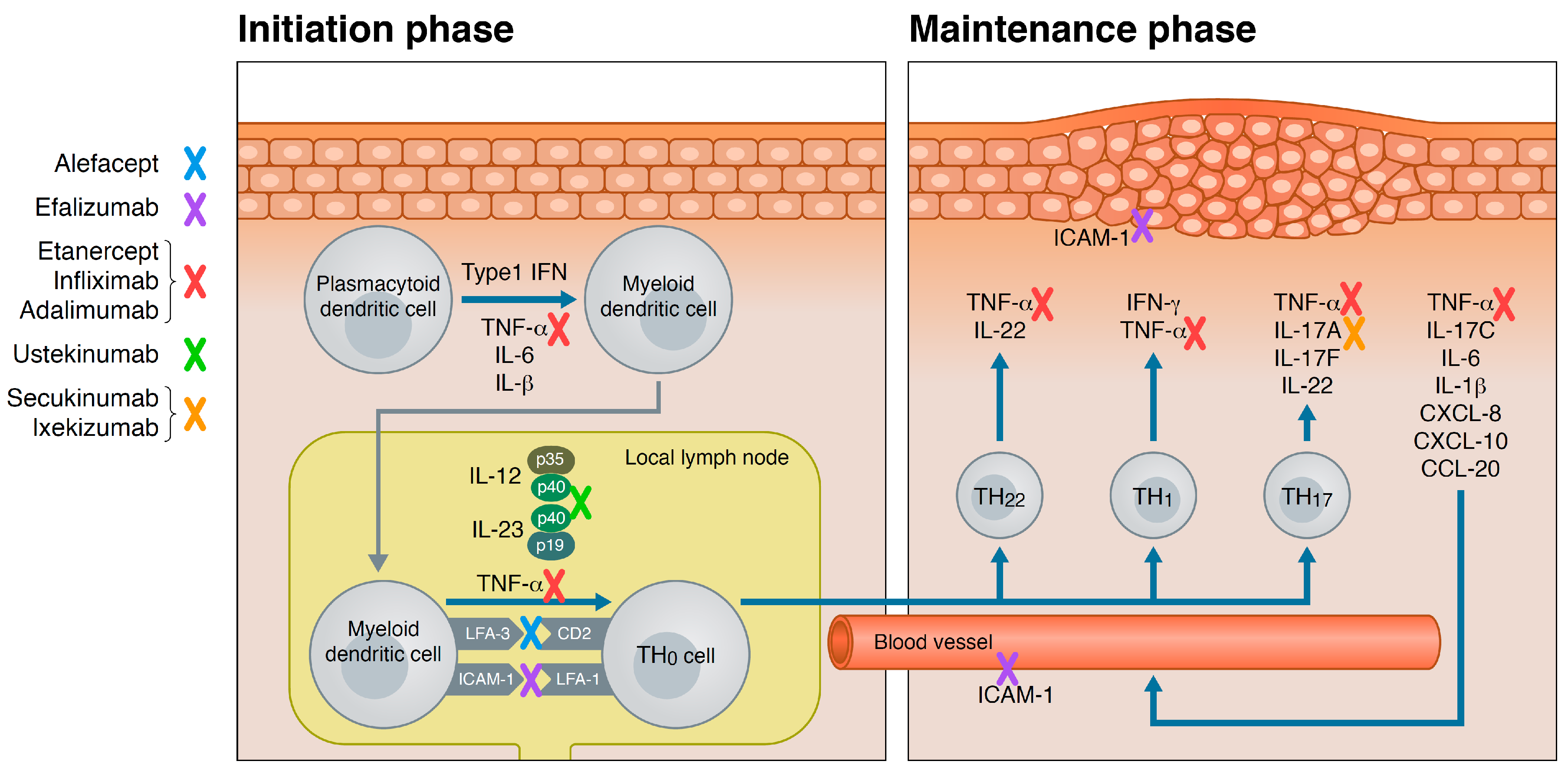
3. Psoriasis Pathogenesis
4. Biologics for Psoriasis
4.1. T-Cell Targeted Biologics
4.1.1. Alefacept
4.1.2. Efalizumab
4.2. Tumor-Necrosis-Factor (TNF)-α Inhibitors
4.2.1. Etanercept
4.2.2. Infliximab
4.2.3. Adalimumab
4.3. IL-12/IL-23 Inhibitor
Ustekinumab
4.4. IL-17 Inhibitors
4.4.1. Secukinumab
4.4.2. Ixekizumab
4.4.3. Brodalumab
4.5. Upcoming Biologics
5. Biosimilars
6. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| PsA | Psoriasis arthritis |
| PASI | Psoriasis area severity index |
| TNF | Tumor necrosis factor |
| IL | Interleukin |
| DNA | Deoxyribonucleic acid |
| IFN | Interferon |
| CD | Cluster of differentiation |
| CXCL | Chemokine (C–X–C motif) ligand |
| CCL | Chemokine (C–C motif) ligand |
| FDA | Food and Drug Administration |
| PGA | Physician Global Assessment |
| LFA | Lymphocyte function–associated antigen |
| Ig | Immunoglobulin |
| APC | Antigen presenting cell |
| I.M. | Intra-muscular |
| I.V. | Intra-venous |
| ICAM | Intercellular Adhesion Molecule |
| PCL | Progressive multifocal leukoencephalopathy |
| ADA | Anti-drug-antibodies |
| TB | Tuberculosis |
References
- Bo, K.; Thoresen, M.; Dalgard, F. Smokers report more psoriasis, but not atopic dermatitis or hand eczema: Results from a norwegian population survey among adults. Dermatology 2008, 216, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.F.; Wang, T.S.; Hung, S.T.; Tsai, P.I.; Schenkel, B.; Zhang, M.; Tang, C.H. Epidemiology and comorbidities of psoriasis patients in a national database in taiwan. J. Dermatol. Sci. 2011, 63, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Madland, T.M.; Apalset, E.M.; Johannessen, A.E.; Rossebo, B.; Brun, J.G. Prevalence, disease manifestations, and treatment of psoriatic arthritis in western norway. J. Rheumatol. 2005, 32, 1918–1922. [Google Scholar] [PubMed]
- Zachariae, H.; Zachariae, R.; Blomqvist, K.; Davidsson, S.; Molin, L.; Mork, C.; Sigurgeirsson, B. Quality of life and prevalence of arthritis reported by 5795 members of the nordic psoriasis associations. Data from the nordic quality of life study. Acta Derm. Venereol. 2002, 82, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Persson, P.G.; Leijonmarck, C.E.; Bernell, O.; Hellers, G.; Ahlbom, A. Risk indicators for inflammatory bowel disease. Int. J. Epidemiol. 1993, 22, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Boffetta, P.; Gridley, G.; Lindelof, B. Cancer risk in a population-based cohort of patients hospitalized for psoriasis in sweden. J. Investig. Dermatol. 2001, 117, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Hjuler, K.F.; Bottcher, M.; Vestergaard, C.; Deleuran, M.; Raaby, L.; Botker, H.E.; Iversen, L.; Kragballe, K. Increased prevalence of coronary artery disease in severe psoriasis and severe atopic dermatitis. Am. J. Med. 2015, 128, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Hjuler, K.F.; Gormsen, L.C.; Vendelbo, M.H.; Egeberg, A.; Nielsen, J.; Iversen, L. Increased global arterial and subcutaneous adipose tissue inflammation in patients with moderate-to-severe psoriasis. Br. J. Dermatol. 2017, 176, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; Azfar, R.S.; Shin, D.B.; Neimanns, A.L.; Troxel, A.B.; Gelfand, J.M. Patients with severe psoriasis are at increased risk of cardiovascular mortality: Cohort study using the general practice research database. Eur. Heart J. 2010, 31, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B.; Gelfand, J.M. Prevalence of cardiovascular risk factors in patients with psoriasis. J. Am. Acad. Dermatol. 2006, 55, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B. Risk of myocardial infarction in patients with psoriasis. JAMA 2006, 296, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Abuabara, K.; Azfar, R.S.; Shin, D.B.; Neimann, A.L.; Troxel, A.B.; Gelfand, J.M. Cause-specific mortality in patients with severe psoriasis: A population-based cohort study in the uk. Br. J. Dermatol. 2010, 163, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Saurat, J.H.; Stingl, G.; Dubertret, L.; Papp, K.; Langley, R.G.; Ortonne, J.P.; Unnebrink, K.; Kaul, M.; Camez, A.; Investigators, C.S. Efficacy and safety results from the randomized controlled comparative study of adalimumab vs. Methotrexate vs. Placebo in patients with psoriasis (champion). Br. J. Dermatol. 2008, 158, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.B.; Mrowietz, U.; von Kiedrowski, R.; Niesmann, J.; Wilsmann-Theis, D.; Ghoreschi, K.; Zschocke, I.; Falk, T.M.; Blodorn-Schlicht, N.; Reich, K. An intensified dosing schedule of subcutaneous methotrexate in patients with moderate to severe plaque-type psoriasis (metop): A 52 week, multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 528–537. [Google Scholar] [CrossRef]
- Dauden, E.; Puig, L.; Ferrandiz, C.; Sanchez-Carazo, J.L.; Hernanz-Hermosa, J.M.; Spanish Psoriasis Group of the Spanish Academy of Dermatology and Venereology. Consensus document on the evaluation and treatment of moderate-to-severe psoriasis: Psoriasis group of the spanish academy of dermatology and venereology. J. Eur. Acad. Dermatol. Venereol. 2016, 30 (Suppl. 2), 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mueller, W.; Herrmann, B. Cyclosporin a for psoriasis. N. Engl. J. Med. 1979, 301, 555. [Google Scholar] [PubMed]
- Wrone-Smith, T.; Nickoloff, B.J. Dermal injection of immunocytes induces psoriasis. J. Clin. Investig. 1996, 98, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. What Are “Biologics” Questions and Answers. Available online: http://www.webcitation.org/6rGBDZ9br (accessed on 16 June 2017).
- Torres, T.; Filipe, P. Small molecules in the treatment of psoriasis. Drug Dev. Res. 2015, 76, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Morrow, T.; Felcone, L.H. Defining the difference: What makes biologics unique. Biotechnol. Healthc. 2004, 1, 24–29. [Google Scholar] [PubMed]
- World Health Organization. International Nonproprietary Names (inn) for Biological and Biotechnological Substances. Available online: http://www.webcitation.org/6rGAZvnd2 (accessed on 16 June 2017).
- Di Cesare, A.; Di Meglio, P.; Nestle, F.O. The il-23/th17 axis in the immunopathogenesis of psoriasis. J. Investig. Dermatol. 2009, 129, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.G.; Wang, T.; Zheng, J.; et al. Pivotal role of dermal il-17-producing gammadelta t cells in skin inflammation. Immunity 2011, 35, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Mahil, S.K.; Capon, F.; Barker, J.N. Update on psoriasis immunopathogenesis and targeted immunotherapy. Semin. Immunopathol. 2016, 38, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Ortega, C.; Fernandez, S.; Carrillo, J.M.; Romero, P.; Molina, I.J.; Moreno, J.C.; Santamaria, M. Il-17-producing CD8(+) t lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete th17-related cytokines. J. Leukoc. Biol. 2009, 86, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.; Fritz, Y.; Dawes, S.M.; Diaconu, D.; Al-Attar, P.M.; Guzman, A.M.; Chen, C.S.; Fu, W.; Gudjonsson, J.E.; McCormick, T.S.; et al. Keratinocyte overexpression of il-17c promotes psoriasiform skin inflammation. J. Immunol. 2013, 190, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.; Reich, K.; Lebwohl, M.; van de Kerkhof, P.; Paul, C.; Menter, A.; Cameron, G.S.; Erickson, J.; Zhang, L.; Secrest, R.J.; et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (uncover-2 and uncover-3): Results from two phase 3 randomised trials. Lancet 2015, 386, 541–551. [Google Scholar] [CrossRef]
- Puig, L. Pasi90 response: The new standard in therapeutic efficacy for psoriasis. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Brimhall, A.K.; King, L.N.; Licciardone, J.C.; Jacobe, H.; Menter, A. Safety and efficacy of alefacept, efalizumab, etanercept and infliximab in treating moderate to severe plaque psoriasis: A meta-analysis of randomized controlled trials. Br. J. Dermatol. 2008, 159, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Jenneck, C.; Novak, N. The safety and efficacy of alefacept in the treatment of chronic plaque psoriasis. Ther. Clin. Risk Manag. 2007, 3, 411–420. [Google Scholar] [PubMed]
- Krueger, G.G. The remittive effects of alefacept. J. Cutan. Med. Surg. 2004, 8 (Suppl. 2), 10–13. [Google Scholar] [CrossRef] [PubMed]
- Krueger, G.G.; Papp, K.A.; Stough, D.B.; Loven, K.H.; Gulliver, W.P.; Ellis, C.N.; Alefacept Clinical Study Group. A randomized, double-blind, placebo-controlled phase iii study evaluating efficacy and tolerability of 2 courses of alefacept in patients with chronic plaque psoriasis. J. Am. Acad. Dermatol. 2002, 47, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Christophers, E.; Langley, R.; Ortonne, J.P.; Roberts, J.; Griffiths, C.E.; Alefacept Clinical Study Group. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch. Dermatol. 2003, 139, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Al-Suwaidan, S.N.; Feldman, S.R. Clearance is not a realistic expectation of psoriasis treatment. J. Am. Acad. Dermatol. 2000, 42, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.S.; Gustafson, C.J.; Sandoval, L.F.; Davis, S.A.; Feldman, S.R. Cost effectiveness of biologic therapies for plaque psoriasis. Am. J. Clin. Dermatol. 2013, 14, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Jullien, D.; Prinz, J.C.; Langley, R.G.B.; Caro, I.; Dummer, W.; Joshi, A.; Dedrick, R.; Natta, P. T-cell modulation for the treatment of chronic plaque psoriasis with efalizumab (raptiva (tm)): Mechanisms of action. Dermatology 2004, 208, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, M.P. Efalizumab in the treatment of psoriasis: Mode of action, clinical indications, efficacy, and safety. Clin. Dermatol. 2008, 26, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Dubertret, L.; Sterry, W.; Bos, J.D.; Chimenti, S.; Shumack, S.; Larsen, C.G.; Shear, N.H.; Papp, K.A.; CLEAR Multinational Study Group. Clinical experience acquired with the efalizumab (raptiva) (clear) trial in patients with moderate-to-severe plaque psoriasis: Results from a phase iii international randomized, placebo-controlled trial. Br. J. Dermatol. 2006, 155, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Gordon, K.; Carey, W.; Hamilton, T.; Glazer, S.; Caro, I.; Li, N.; Gulliver, W. Efficacy and safety observed during 24 weeks of efalizumab therapy in patients with moderate to severe plaque psoriasis. Arch. Dermatol. 2005, 141, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.; Menter, A.; Hamilton, T.; Caro, I.; Xing, B.; Gottlieb, A.B. Efalizumab: Results of a 3-year continuous dosing study for the long-term control of psoriasis. Br. J. Dermatol. 2008, 158, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Seminara, N.M.; Gelfand, J.M. Assessing long-term drug safety: Lessons (re) learned from raptiva. Semin. Cutan. Med. Surg. 2010, 29, 16–19. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Fda Statement on the Voluntary Withdrawal of Raptiva from the U.S. Market. Available online: http://www.webcitation.org/6rGBTfcbt (accessed on 16 June 2017).
- Mease, P.J.; Goffe, B.S.; Metz, J.; VanderStoep, A.; Finck, B.; Burge, D.J. Etanercept in the treatment of psoriatic arthritis and psoriasis: A randomised trial. Lancet 2000, 356, 385–390. [Google Scholar] [CrossRef]
- Mease, P.J.; Kivitz, A.J.; Burch, F.X.; Siegel, E.L.; Cohen, S.B.; Ory, P.; Salonen, D.; Rubenstein, J.; Sharp, J.T.; Tsuji, W. Etanercept treatment of psoriatic arthritis: Safety, efficacy, and effect on disease progression. Arthritis Rheumatol. 2004, 50, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Prnewswire. Fda Approves Expanded Use of Enbrel® (Etanercept) to Treat Children with Chronic Moderate-to-Severe Plaque Psoriasis. Available online: http://www.webcitation.org/6rGBpHvS0 (accessed on 16 June 2017).
- Tyring, S.; Gottlieb, A.; Papp, K.; Gordon, K.; Leonardi, C.; Wang, A.; Lalla, D.; Woolley, M.; Jahreis, A.; Zitnik, R.; et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: Double-blind placebo-controlled randomised phase iii trial. Lancet 2006, 367, 29–35. [Google Scholar] [CrossRef]
- Leonardi, C.L.; Powers, J.L.; Matheson, R.T.; Goffe, B.S.; Zitnik, R.; Wang, A.; Gottlieb, A.B.; Etanercept Psoriasis Study Group. Etanercept as monotherapy in patients with psoriasis. N. Engl. J. Med. 2003, 349, 2014–2022. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Tyring, S.; Lahfa, M.; Prinz, J.; Griffiths, C.E.M.; Nakanishi, A.M.; Zitnik, R.; van de Kerkhof, P.C.M.; Grp, E.P.S. A global phase iii randomized controlled trial of etanercept in psoriasis: Safety, efficacy, and effect of dose reduction. Br. J. Dermatol. 2005, 152, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Tyring, S.; Gordon, K.B.; Poulin, Y.; Langley, R.G.; Gottlieb, A.B.; Dunn, M.; Jahreis, A. Long-term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch. Dermatol. 2007, 143, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Bachelez, H.; van de Kerkhof, P.C.; Strohal, R.; Kubanov, A.; Valenzuela, F.; Lee, J.H.; Yakusevich, V.; Chimenti, S.; Papacharalambous, J.; Proulx, J.; et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: A phase 3 randomised non-inferiority trial. Lancet 2015, 386, 552–561. [Google Scholar] [CrossRef]
- Gall, J.S.; Kalb, R.E. Infliximab for the treatment of plaque psoriasis. Biologics 2008, 2, 115–124. [Google Scholar] [PubMed]
- Lucka, T.C.; Pathirana, D.; Sammain, A.; Bachmann, F.; Rosumeck, S.; Erdmann, R.; Schmitt, J.; Orawa, H.; Rzany, B.; Nast, A. Efficacy of systemic therapies for moderate-to-severe psoriasis: A systematic review and meta-analysis of long-term treatment. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Feldman, S.R.; Weinstein, G.D.; Papp, K.; Evans, R.; Guzzo, C.; Li, S.; Dooley, L.T.; Arnold, C.; Gottlieb, A.B. A randomized comparison of continuous vs. Intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J. Am. Acad. Dermatol. 2007, 56, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Nestle, F.O.; Papp, K.; Ortonne, J.P.; Evans, R.; Guzzo, C.; Li, S.; Dooley, L.T.; Griffiths, C.E.M.; Investigators, E.S. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: A phase iii, multicentre, double-blind trial. Lancet 2005, 366, 1367–1374. [Google Scholar] [CrossRef]
- Yang, H.Z.; Wang, K.; Jin, H.Z.; Gao, T.W.; Xiao, S.X.; Xu, J.H.; Wang, B.X.; Zhang, F.R.; Li, C.Y.; Liu, X.M.; et al. Infliximab monotherapy for chinese patients with moderate to severe plaque psoriasis: A randomized, double-blind, placebo-controlled multicenter trial. Chin. Med. J. (Engl.) 2012, 125, 1845–1851. [Google Scholar] [PubMed]
- Torii, H.; Nakagawa, H.; Japanese Infliximab Study Ivestigators. Infliximab monotherapy in japanese patients with moderate-to-severe plaque psoriasis and psoriatic arthritis. A randomized, double-blind, placebo-controlled multicenter trial. J. Dermatol. Sci. 2010, 59, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Spertino, J.; Lopez-Ferrer, A.; Vilarrasa, E.; Puig, L. Long-term study of infliximab for psoriasis in daily practice: Drug survival depends on combined treatment, obesity and infusion reactions. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.B.; Evans, R.; Li, S.; Dooley, L.T.; Guzzo, C.A.; Baker, D.; Bala, M.; Marano, C.W.; Menter, A. Infliximab induction therapy for patients with severe plaque-type psoriasis: A randomized, double-blind, placebo-controlled trial. J. Am. Acad. Dermatol. 2004, 51, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, L.; Ron, Y.; Kivity, S.; Ben-Horin, S.; Israeli, E.; Fraser, G.M.; Dotan, I.; Chowers, Y.; Confino-Cohen, R.; Weiss, B. Infliximab-related infusion reactions: Systematic review. J. Crohn’s Colitis 2015, 9, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, J.; Fu, L.; Dong, S.; Ge, Y.; Zhang, J.; Huang, B.; Wang, Q.; Wang, Z. Effectiveness and risk associated with infliximab alone and in combination with immunosuppressors for crohn’s disease: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 4846–4854. [Google Scholar] [PubMed]
- Mease, P.J.; Gladman, D.D.; Ritchlin, C.T.; Ruderman, E.M.; Steinfeld, S.D.; Choy, E.H.; Sharp, J.T.; Ory, P.A.; Perdok, R.J.; Weinberg, M.A.; et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: Results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheumatol. 2005, 52, 3279–3289. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Tyring, S.K.; Gordon, K.; Kimball, A.B.; Leonardi, C.L.; Langley, R.G.; Strober, B.E.; Kaul, M.; Gu, Y.; Okun, M.; et al. Adalimumab therapy for moderate to severe psoriasis: A randomized, controlled phase iii trial. J. Am. Acad. Dermatol. 2008, 58, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.; Papp, K.; Poulin, Y.; Gu, Y.H.; Rozzo, S.; Sasso, E.H. Long-term efficacy and safety of adalimumab in patients with moderate to severe psoriasis treated continuously over 3 years: Results from an open-label extension study for patients from reveal. J. Am. Acad. Dermatol. 2012, 66, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Asahina, A.; Nakagawa, H.; Etoh, T.; Ohtsuki, M.; Grp, A.M.-S. Adalimumab in japanese patients with moderate to severe chronic plaque psoriasis: Efficacy and safety results from a phase ii/iii randomized controlled study. J. Dermatol. 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Gu, J.; Zheng, J.; Zheng, M.; Wang, G.; Xi, L.Y.; Hao, F.; Liu, X.M.; Sun, Q.N.; Wang, Y.; et al. Efficacy and safety of adalimumab in chinese patients with moderate-to-severe plaque psoriasis: Results from a phase 3, randomized, placebo-controlled, double-blind study. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Nast, A.; Gisondi, P.; Ormerod, A.D.; Saiag, P.; Smith, C.; Spuls, P.I.; Arenberger, P.; Bachelez, H.; Barker, J.; Dauden, E.; et al. European S3-guidelines on the systemic treatment of psoriasis vulgaris—Update 2015—Short version—EDF in cooperation with EADV and IPC. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2277–2294. [Google Scholar] [CrossRef] [PubMed]
- Raaby, L.; Zachariae, C.; Ostensen, M.; Heickendorff, L.; Thielsen, P.; Gronbaek, H.; Skov, L.; Kyvsgaard, N.; Madsen, J.T.; Heidenheim, M.; et al. Methotrexate use and monitoring in patients with psoriasis: A consensus report based on a danish expert meeting. Acta Derm. Venereol. 2017, 97, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Thaci, D.; Ortonne, J.P.; Chimenti, S.; Ghislain, P.D.; Arenberger, P.; Kragballe, K.; Saurat, J.H.; Khemis, A.; Sprogel, P.; Esslinger, H.U.; et al. A phase iiib, multicentre, randomized, double-blind, vehicle-controlled study of the efficacy and safety of adalimumab with and without calcipotriol/betamethasone topical treatment in patients with moderate to severe psoriasis: The believe study. Br. J. Dermatol. 2010, 163, 402–411. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.C.; Thio, H.B.; de Kort, W.J.; Opmeer, B.C.; van der Stok, H.M.; de Jong, E.M.; Horvath, B.; Busschbach, J.J.; Nijsten, T.E.; Spuls, P.I. A prospective randomized controlled trial comparing infliximab and etanercept in patients with moderate-to-severe chronic plaque-type psoriasis: The psoriasis infliximab vs. Etanercept comparison evaluation (piece) study. Br. J. Dermatol. 2017, 176, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Zhang, Z.; Wozel, G.; Meurer, M.; Kirch, W. Efficacy and tolerability of biologic and nonbiologic systemic treatments for moderate-to-severe psoriasis: Meta-analysis of randomized controlled trials. Br. J. Dermatol. 2008, 159, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.; Armstrong, A.W. Anti-drug antibodies in psoriasis: A critical evaluation of clinical significance and impact on treatment response. Expert. Rev. Clin. Immunol. 2013, 9, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Kui, R.; Gal, B.; Gaal, M.; Kiss, M.; Kemeny, L.; Gyulai, R. Presence of antidrug antibodies correlates inversely with the plasma tumor necrosis factor (tnf)-alpha level and the efficacy of tnf-inhibitor therapy in psoriasis. J. Dermatol. 2016, 43, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Prescribing Information, Enbrel (Etanercept). Available online: http://www.webcitation.org/6rGCfL3tR (accessed on 16 June 2017).
- Prescribing Information, Remicade (Infliximab). Available online: http://www.webcitation.org/6rGDV99FL (accessed on 16 June 2017).
- Prescribing Information, Humira (Adalimumab). Available online: http://www.webcitation.org/6rGCyzYce (accessed on 16 June 2017).
- Harris, J.; Keane, J. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin. Exp. Immunol. 2010, 161, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ergun, T.; Seckin, D.; Baskan Bulbul, E.; Onsun, N.; Ozgen, Z.; Unalan, P.; Alpsoy, E.; Karakurt, S. The risk of tuberculosis in patients with psoriasis treated with anti-tumor necrosis factor agents. Int. J. Dermatol. 2015, 54, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Report from The Danish Council for the Use of Expensive Hospital Medicines. Available online: http://www.regioner.dk/media/1843/bgn-pso-123452401591.pdf (accesseed on 31 October 2017).
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.H.; Li, S.; Dooley, L.T.; Gordon, K.B.; Investigators, P.S. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (phoenix 1). Lancet 2008, 371, 1665–1674. [Google Scholar] [CrossRef]
- Papp, K.A.; Langley, R.G.; Lebwohl, M.; Krueger, G.G.; Szapary, P.; Yeilding, N.; Guzzo, C.; Hsu, M.C.; Wang, Y.H.; Li, S.; et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (phoenix 2). Lancet 2008, 371, 1675–1684. [Google Scholar] [CrossRef]
- Igarashi, A.; Kato, T.; Kato, M.; Song, M.; Nakagawa, H.; Grp, J.U.S. Efficacy and safety of ustekinumab in japanese patients with moderate-to-severe plaque-type psoriasis: Long-term results from a phase 2/3 clinical trial. J. Dermatol. 2012, 39, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.F.; Ho, J.C.; Song, M.; Szapary, P.; Guzzo, C.; Shen, Y.K.; Li, S.; Kim, K.J.; Kim, T.Y.; Choi, J.H.; et al. Efficacy and safety of ustekinumab for the treatment of moderate-to-severe psoriasis: A phase iii, randomized, placebo-controlled trial in taiwanese and korean patients (pearl). J. Dermatol. Sci. 2011, 63, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zheng, M.; Song, M.; Shen, Y.K.; Chan, D.; Szapary, P.O.; Wang, B.; Investigators, L. Efficacy and safety of ustekinumab in chinese patients with moderate to severe plaque-type psoriasis: Results from a phase 3 clinical trial (lotus). J. Drugs Dermatol. 2013, 12, 166–174. [Google Scholar] [PubMed]
- Gniadecki, R.; Bang, B.; Bryld, L.E.; Iversen, L.; Lasthein, S.; Skov, L. Comparison of long-term drug survival and safety of biologic agents in patients with psoriasis vulgaris. Br. J. Dermatol. 2015, 172, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Van den Reek, J.M.; Zweegers, J.; Kievit, W.; Otero, M.E.; van Lumig, P.P.; Driessen, R.J.; Ossenkoppele, P.M.; Njoo, M.D.; Mommers, J.M.; Koetsier, M.I.; et al. ‘Happy’ drug survival of adalimumab, etanercept and ustekinumab in psoriasis in daily practice care: Results from the biocapture network. Br. J. Dermatol. 2014, 171, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.B.; Smith, C.H.; Yiu, Z.Z.N.; Ashcroft, D.M.; Barker, J.N.W.N.; Burden, A.D.; Lunt, M.; McElhone, K.; Ormerod, A.D.; Owen, C.M.; et al. Differential drug survival of biologic therapies for the treatment of psoriasis: A prospective observational cohort study from the british association of dermatologists biologic interventions register (badbir). J. Investig. Dermatol. 2015, 135, 2632–2640. [Google Scholar] [CrossRef] [PubMed]
- Clemmensen, A.; Spon, M.; Skov, L.; Zachariae, C.; Gniadecki, R. Responses to ustekinumab in the anti-tnf agent-naive vs. Anti-tnf agent-exposed patients with psoriasis vulgaris. J. Eur. Acad. Dermatol. 2011, 25, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.B.; Papp, K.A.; Wasfi, Y.; Chan, D.; Bissonnette, R.; Sofen, H.; Yeilding, N.; Li, S.; Szapary, P.; Gordon, K.B.; et al. Long-term efficacy of ustekinumab in patients with moderate-to-severe psoriasis treated for up to 5 years in the phoenix 1 study. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.G.; Lebwohl, M.; Krueger, G.G.; Szapary, P.O.; Wasfi, Y.; Chan, D.; Hsu, M.C.; You, Y.; Poulin, Y.; Korman, N.; et al. Long-term efficacy and safety of ustekinumab, with and without dosing adjustment, in patients with moderate-to-severe psoriasis: Results from the phoenix 2 study through 5 years of follow-up. Br. J. Dermatol. 2015, 172, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.; Strober, B.E.; van de Kerkhof, P.; Ho, V.; Fidelus-Gort, R.; Yeilding, N.; Guzzo, C.; Xia, Y.; Zhou, B.; Li, S.; et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N. Engl. J. Med. 2010, 362, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Papp, K.A.; Langley, R.G.; Ho, V.; Kimball, A.B.; Guzzo, C.; Yeilding, N.; Szapary, P.O.; Fakharzadeh, S.; Li, S.; et al. Long-term safety experience of ustekinumab in patients with moderate to severe psoriasis (part ii of ii): Results from analyses of infections and malignancy from pooled phase ii and iii clinical trials. J. Am. Acad. Dermatol. 2012, 66, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Prescribing Information, Stelara (Ustekinumab). Available online: http://www.webcitation.org/6rKr49YYM (accessed on 19 June 2017).
- MacLennan, C.; Fieschi, C.; Lammas, D.A.; Picard, C.; Dorman, S.E.; Sanal, O.; MacLennan, J.M.; Holland, S.M.; Ottenhoff, T.H.M.; Casanova, J.L.; et al. Interleukin (il)-12 and il-23 are key cytokines for immunity against salmonella in humans. J. Infect. Dis. 2004, 190, 1755–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.; Roche, L.; Horgan, M.; Ahmad, K.; Hackett, C.; Ramsay, B. Peritoneal tuberculosis in the setting of ustekinumab treatment for psoriasis. JAAD Case Rep. 2017, 3, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, B.W.; Kavanaugh, A.; Reich, K. Interleukin-17a: A unique pathway in immune-mediated diseases: Psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology 2014, 141, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Prinz, J.C.; Gottlieb, A.B.; Kingo, K.; Sofen, H.; Ruer-Mulard, M.; Singh, V.; Pathan, R.; Papavassilis, C.; Cooper, S.; et al. Secukinumab administration by pre-filled syringe: Efficacy, safety and usability results from a randomized controlled trial in psoriasis (feature). Br. J. Dermatol. 2015, 172, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Lacour, J.P.; Tedremets, L.; Kreutzer, K.; Jazayeri, S.; Adams, S.; Guindon, C.; You, R.; Papavassilis, C.; Grp, J.S. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: A randomized, controlled trial (juncture). J. Eur. Acad. Dermatol. 2015, 29, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Reich, K.; Tsai, T.F.; Tyring, S.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Vender, R.; Hugot, S.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate-to-severe plaque psoriasis up to 1 year: Results from the clear study. J. Am. Acad. Dermatol. 2017, 76, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Thaci, D.; Blauvelt, A.; Reich, K.; Tsai, T.F.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Hugot, S.; You, R.Q.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: Clear, a randomized controlled trial. J. Am. Acad. Dermatol. 2015, 73, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Van de Kerkhof, P.C.; Griffiths, C.E.; Reich, K.; Leonardi, C.L.; Blauvelt, A.; Tsai, T.F.; Gong, Y.; Huang, J.; Papavassilis, C.; Fox, T. Secukinumab long-term safety experience: A pooled analysis of 10 phase ii and iii clinical studies in patients with moderate to severe plaque psoriasis. J. Am. Acad. Dermatol. 2016, 75, 83–98. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administartion. Fda Approves New Psoriasis Drug Taltz. Available online: http://www.webcitation.org/6rGDjK5VG (accessed on 16 June 2017).
- Mease, P.J.; van der Heijde, D.; Ritchlin, C.T.; Okada, M.; Cuchacovich, R.S.; Shuler, C.L.; Lin, C.Y.; Braun, D.K.; Lee, C.H.; Gladman, D.D.; et al. Ixekizumab, an interleukin-17a specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: Results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase iii trial spirit-p1. Ann. Rheum. Dis. 2017, 76, 79–87. [Google Scholar] [PubMed]
- Nash, P.; Kirkham, B.; Okada, M.; Rahman, P.; Combe, B.; Burmester, G.R.; Adams, D.H.; Kerr, L.; Lee, C.; Shuler, C.L.; et al. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: Results from the 24-week randomised, double-blind, placebo-controlled period of the spirit-p2 phase 3 trial. Lancet 2017. [Google Scholar] [CrossRef]
- Gordon, K.B.; Blauvelt, A.; Papp, K.A.; Langley, R.G.; Luger, T.; Ohtsuki, M.; Reich, K.; Amato, D.; Ball, S.G.; Braun, D.K.; et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N. Engl. J. Med. 2016, 375, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Saeki, H.; Nakagawa, H.; Ishii, T.; Morisaki, Y.; Aoki, T.; Berclaz, P.Y.; Heffernan, M. Efficacy and safety of open-label ixekizumab treatment in japanese patients with moderate-to-severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis. J. Eur. Acad. Dermatol. 2015, 29, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Saeki, H.; Nakagawa, H.; Nakajo, K.; Ishii, T.; Morisaki, Y.; Aoki, T.; Cameron, G.S.; Osuntokun, O.O.; Grp, J.I.S. Efficacy and safety of ixekizumab treatment for japanese patients with moderate to severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis: Results from a 52-week, open-label, phase 3 study (uncover-j). J. Dermatol. 2017, 44, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Papp, K.A.; Griffiths, C.E.; Puig, L.; Weisman, J.; Dutronc, Y.; Kerr, L.F.; Ilo, D.; Mallbris, L.; Augustin, M. Efficacy and safety of switching to ixekizumab in etanercept non-responders: A subanalysis from two phase iii randomized clinical trials in moderate-to-severe plaque psoriasis (uncover-2 and -3). Am. J. Clin. Dermatol. 2017, 18, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Warren, R.B.; Langley, R.G.; Merola, J.F.; Kerr, L.N.; Dennehy, E.B.; Shrom, D.; Amato, D.; Okubo, Y.; Reich, K. Efficacy of ixekizumab compared to etanercept and placebo in patients with moderate-to-severe plaque psoriasis and non-pustular palmoplantar involvement: Results from three phase 3 trials (uncover-1, uncover-2 and uncover-3). J. Eur. Acad. Dermatol. Venereol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W.; Lynde, C.W.; McBride, S.R.; Stahle, M.; Edson-Heredia, E.; Zhu, B.; Amato, D.; Nikai, E.; Yang, F.E.; Gordon, K.B. Effect of ixekizumab treatment on work productivity for patients with moderate-to-severe plaque psoriasis: Analysis of results from 3 randomized phase 3 clinical trials. JAMA Dermatol. 2016, 152, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Strober, B.; Leonardi, C.; Papp, K.A.; Mrowietz, U.; Ohtsuki, M.; Bissonnette, R.; Ferris, L.K.; Paul, C.; Lebwohl, M.; Braun, D.K.; et al. Short- and long-term safety outcomes with ixekizumab from 7 clinical trials in psoriasis: Etanercept comparisons and integrated data. J. Am. Acad. Dermatol. 2017, 76, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Structure and signalling in the il-17 receptor family (vol 9, pg 556, 2009). Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.A.; Towne, J.E.; Kricorian, G.; Klekotka, P.; Gudjonsson, J.E.; Krueger, J.G.; Russell, C.B. The emerging role of il-17 in the pathogenesis of psoriasis: Preclinical and clinical findings. J. Investig. Dermatol. 2013, 133, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, S.; Watanabe, H.; Kawaguchi, M.; Suzuki, T.; Matsukura, S.; Homma, T.; Howell, B.G.; Hizawa, N.; Mitsuya, T.; Huang, S.K.; et al. Involvement of il-17f via the induction of il-6 in psoriasis. Arch. Dermatol. Res. 2010, 302, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Kawaguchi, M.; Fujishima, S.; Ogura, M.; Matsukura, S.; Takeuchi, H.; Ohba, M.; Sueki, H.; Kokubu, F.; Hizawa, N.; et al. Functional characterization of il-17f as a selective neutrophil attractant in psoriasis. J. Investig. Dermatol. 2009, 129, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, T.; Ljungberg, C.; Kjellerup, R.; Iversen, L.; Johansen, C. Il-17f regulates psoriasis-associated genes through i kappa b zeta. J. Investig. Dermatol. 2016, 136, S220. [Google Scholar] [CrossRef]
- Johansen, C.; Usher, P.A.; Kjellerup, R.B.; Lundsgaard, D.; Iversen, L.; Kragballe, K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br. J. Dermatol. 2009, 160, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Reich, K.; Paul, C.; Blauvelt, A.; Baran, W.; Bolduc, C.; Toth, D.; Langley, R.G.; Cather, J.; Gottlieb, A.B.; et al. A prospective phase iii, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br. J. Dermatol. 2016, 175, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Niiro, H.; Ootaki, K.; Japanese Brodalumab Study Group. Brodalumab, a human anti-interleukin-17-receptor antibody in the treatment of japanese patients with moderate-to-severe plaque psoriasis: Efficacy and safety results from a phase ii randomized controlled study. J. Dermatol. Sci. 2016, 81, 44–52. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Fda Approves New Psoriasis Drug. Available online: http://www.webcitation.org/6rGE8u2CY (accessed on 16 June 2017).
- European Medicines Agency. Kyntheum. Available online: http://www.webcitation.org/6rGERhJc2 (accessed on 16 June 2017).
- Kyowa Kirin. Lumicef® Approved in Japan. Available online: http://www.webcitation.org/6rGEdu4hs (accessed on 16 June 2017).
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and il-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.Y.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.F.; Blauvelt, A.; Gong, Y.K.; Huang, J.Q.; Fox, T. Secukinumab treatment shows no evidence for reactivation of previous or latent tb infection in subjects with psoriasis: A pooled phase 3 safety analysis. J. Am. Acad. Dermatol. 2015, 72, Ab251. [Google Scholar]
- Forlow, S.B.; Schurr, J.R.; Kolls, J.K.; Bagby, G.J.; Schwarzenberger, P.O.; Ley, K. Increased granulopoiesis through interleukin-17 and granulocyte colony-stimulating factor in leukocyte adhesion molecule-deficient mice. Blood 2001, 98, 3309–3314. [Google Scholar] [CrossRef] [PubMed]
- Prescribing Information, Brodalumab. Available online: http://www.webcitation.org/6rKuHGiye (accesseed on 19 June 2017).
- Kulig, P.; Musiol, S.; Freiberger, S.N.; Schreiner, B.; Gyulveszi, G.; Russo, G.; Pantelyushin, S.; Kishihara, K.; Alessandrini, F.; Kundig, T.; et al. Il-12 protects from psoriasiform skin inflammation. Nat. Commun. 2016, 7, 13466. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.P.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S.; et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N. Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Papp, K.A.; Griffiths, C.E.; Randazzo, B.; Wasfi, Y.; Shen, Y.K.; Li, S.; Kimball, A.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase iii, double-blinded, placebo- and active comparator-controlled voyage 1 trial. J. Am. Acad. Dermatol. 2017, 76, 405–417. [Google Scholar] [PubMed]
- Reich, K.; Armstrong, A.W.; Foley, P.; Song, M.; Wasfi, Y.; Randazzo, B.; Li, S.; Shen, Y.K.; Gordon, K.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase iii, double-blind, placebo- and active comparator-controlled voyage 2 trial. J. Am. Acad. Dermatol. 2017, 76, 418–431. [Google Scholar] [PubMed]
- Weise, M.; Bielsky, M.C.; De Smet, K.; Ehmann, F.; Ekman, N.; Giezen, T.J.; Gravanis, I.; Heim, H.K.; Heinonen, E.; Ho, K.; et al. Biosimilars: What clinicians should know. Blood 2012, 120, 5111–5117. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Puig, L.; Chimenti, S.; Vender, R.; Rajagopalan, M.; Romiti, R.; Skov, L.; Zachariae, C.; Young, H.; Prens, E.; et al. Biosimilars for psoriasis: Clinical studies to determine similarity. Br. J. Dermatol. 2016, 177, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, K.K.; Olsen, I.C.; Goll, G.L.; Lorentzen, M.; Bolstad, N.; Haavardsholm, E.A.; Lundin, K.E.A.; Mork, C.; Jahnsen, J.; Kvien, T.K.; et al. Switching from originator infliximab to biosimilar ct-p13 compared with maintained treatment with originator infliximab (nor-switch): A 52-week, randomised, double-blind, non-inferiority trial. Lancet 2017, 389, 2304–2314. [Google Scholar] [CrossRef]


{kind=link}
| Biologic Drug | Target | Administration | Treatment Algorithm | Stage of Development | Approved for Psoriasis Arthritis | Withdrawn |
|---|---|---|---|---|---|---|
| Alefacept | LFA 1-3 | Intra-muscular | 15 mg once weekly for 12 weeks | Approved 2003 | 2011 | |
| Efalizumab | CD 211a | Subcutaneous | 0.7 mg/kg initial dose, then 1 mg/kg (max 200 mg) once weekly | Approved 2003 | 2009 | |
| Etanercept | TNF 3-α | Subcutaneous | 50 mg twice weekly for 12 weeks, then 50 mg once weekly | Approved 2004 | + | |
| Infliximab | TNF-α | Intra-venous | 5 mg/kg on week 0, 2 and 6, then every 8 weeks | Approved 2006 | + | |
| Adalimumab | TNF-α | Subcutaneous | 80 mg initial dose, then 40 mg every 2 weeks, starting one week after initial dose | Approved 2008 | + | |
| Ustekinumab | IL-12/IL-23 p40 | Subcutaneous | 45 mg (≤100 kg) or 90 mg (>100 kg) on week 0 and 4, then every 12 weeks | Approved 2009 | + | |
| Secukinumab | IL-17A | Subcutaneous | 300 mg on week 0, 1, 2, 3, and 4 followed by 300 mg every 4 weeks | Approved 2015 | + | |
| Ixekizumab | IL-17A | Subcutaneous | 160 mg week 0, then 80 mg week 2, 4, 6, 8, 10, 12, then 80 mg every 4 weeks | Approved 2016 | ||
| Brodalumab | IL-17A receptor | Subcutaneous | 210 mg on week 0, 1, and 2, then every 2 weeks | Approved 2017 |
| Biologic | Type | Target | Stage of Development | www.clinicaltrial.gov |
|---|---|---|---|---|
| Guselkumab | Human Ig 1G1 monoclonal antibody | IL 2-23p19 | Approved | |
| Tildrakizumab | Humanized Ig 1G1 monoclonal antibody | IL-23p19 | Phase III | NCT01722331 NCT01729754 |
| Risankizumab | Humanized IgG1 monoclonal antibody | IL-23p19 | Phase III | NCT03047395 NCT02672852 NCT02684370 NCT02694523 NCT02684357 |
| Certolizumab Pegol | PEGylated Fab’ fragment of a humanized IgG1 monoclonal antibody | TNF-α | Phase III | NCT02326298 NCT02326272 NCT02346240 |
| Bimekizumab | Humanized IgG1 monoclonal antibody | IL-17A and IL-17F | Phase II | NCT03025542 NCT03010527 NCT02905006 |
| Neihulizumab | Humanized monoclonal antibody | CD 3162 on T-cells | Phase II | NCT02223039 NCT01855880 |
| CJM112 | Human monoclonal antibody | IL-17A | Phase II | NCT01828086 |
| Namilumab | Human IgG1 monoclonal antibody | GM-CSF 4 | Phase II | NCT02129777 |
| Mirikizumab | Humanized monoclonal antibody | IL-23p19 | Phase II | NCT02899988 |
| TAB08 | Humanized IgG4 monoclonal antibody | CD28 on T-cells | Phase II | NCT02796053 |
| GSK2831781 | Humanized antibody dependent cell cytotoxicity enhanced monoclonal afucosylated IgG1antibody | Lymphocyte activation gene-3 | Phase I | NCT02195349 |
| T1h | Humanized IgG1 monoclonal antibody | CD6 | Phase I | NCT02649270 |
| MSB0010841 | Nanobody | IL-17A and IL-17F | Phase I | NCT02156466 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rønholt, K.; Iversen, L. Old and New Biological Therapies for Psoriasis. Int. J. Mol. Sci. 2017, 18, 2297. https://doi.org/10.3390/ijms18112297
Rønholt K, Iversen L. Old and New Biological Therapies for Psoriasis. International Journal of Molecular Sciences. 2017; 18(11):2297. https://doi.org/10.3390/ijms18112297
Chicago/Turabian StyleRønholt, Kirsten, and Lars Iversen. 2017. "Old and New Biological Therapies for Psoriasis" International Journal of Molecular Sciences 18, no. 11: 2297. https://doi.org/10.3390/ijms18112297
APA StyleRønholt, K., & Iversen, L. (2017). Old and New Biological Therapies for Psoriasis. International Journal of Molecular Sciences, 18(11), 2297. https://doi.org/10.3390/ijms18112297