Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation




Abstract
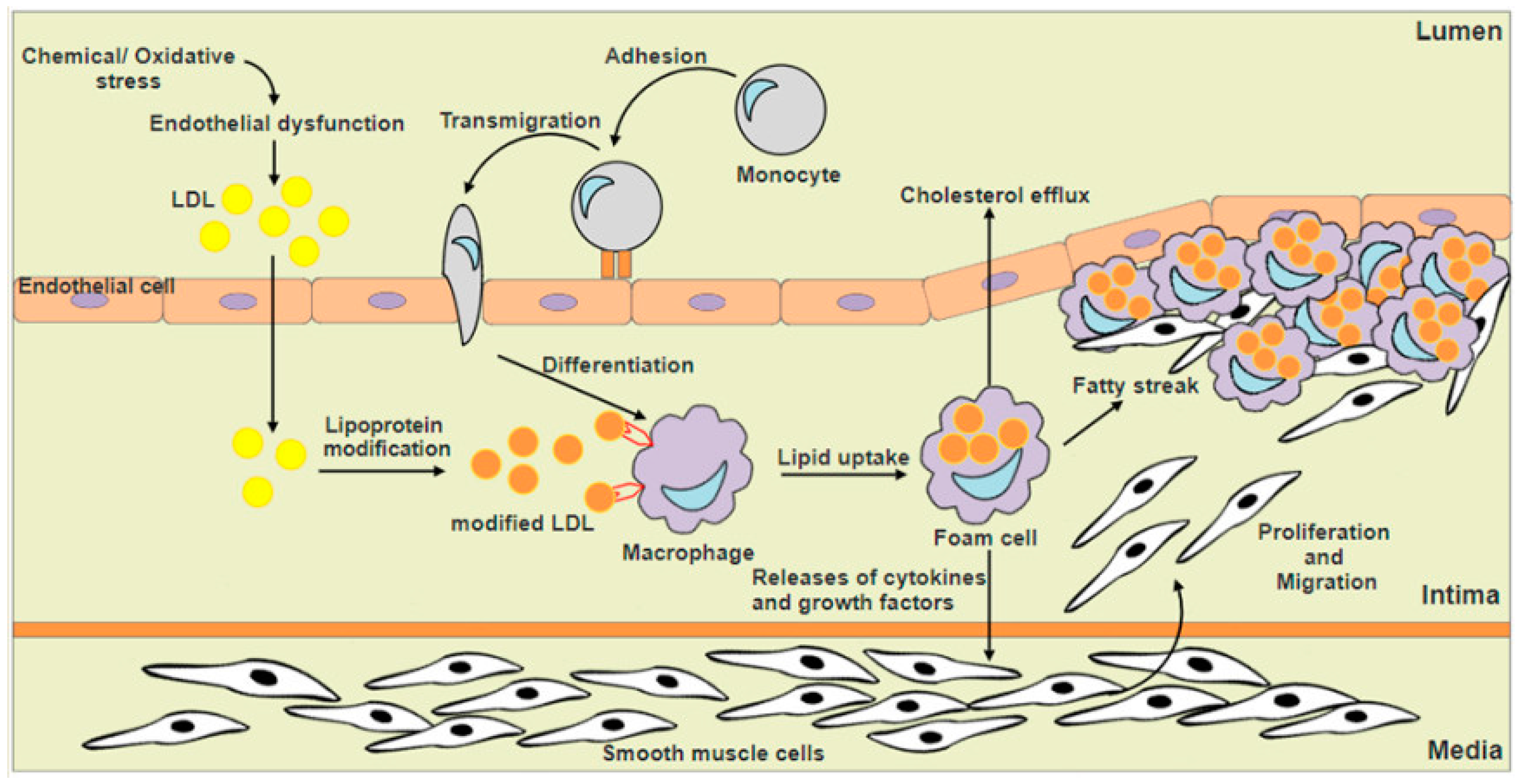
1. Introduction
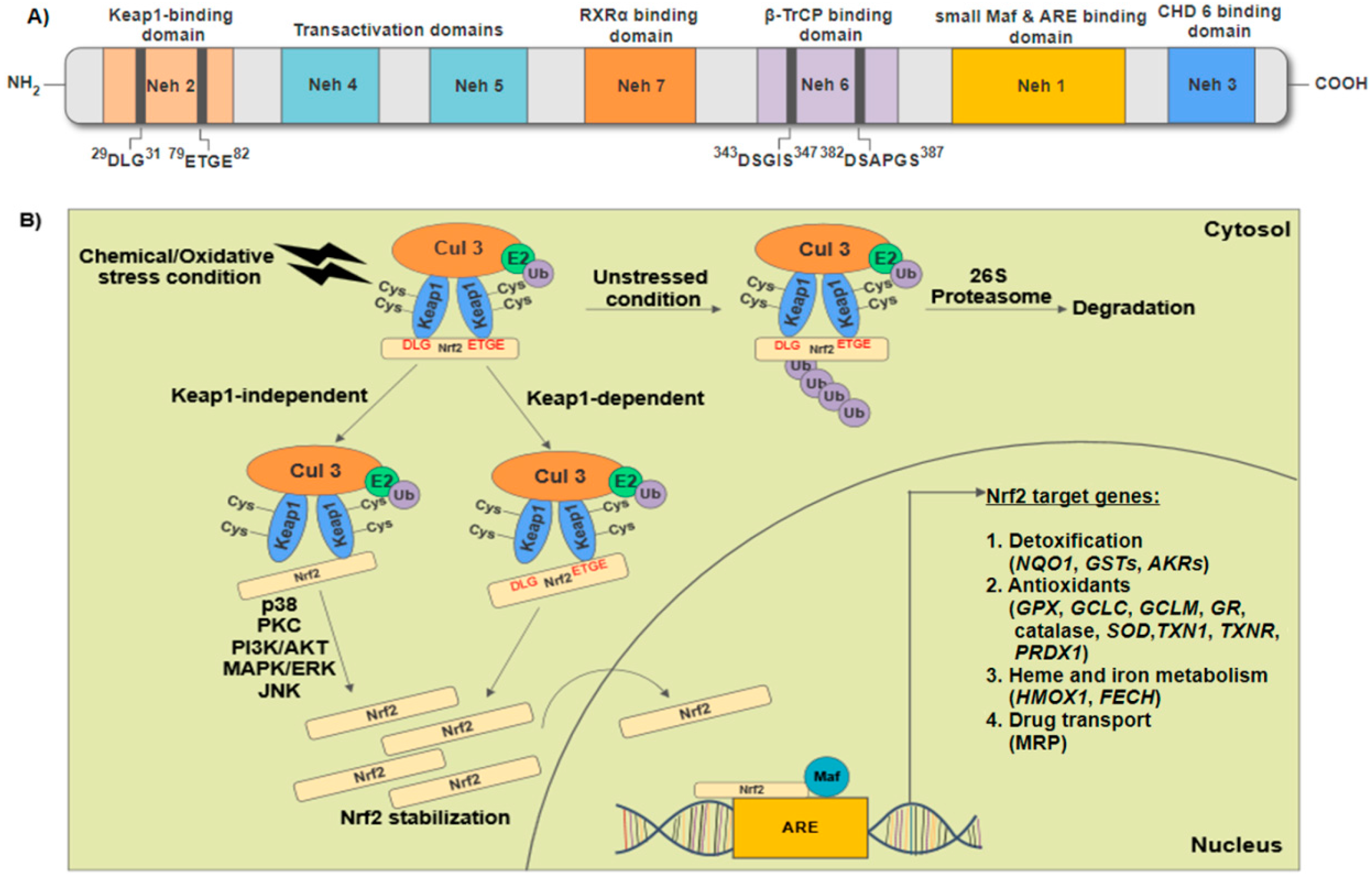
2. Structural Features of Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) and Regulation of Nrf2-Keap1/Antioxidant Response Elements (ARE) Signaling
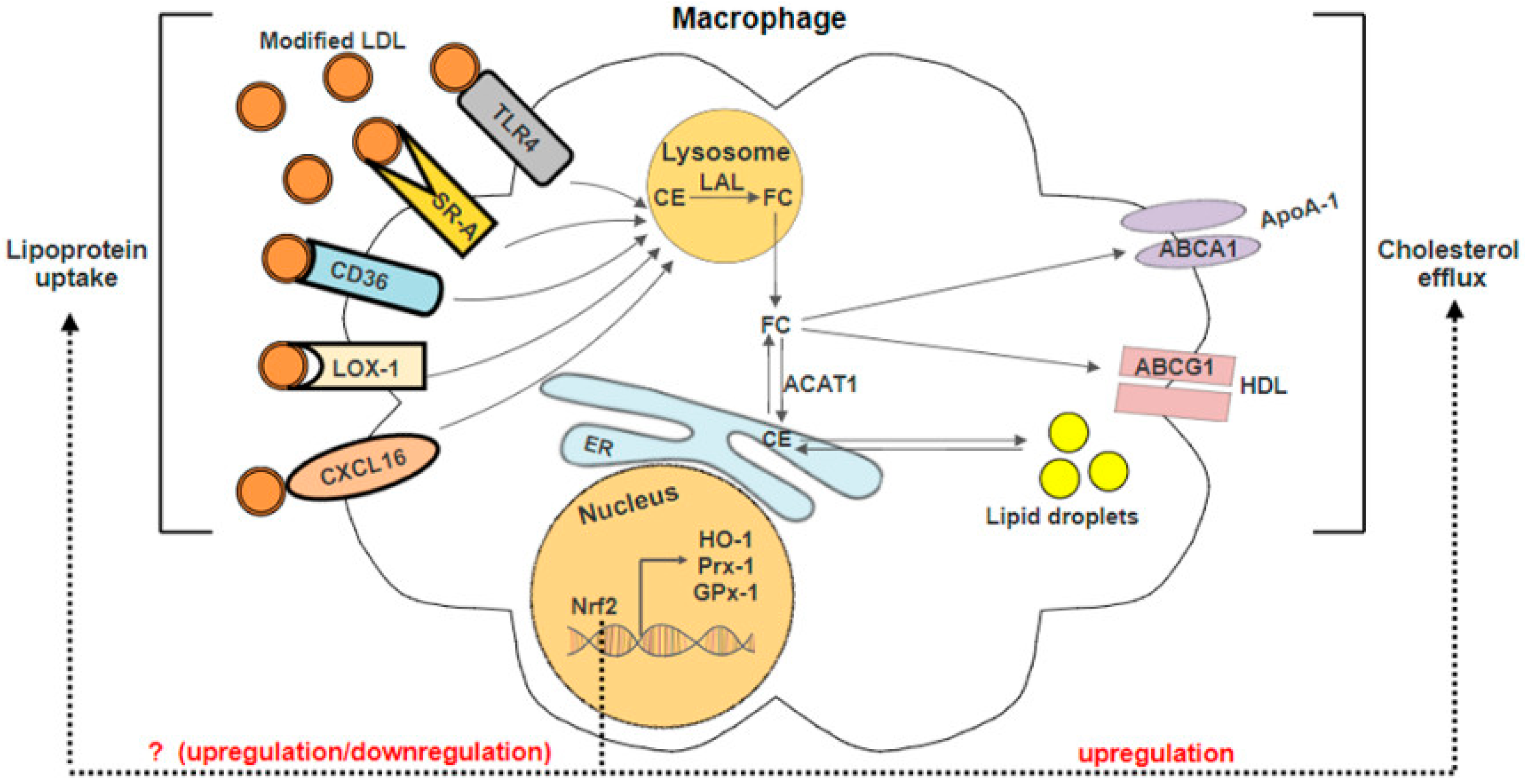
3. Nrf2 and Macrophage Foam Cells Formation
3.1. Nrf2-Regulated Antioxidant Genes
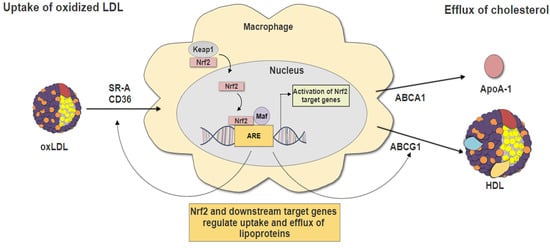
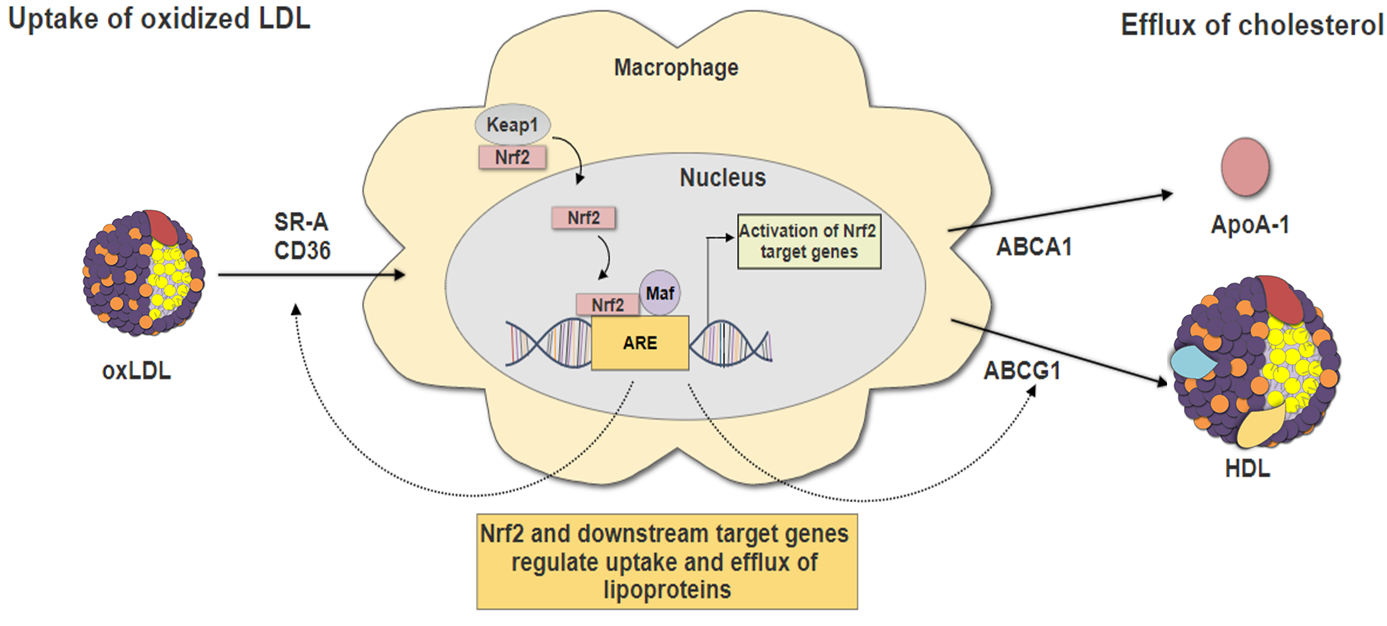
3.2. Nrf2 in Lipid Uptake
3.3. Nrf2 in Cholesterol Efflux
4. Recent Insights of Nrf2 in Macrophage Foam Cells Formation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Health Estimates 2015: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2015. Available online: http://www.who.int/healthinfo/global_burden_disease/estimates/en/index1.html (accessed on 18 June 2017).
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Pashkow, F.J. Oxidative stress and inflammation in heart disease: Do antioxidants have a role in treatment and/or prevention? Int. J. Inflamm. 2011, 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, D.P.; Gotto, A.M. Biological relevance of inflammation and oxidative stress in the pathogenesis of arterial diseases. Am. J. Pathol. 2013, 182, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, M.; Garbin, U.; Agostoni, P.; Fusaro, M.; Pasini, A.F.; Nava, C.; Keta, D.; Turri, M.; Zardini, P.; Vassanelli, C.; et al. Plasma levels of oxidized-low-density lipoproteins are higher in patients with unstable angina and correlated with angiographic coronary complex plaques. Atherosclerosis 2006, 185, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ehara, S.; Ueda, M.; Naruko, T.; Haze, K.; Itoh, A.; Otsuka, M.; Komatsu, R.; Matsuo, T.; Itabe, H.; Takano, T.; et al. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation 2001, 103, 1955–1960. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.J.; Linton, E.F.; Vickers, K.C. The role of lipids and lipoproteins in atherosclerosis. In The Role of Lipids and Lipoproteins in Atherosclerosis; de Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Oishi, Y.; Manabe, I. Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech. Dis. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Howden, R. Nrf2 and cardiovascular defense. Oxid. Med. Cell. Longev. 2013, 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, A.-K.; Inkala, M.; Partanen, M.E.; Lappalainen, J.P.; Kansanen, E.; Mäkinen, P.I.; Heinonen, S.E.; Laitinen, H.M.; Heikkilä, J.; Vatanen, T.; et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc. Res. 2013, 98, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor keap1 functions as an adaptor for cul3-based e3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Nrf2–keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef] [PubMed]
- Zgórzyńska, E.; Dziedzic, B.; Gorzkiewicz, A.; Stulczewski, D.; Bielawska, K.; Su, K.-P.; Walczewska, A. Omega-3 polyunsaturated fatty acids improve the antioxidative defense in rat astrocytes via an Nrf2-dependent mechanism. Pharmacol. Rep. 2017, 69, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Majkova, Z.; Layne, J.; Sunkara, M.; Morris, A.J.; Toborek, M.; Hennig, B. Omega-3 fatty acid oxidation products prevent vascular endothelial cell activation by coplanar polychlorinated biphenyls. Toxicol. Appl. Pharmacol. 2011, 251, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, J.; Sekhar, K.R.; Yin, H.; Yared, N.F.; Schneider, S.N.; Sasi, S.; Dalton, T.P.; Anderson, M.E.; Chan, J.Y.; et al. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between keap1 and cullin3. J. Biol. Chem. 2007, 282, 2529–2537. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2–keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [PubMed]
- Zakkar, M.; Van der Heiden, K.; Luong, L.A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, S.; Lim, J.H.; Lee, C.; Choi, H.C.; Woo, C.H. Laminar flow activation of erk5 protein in vascular endothelium leads to atheroprotective effect via nf-e2-related factor 2 (Nrf2) activation. J. Biol. Chem. 2012, 287, 40722–40731. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K.J.; Lewis, K.N.; Price, N.L.; Chang, J.W.; Perez, E.; Cascajo, M.V.; Tamashiro, K.L.; Poosala, S.; Csiszar, A.; Ungvari, Z.; et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc. Natl. Acad. Sci. USA 2008, 105, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Z.; Chen, H.; Zhou, S.; Zheng, W.; Liu, G.; Wei, Y.; Cai, H.; Liu, D.; Liang, C. Endothelium-specific overexpression of class III deacetylase sirt1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008, 80, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Gao, X.; Wei, W. The crosstalk between sirt1 and keap1/Nrf2/are anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-β1 expressions in rat glomerular mesangial cells. Exp. Cell Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, H.; Zhou, J.S.; Cao, J.F.; Zhou, X.B.; Choi, A.M.K.; Chen, Z.H.; Shen, H.H. Caveolin-1 inhibits expression of antioxidant enzymes through direct interaction with nuclear erythroid 2 p45-related factor-2 (Nrf2). J. Biol. Chem. 2012, 287, 20922–20930. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jia, Z.; Zhang, L.; Yamamoto, M.; Misra, H.P.; Trush, M.A.; Li, Y. Antioxidants and phase 2 enzymes in macrophages: Regulation by Nrf2 signaling and protection against oxidative and electrophilic stress. Exp. Biol. Med. 2008, 233, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Gupte, A.A.; Ji, R.; Ramirez, M.R.; Minze, L.J.; Liu, J.Z.; Arredondo, M.; Ren, Y.; Deng, T.; Wang, J.; et al. Myeloid deletion of nuclear factor erythroid 2-related factor 2 increases atherosclerosis and liver injury. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates apoe-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar] [CrossRef] [PubMed]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. Nf-e2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Ito, K.; Hosoya, T.; Mimura, J.; Maruyama, A.; Noguchi, N.; Yagami, K.-I.; Morito, N.; Takahashi, S.; Maher, J.M.; et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic. Biol. Med. 2012, 53, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Maltese, G.; Psefteli, P.M.; Rizzo, B.; Srivastava, S.; Gnudi, L.; Mann, G.E.; Siow, R.C.M. The anti-ageing hormone klotho induces Nrf2-mediated antioxidant defences in human aortic smooth muscle cells. J. Cell. Mol. Med. 2017, 21, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.-M.; Tzieply, N.; Schmidt, M.V.; von Knethen, A.; Namgaladze, D.; Yamamoto, M.; Brüne, B. Antioxidant signaling via Nrf2 counteracts lipopolysaccharide-mediated inflammatory responses in foam cell macrophages. Free Radic. Biol. Med. 2011, 50, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/are pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of cd36 and stress protein expression in murine macrophages: Activation by oxidatively modified ldl and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, J.; Huang, E.; Gao, S.; Li, H.; Lu, J.; Tian, K.; Little, P.J.; Shen, X.; Xu, S.; et al. Tanshinone iia suppresses cholesterol accumulation in human macrophages: Role of heme oxygenase-1. J. Lipid Res. 2014, 55, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Tang, S.L.; Liu, X.Y.; Zhao, G.J.; Ouyang, X.P.; Lv, Y.C.; He, P.P.; Yao, F.; Chen, W.J.; Tang, Y.Y.; et al. Tertiary-butylhydroquinone upregulates expression of atp-binding cassette transporter a1 via nuclear factor e2-related factor 2/heme oxygenase-1 signaling in thp-1 macrophage-derived foam cells. Circ. J. 2013, 77, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Mo, Z.; Yin, K.; Zhao, G.; Lv, Y.; Ouyang, X.; Jiang, Z.; Fu, Y.; Tang, C. Epigallocatechin-3-gallate prevents tnf-α-induced nf-κb activation thereby upregulating abca1 via the Nrf2/keap1 pathway in macrophage foam cells. Int. J. Mol. Med. 2012, 29, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Ryoo, I.; Choi, H.; Choi, B.; Kim, S.T.; Heo, T.H.; Lee, J.Y.; Park, P.-H.; Kwak, M.K. Nrf2 signaling negatively regulates phorbol-12-myristate-13-acetate (pma)-induced differentiation of human monocytic u937 cells into pro-inflammatory macrophages. PLoS ONE 2015, 10, e0134235. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Rangasamy, T.; Thimmulappa, R.K.; Lee, H.; Osburn, W.O.; Brigelius-Flohé, R.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of gpx in lungs, is regulated by Nrf2. Am. J. Respir. Cell Mol. Biol. 2006, 35, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Jyrkkänen, H.K.; Kansanen, E.; Inkala, M.; Kivelä, A.M.; Hurttila, H.; Heinonen, S.E.; Goldsteins, G.; Jauhiainen, S.; Tiainen, S.; Makkonen, H.; et al. Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ. Res. 2008, 103, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Sugawara, D.; Wang, X.; Suzuki, K.; Itabe, H.; Maruyama, Y.; Lusis, A.J. Heme oxygenase-1 inhibits atherosclerotic lesion formation in LDL-receptor knockout mic. Circ. Res. 2001, 88, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Lee, T.S.; Lee, F.Y.; Pai, R.C.; Chau, L.Y. Expression of heme oxygenase-1 in atherosclerotic lesions. Am. J. Pathol. 1998, 152, 711–720. [Google Scholar] [PubMed]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Sugawara, D.; Goto, J.; Watanabe, Y.; Kawamura, K.; Shiomi, M.; Itabe, H.; Maruyama, Y. Heme oxygenase-1 inhibits atherogenesis in watanabe heritable hyperlipidemic rabbits. Circulation 2001, 104, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Navab, M.; Lusis, A.J. Vasculitis, atherosclerosis, and altered HDL composition in heme-oxygenase-1-knockout mice. Int. J. Hypertens. 2012, 2012, 948203. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.Y.; Yang, Y.S.; Tsai, M.L.; Lee, C.H.; Chang, C.J.; Shiau, A.L.; Wu, C.L. Adenovirus-mediated prothymosin α gene transfer inhibits the development of atherosclerosis in apoe-deficient mice. Int. J. Biol. Sci. 2014, 10, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, B.H.; Chen, L.; An, W. Overexpression of heme oxygenase-1 protects smooth muscle cells against oxidative injury and inhibits cell proliferation. Cell Res. 2002, 12, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Noordeloos, A.M.; Jeney, V.; Soares, M.P.; Moll, F.; Pasterkamp, G.; Serruys, P.W.; Duckers, H.J. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circ. Res. 2009, 119, 3017–3027. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.P.; Kinter, M. Dual role of peroxiredoxin i in macrophage-derived foam cells. J. Biol. Chem. 2006, 281, 27991–28001. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Yoo, J.Y.; Jeong, S.J.; Choi, J.H.; Lee, M.R.; Lee, M.N.; Lee, J.H.; Kim, H.C.; Jo, H.; Yu, D.Y.; et al. Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein e-deficient mice. Circ. Res. 2011, 109, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Kisucka, J.; Chauhan, A.K.; Patten, I.S.; Yesilaltay, A.; Neumann, C.; van Etten, R.A.; Krieger, M.; Wagner, D.D. Peroxiredoxin1 prevents excessive endothelial activation and early atherosclerosis. Circ. Res. 2008, 103, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jeong, S.J.; Oh, G.T. Peroxiredoxin 1 have protective role in the vascular disease by regulating macrophages. Atherosclerosis 2016, 252, e226. [Google Scholar] [CrossRef]
- Jeong, S.J.; Kim, S.; Park, J.G.; Jung, I.; Lee, M.N.; Jeon, S.; Kweon, H.Y.; Yu, D.Y.; Lee, S.H.; Jang, Y.; et al. Prdx1 (peroxiredoxin 1) deficiency reduces cholesterol efflux via impaired macrophage lipophagic flux. Autophagy 2017. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R. Tissue-specific functions of individual glutathione peroxidases. Free Radic. Biol. Med. 1999, 27, 951–965. [Google Scholar] [CrossRef]
- Espinola-Klein, C.; Rupprecht, H.J.; Bickel, C.; Schnabel, R.; Genth-Zotz, S.; Torzewski, M.; Lackner, K.; Munzel, T.; Blankenberg, S. Glutathione peroxidase-1 activity, atherosclerotic burden, and cardiovascular prognosis. Am. J. Cardiol. 2007, 99, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Torzewski, M.; Ochsenhirt, V.; Kleschyov, A.L.; Oelze, M.; Daiber, A.; Li, H.; Rossmann, H.; Tsimikas, S.; Reifenberg, K.; Cheng, F.; et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein e-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.; Stefanovic, N.; Pete, J.; Calkin, A.C.; Giunti, S.; Thallas-Bonke, V.; Jandeleit-Dahm, K.A.; Allen, T.J.; Kola, I.; Cooper, M.E.; et al. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein e-deficient mice. Circulation 2007, 115, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Forgione, M.A.; Cap, A.; Liao, R.; Moldovan, N.I.; Eberhardt, R.T.; Lim, C.C.; Jones, J.; Goldschmidt-Clermont, P.J.; Loscalzo, J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: Abnormalities in vascular and cardiac function and structure. Circ. Res. 2002, 106, 1154–1158. [Google Scholar] [CrossRef]
- Zhang, Y.; Handy, D.E.; Loscalzo, J. Adenosine-dependent induction of glutathione peroxidase 1 in human primary endothelial cells and protection against oxidative stress. Circ. Res. 2005, 96, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger receptors class a-i/ii and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, D.F.; Riazy, M.; Parhar, K.S.; Chen, J.H.; Duronio, V.; Sawamura, T.; Steinbrecher, U.P. Lox-1 augments oxldl uptake by lysopc-stimulated murine macrophages but is not required for OXLDL clearance from plasma. J. Lipid Res. 2009, 50, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class b scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class b scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Marleau, S.; Harb, D.; Bujold, K.; Avallone, R.; Iken, K.; Wang, Y.; Demers, A.; Sirois, M.G.; Febbraio, M.; Silverstein, R.L.; et al. Ep 80317, a ligand of the CD36 scavenger receptor, protects apolipoprotein e-deficient mice from developing atherosclerotic lesions. FASEB J. 2005, 19, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Lian, T.W.; Wang, L.; Lo, Y.H.; Huang, I.J.; Wu, M.J. Fisetin, morin and myricetin attenuate cd36 expression and oxldl uptake in u937-derived macrophages. Biochim. Biophys. Acta 2008, 1781, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Fraser, I.P.; Gordon, S. Murine macrophage scavenger receptor: In vivo expression and function as receptor for macrophage adhesion in lymphoid and non-lymphoid organs. Eur. J. Immunol. 1995, 25, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Naito, M.; Itakura, H.; Ikemoto, S.; Asaoka, H.; Hayakawa, I.; Kanamori, H.; Aburatani, H.; Takaku, F.; Suzuki, H. Human macrophage scavenger receptors: Primary structure, expression, and localization in atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 1990, 87, 9133–9137. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Suzuki, H.; Mori, T.; Matsumoto, A.; Kodama, T.; Takahashi, K. Coexpression of type i and type ii human macrophage scavenger receptors in macrophages of various organs and foam cells in atherosclerotic lesions. Am. J. Pathol. 1992, 141, 591–599. [Google Scholar] [PubMed]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, P.I.; Lappalainen, J.P.; Heinonen, S.E.; Leppänen, P.; Lähteenvuo, M.T.; Aarnio, J.V.; Heikkilä, J.; Turunen, M.P.; Ylä-Herttuala, S. Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc. Res. 2010, 88, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, S.; Vanegas, D.; Kennedy, D.J.; Guy, E.; Nimako, G.; Morton, R.E.; Febbraio, M. Absence of cd36 protects against atherosclerosis in apoe knock-out mice with no additional protection provided by absence of scavenger receptor a i/ii. Cardiovasc. Res. 2008, 78, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Manning-Tobin, J.J.; Moore, K.J.; Seimon, T.A.; Bell, S.A.; Sharuk, M.; Alvarez-Leite, J.I.; de Winther, M.P.J.; Tabas, I.; Freeman, M.W. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.L.; Chen, J.; Hermonat, P.L.; Romeo, F.; Novelli, G. Lectin-like, oxidized low-density lipoprotein receptor-1 (lox-1): A critical player in the development of atherosclerosis and related disorders. Cardiovasc. Res. 2006, 69, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Navarra, T.; Del Turco, S.; Berti, S.; Basta, G. The lectin-like oxidized low-density lipoprotein receptor-1 and its soluble form: Cardiovascular implications. J. Atheroscler. Thromb. 2010, 17, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, X.; Zhao, Y.; Chen, B.; Li, X.; Qi, R. Ginkgolide b reduces lox-1 expression by inhibiting akt phosphorylation and increasing sirt1 expression in oxidized LDL-stimulated human umbilical vein endothelial cells. PLoS ONE 2013, 8, e74769. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sawamura, T.; Renier, G. Glucose enhances human macrophage LOX-1 expression: Role for lox-1 in glucose-induced macrophage foam cell formation. Circ. Res. 2004, 94, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, J.; Taguchi, R.; Yamamoto, A.; Murakami, K. Palmitic acid enhances lectin-like oxidized ldl receptor (lox-1) expression and promotes uptake of oxidized ldl in macrophage cells. Atherosclerosis 2010, 209, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, A.; Rozenberg, I.; Paneni, F.; Camici, G.G.; Shi, Y.; Doerries, C.; Sledzinska, A.; Mocharla, P.; Breitenstein, A.; Lohmann, C.; et al. Endothelial overexpression of lox-1 increases plaque formation and promotes atherosclerosis in vivo. Eur. Heart J. 2014, 35, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Ranalletta, M.; Wang, N.; Han, S.; Terasaka, N.; Li, R.; Welch, C.; Tall, A.R. Combined deficiency of abca1 and abcg1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Investig. 2007, 117, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Zhu, X.; Gao, C.; Shewale, S.; Cao, Q.; Liu, M.; Boudyguina, E.; Gebre, A.K.; Wilson, M.D.; Brown, A.L.; et al. Myeloid cell-specific abca1 deletion has minimal impact on atherogenesis in atherogenic diet-fed LDL receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Fan, B.; Ma, A.; Shaul, P.W.; Zhu, H. Inhibition of abca1 protein degradation promotes HDL cholesterol efflux capacity and rct and reduces atherosclerosis in mice. J. Lipid Res. 2015, 56, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Van Eck, M.; Singaraja, R.R.; Ye, D.; Hildebrand, R.B.; James, E.R.; Hayden, M.R.; van Berkel, T.J.C. Macrophage ATP-binding cassette transporter a1 overexpression inhibits atherosclerotic lesion progression in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Lammers, B.; Out, R.; Hildebrand, R.B.; Quinn, C.M.; Williamson, D.; Hoekstra, M.; Meurs, I.; van Berkel, T.J.C.; Jessup, W.; van Eck, M. Independent protective roles for macrophage abcg1 and apoe in the atherosclerotic lesion development. Atherosclerosis 2009, 205, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Meurs, I.; Lammers, B.; Zhao, Y.; Out, R.; Hildebrand, R.B.; Hoekstra, M.; van Berkel, T.J.C.; van Eck, M. The effect of abcg1 deficiency on atherosclerotic lesion development in ldl receptor knockout mice depends on the stage of atherogenesis. Atherosclerosis 2012, 221, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.W.; Wang, Q.; Ma, X.; Li, X.X.; Liu, X.H.; Xiao, J.; Liao, D.F.; Xiang, J.; Tang, C.K. Tgf-beta1 up-regulates expression of abca1, abcg1 and sr-bi through liver x receptor α signaling pathway in thp-1 macrophage-derived foam cells. J. Atheroscler. Thromb. 2010, 17, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C.W.; Wagner, E.M.; Basso, F.; Amar, M.J.; Freeman, L.A.; Shamburek, R.D.; Knapper, C.L.; Syed, J.; Wu, J.; Vaisman, B.L.; et al. Abca1 overexpression in the liver of ldlr-ko mice leads to accumulation of pro-atherogenic lipoproteins and enhanced atherosclerosis. J. Biol. Chem. 2006, 281, 33053–33065. [Google Scholar] [CrossRef] [PubMed]
- Baldán, Á.; Pei, L.; Lee, R.; Tarr, P.; Tangirala, R.K.; Weinstein, M.M.; Frank, J.; Li, A.C.; Tontonoz, P.; Edwards, P.A. Impaired development of atherosclerosis in hyperlipidemic ldlr−/− and apoe−/− mice transplanted with abcg1−/− bone marrow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2301–2307. [Google Scholar] [CrossRef] [PubMed]
- Ranalletta, M.; Wang, N.; Han, S.; Yvan-Charvet, L.; Welch, C.; Tall, A.R. Decreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with abcg1−/− bone marrow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2308–2315. [Google Scholar] [CrossRef] [PubMed]
- Out, R.; Jessup, W.; Le Goff, W.; Hoekstra, M.; Gelissen, I.C.; Zhao, Y.; Kritharides, L.; Chimini, G.; Kuiper, J.; Chapman, M.J.; et al. Coexistence of foam cells and hypocholesterolemia in mice lacking the abc transporters a1 and g1. Circ. Res. 2008, 102, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Murphy, A.J.; Wang, M.; Pagler, T.A.; Vengrenyuk, Y.; Kappus, M.S.; Gorman, D.J.; Nagareddy, P.R.; Zhu, X.; Abramowicz, S.; et al. Deficiency of abca1 and abcg1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res. 2013, 112. [Google Scholar] [CrossRef] [PubMed]
- Costet, P.; Luo, Y.; Wang, N.; Tall, A.R. Sterol-dependent transactivation of the abc1 promoter by the liver x receptor/retinoid x receptor. J. Biol. Chem. 2000, 275, 28240–28245. [Google Scholar] [PubMed]
- Schwartz, K.; Lawn, R.M.; Wade, D.P. Abc1 gene expression and apoa-i-mediated cholesterol efflux are regulated by lxr. Biochem. Biophys. Res. Commun. 2000, 274, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Venkateswaran, A.; Tarr, P.T.; Xenarios, I.; Kudoh, J.; Shimizu, N.; Edwards, P.A. Characterization of the human abcg1 gene: Liver x receptor activates an internal promoter that produces a novel transcript encoding an alternative form of the protein. J. Biol. Chem. 2001, 276, 39438–39447. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.H.; Chen, W.Q.; Shen, Z.Y. Urolithin a shows anti-atherosclerotic activity via activation of class b scavenger receptor and activation of nef2 signaling pathway. Pharmacol. Rep. 2017. [Google Scholar] [CrossRef]
- Jongstra-Bilen, J.; Zhang, C.X.; Wisnicki, T.; Li, M.K.; White-Alfred, S.; Ilaalagan, R.; Ferri, D.M.; Deonarain, A.; Wan, M.H.; Hyduk, S.J.; et al. Oxidized low-density lipoprotein loading of macrophages downregulates tlr-induced proinflammatory responses in a gene-specific and temporal manner through transcriptional control. J. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen sulfide induces keap1 s-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Chen, M.; Xie, X.; Wang, X.; Kherada, N.; Desikan, R.; Mihai, G.; Burns, P.; Sun, Q.; Rajagopalan, S. Lipoicmethylenedioxyphenol reduces experimental atherosclerosis through activation of Nrf2 signaling. PLoS ONE 2016, 11, e0148305. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, W.; Xu, H.; Sun, Y.; Sun, J.; Jiang, Y.; Yao, J.; Tian, Y. Non-lethal sonodynamic therapy inhibits atherosclerotic plaque progression in APOE−/− mice and attenuates OX-LDL-mediated macrophage impairment by inducing heme oxygenase-1. Cell. Physiol. Biochem. 2017, 41, 2432–2446. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Experimental Model/Cell Line | Study Finding | Properties | Source |
|---|---|---|---|---|
| Antioxidant genes | HASMC | Nrf2−/−, ↓HO-1 & Prx-1 | Anti-atherogenic | [39] |
| RAW264.7 & Nrf2−/− mice | Nrf2−/−, ↓HO-1, ↑IL-1β & IL-6 | Anti-atherogenic | [40] | |
| HAECs, HMECs, Human mesangial cells & U937 cells | Nrf2+/+, ↑HO-1 & GPx, ↑intracellular GSH level, ↓MCP-1 & VCAM-1, ↓adhesion activity | Anti-atherogenic | [41] | |
| Mouse peritoneal macrophages & SMCs | Nrf2+/−, ↑ stress protein A170, HO-1 & Prx-1 | Anti-atherogenic | [42] | |
| Cholesterol uptake receptors | LDLR−/− mice | Nrf2−/−, ↑atherosclerotic lesions, ↑uptake of acetylated and malondialdehyde-modified LDLs, ↑expression of TLR4, SR-A, LOX-1 & CXCL16 | Anti-atherogenic | [13] |
| ApoE−/− mice | Nrf2−/−, ↓CD36, ↓ cholesterol influx | Pro-atherogenic | [37] | |
| ApoE−/− mice | Nrf2−/−, ↓atherosclerotic plaques, ↓uptake of acLDL, ↓expression of CD36 | Pro-atherogenic | [36] | |
| Mouse peritoneal macrophages | Nrf2+/+, ↑CD36 | Pro-atherogenic | [42] | |
| Cholesterol efflux receptors | THP-1 cells & primary human macrophages | Tan-induced Nrf2 activation, ↑HO-1, ↓SR-A, ↑ABCA1 & ABCG1 | Anti-atherogenic | [43] |
| THP-1 cells | tBHQ-induced Nrf2 & HO-1 activation, ↑ABCA1, ↑cholesterol efflux | Anti-atherogenic | [44] | |
| THP-1 cells | EGCG-induced Nrf2 activation, ↓TNF-α-induced NF-κB activation, ↑ABCA1 | Anti-atherogenic | [45] | |
| Proinflammatory cytokines & others mediators | U937 cells | Nrf2−/−, ↑IL-1β, IL-6 & TNFα, ↑MCP-1, ↑ROS & ER stress markers expression | Anti-atherogenic | [46] |
| LDLR−/− mice | Nrf2−/−, ↑ MCP-1, IL-6 & TNF-α | Anti-atherogenic | [13] | |
| ApoE−/− mice | Nrf2−/−, ↓atherosclerotic lesions, ↓cholesterol crystal-induced IL-1 production | Pro-atherogenic | [11] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ooi, B.K.; Goh, B.H.; Yap, W.H. Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. Int. J. Mol. Sci. 2017, 18, 2336. https://doi.org/10.3390/ijms18112336
Ooi BK, Goh BH, Yap WH. Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. International Journal of Molecular Sciences. 2017; 18(11):2336. https://doi.org/10.3390/ijms18112336
Chicago/Turabian StyleOoi, Bee Kee, Bey Hing Goh, and Wei Hsum Yap. 2017. "Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation" International Journal of Molecular Sciences 18, no. 11: 2336. https://doi.org/10.3390/ijms18112336
APA StyleOoi, B. K., Goh, B. H., & Yap, W. H. (2017). Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. International Journal of Molecular Sciences, 18(11), 2336. https://doi.org/10.3390/ijms18112336