TGF-?1 in Vascular Wall Pathology: Unraveling Chronic Venous Insufficiency Pathophysiology


Abstract
:1. Introduction
2. TGF-β1 Family and Function
3. TGF-β1 Signaling Pathways: Major Components and Regulation
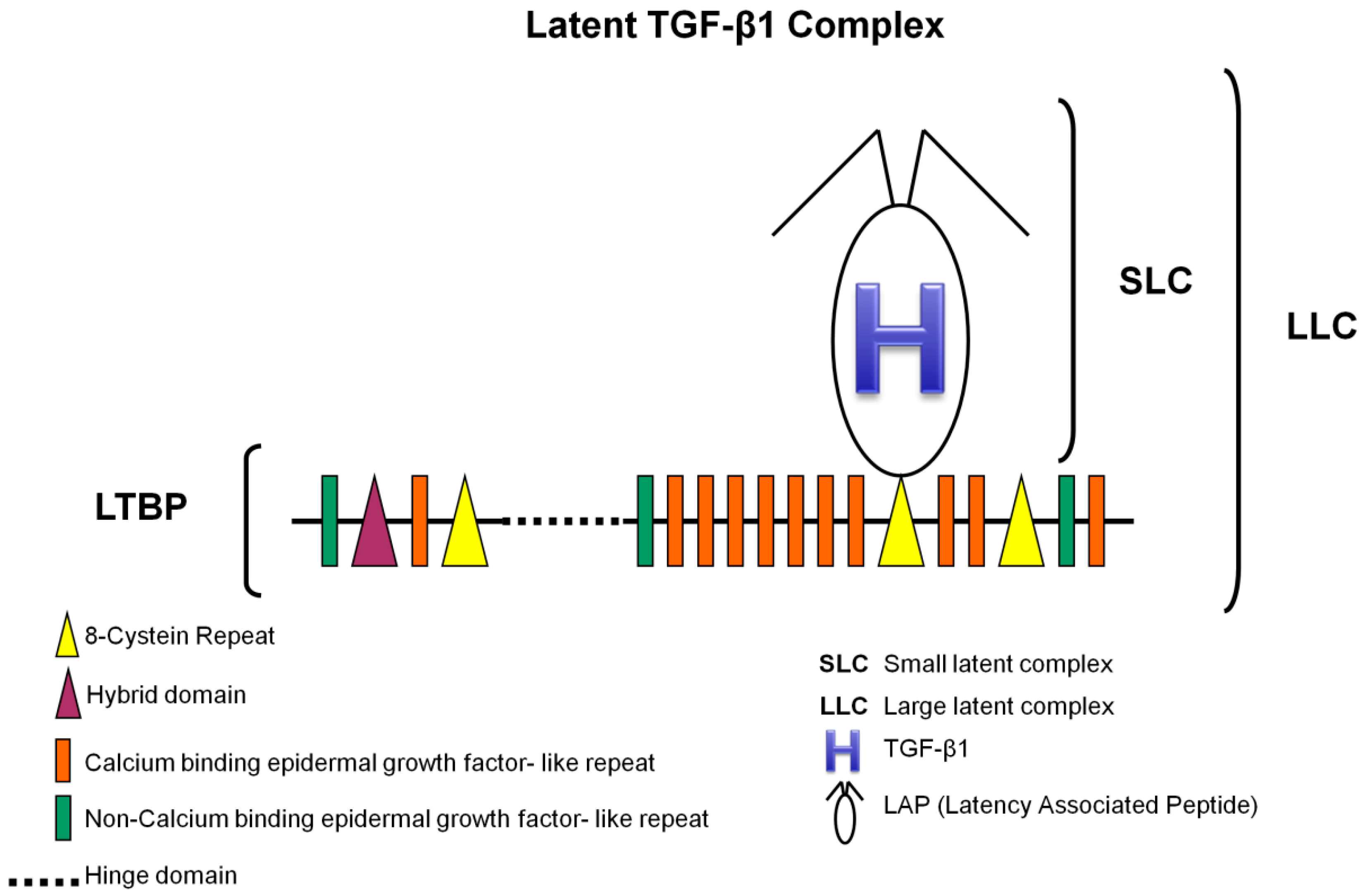
3.1. TGF-β1 Latent Complexes
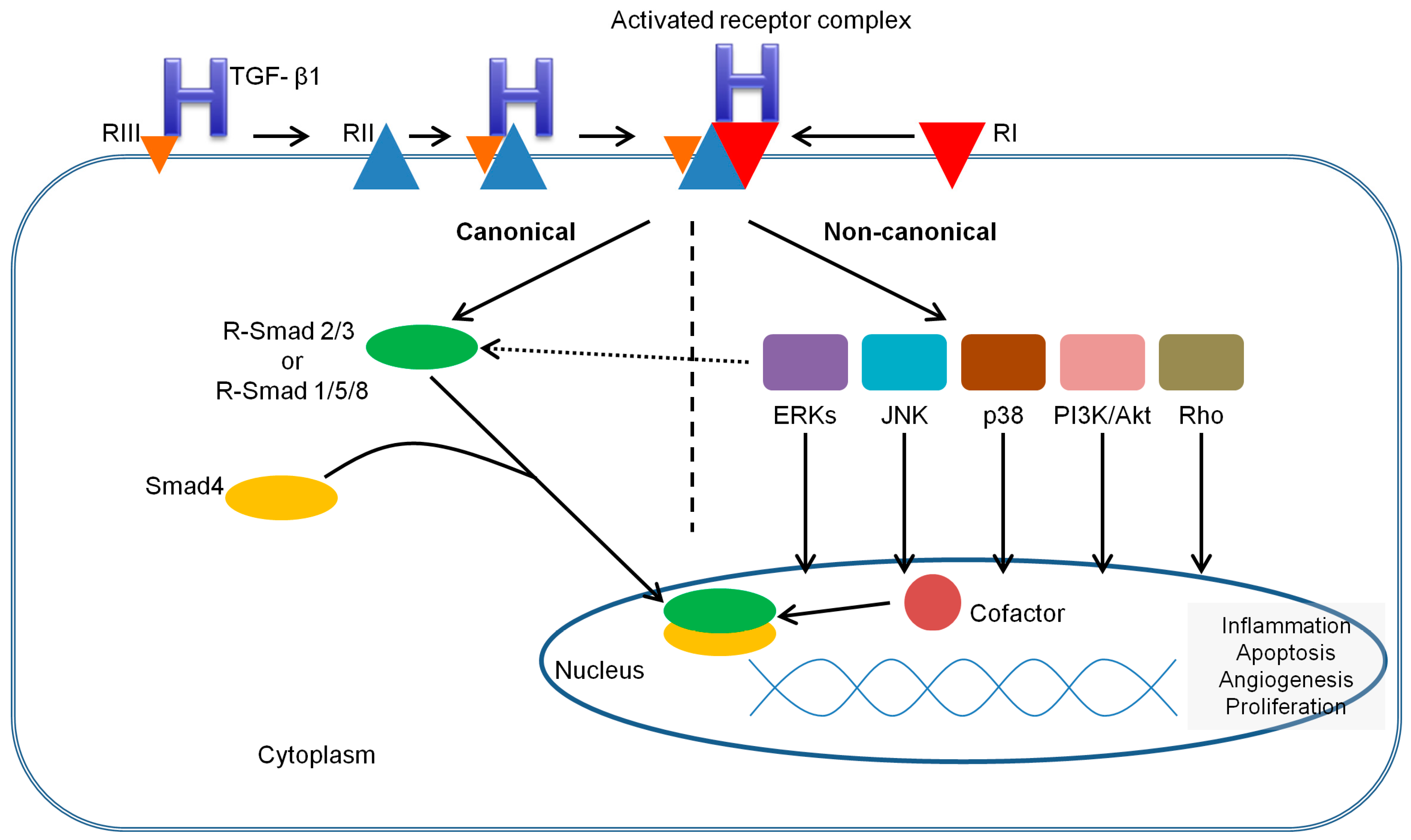
3.2. TGF-β Receptors and Smads
3.3. TGF-β1 Signaling Pathways
3.4. Regulation of TGF-β1 Signaling
4. The Role of TGF-β1 Signaling Pathways in Vessel Wall Pathological Processes
4.1. TGF-β1 and the Vascular Wall Shear Stress
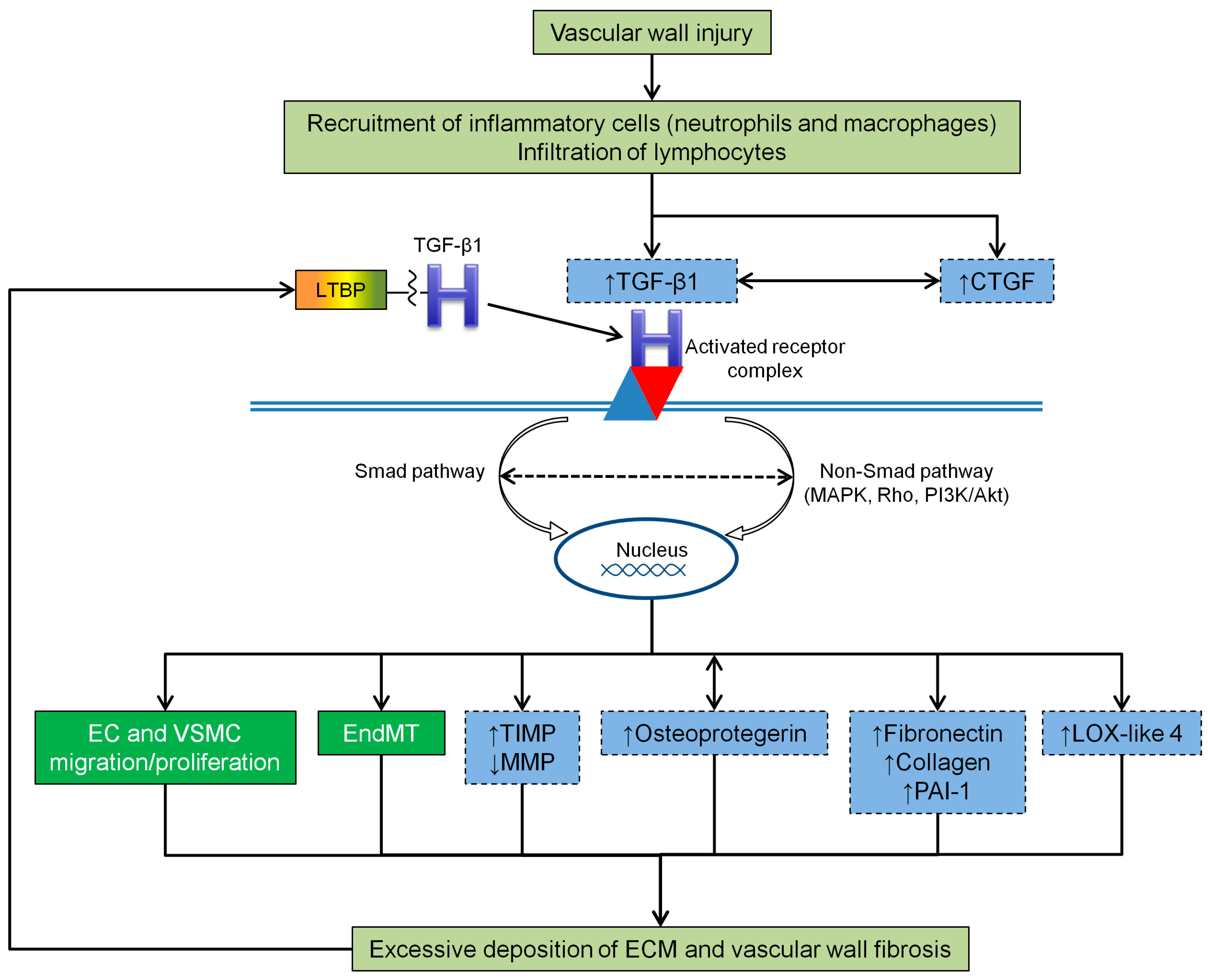
4.2. TGF-β1 and Vascular Wall Fibrosis
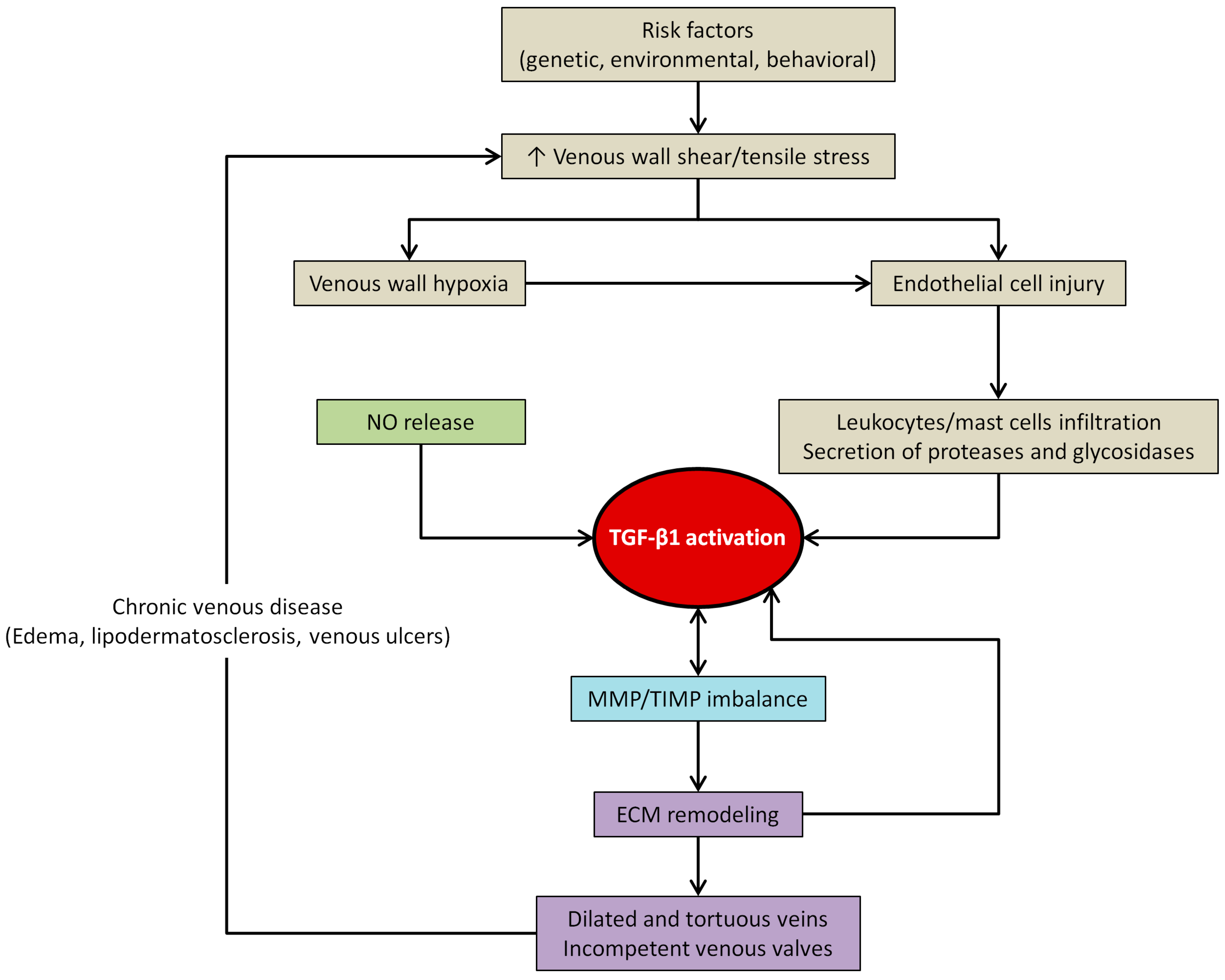
4.3. TGF-β1 and Venous Wall Abnormal Morphology and Functioning
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eklof, B.; Perrin, M.; Delis, K.T.; Rutherford, R.B.; Gloviczki, P.; American Venous Forum; European Venous Forum; International Union of Phlebology; American College of Phlebology; International Union of Angiology. Updated terminology of chronic venous disorders: The VEIN-TERM transatlantic interdisciplinary consensus document. J. Vasc. Surg. 2009, 49, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Beebe-Dimmer, J.L.; Pfeifer, J.R.; Engle, J.S.; Schottenfeld, D. The epidemiology of chronic venous insufficiency and varicose veins. Ann. Epidemiol. 2005, 15, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Robertson, L.; Evans, C.; Fowkes, F.G. Epidemiology of chronic venous disease. Phlebology 2008, 23, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.J.; You, R.; Rameshwar, P.; Gorti, R.; DeFouw, D.O.; Phillips, C.K.; Padberg, F.T., Jr.; Silva, M.B., Jr.; Simonian, G.T.; Hobson, R.W., II; et al. Dermal tissue fibrosis in patients with chronic venous insufficiency is associated with increased transforming growth factor-beta1 gene expression and protein production. J. Vasc. Surg. 1999, 30, 1129–1145. [Google Scholar] [CrossRef]
- Eberhardt, R.T.; Raffetto, J.D. Chronic venous insufficiency. Circulation 2005, 111, 2398–2409. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, P.H.; Maricq, H.R.; Biro, C.; Poncot-Makinen, C.O.; Franco, A. Prevalence, risk factors, and clinical patterns of chronic venous disorders of lower limbs: A population-based study in France. J. Vasc. Surg. 2004, 40, 650–659. [Google Scholar] [CrossRef] [PubMed]
- McGuckin, M.; Waterman, R.; Brooks, J.; Cherry, G.; Porten, L.; Hurley, S.; Kerstein, M.D. Validation of venous leg ulcer guidelines in the United States and United Kingdom. Am. J. Surg. 2002, 183, 132–137. [Google Scholar] [CrossRef]
- Bergan, J.; Pascarella, L. Venous Anatomy, Physiology, and Pathophysiology. In The Vein Book, 2nd ed.; Bergan, J.J., Bunke-Paquette, N., Eds.; Oxford University Press: New York, NY, USA, 2014; pp. 37–44. [Google Scholar]
- Bergan, J. Molecular mechanisms in chronic venous insufficiency. Ann. Vasc. Surg. 2007, 21, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, L.; Schmid-Schonbein, G.W.; Bergan, J. An animal model of venous hypertension: The role of inflammation in venous valve failure. J. Vasc. Surg. 2005, 41, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Takase, S.; Pascarella, L.; Lerond, L.; Bergan, J.J.; Schmid-Schonbein, G.W. Venous hypertension, inflammation and valve remodeling. Eur. J. Vasc. Endovasc. Surg. 2004, 28, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Woodside, K.J.; Hu, M.; Burke, A.; Murakami, M.; Pounds, L.L.; Killewich, L.A.; Daller, J.A.; Hunter, G.C. Morphologic characteristics of varicose veins: Possible role of metalloproteinases. J. Vasc. Surg. 2003, 38, 162–169. [Google Scholar] [CrossRef]
- Mannello, F.; Ligi, D.; Canale, M.; Raffetto, J.D. Omics profiles in chronic venous ulcer wound fluid: Innovative applications for translational medicine. Expert Rev. Mol. Diagn. 2014, 14, 737–762. [Google Scholar] [CrossRef] [PubMed]
- Yasim, A.; Kilinc, M.; Aral, M.; Oksuz, H.; Kabalci, M.; Eroglu, E.; Imrek, S. Serum concentration of procoagulant, endothelial and oxidative stress markers in early primary varicose veins. Phlebology 2008, 23, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Trovato, M.J.; You, R.; Lal, B.K.; Fasehun, F.; Padberg, F.T., Jr.; Hobson, R.W., II; Duran, W.N.; Pappas, P.J. Role of matrix metalloproteinases 1, 2, and 9 and tissue inhibitor of matrix metalloproteinase-1 in chronic venous insufficiency. J. Vasc. Surg. 2001, 34, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Aravind, B.; Saunders, B.; Navin, T.; Sandison, A.; Monaco, C.; Paleolog, E.M.; Davies, A.H. Inhibitory effect of TIMP influences the morphology of varicose veins. Eur. J. Vasc. Endovasc. Surg. 2010, 40, 754–765. [Google Scholar] [CrossRef] [PubMed]
- MacColl, E.; Khalil, R.A. Matrix Metalloproteinases as Regulators of vein structure and function: Implications in chronic venous disease. J. Pharmacol. Exp. Ther. 2015, 355, 410–428. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, W.; Raffetto, J.D.; Khalil, R.A. Matrix metalloproteinases in remodeling of lower extremity veins and chronic venous disease. Prog. Mol. Boil. Transl. Sci. 2017, 147, 267–299. [Google Scholar]
- Pascual, G.; Mendieta, C.; Garcia-Honduvilla, N.; Corrales, C.; Bellon, J.M.; Bujan, J. TGF-beta1 upregulation in the aging varicose vein. J. Vasc. Res. 2007, 44, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; Goumans, M.J.; Dijke, P. TGF-β and Cardiovascular Disorders. In TGF-β in Human Disease, 1st ed.; Moustakas, A., Miyazawa, K., Eds.; Springer: Tokyo, Japan, 2013; pp. 297–322. [Google Scholar]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Boil. 2013, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rodriguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 2008, 1782, 197–228. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116 Pt 2, 217–224. [Google Scholar] [CrossRef]
- Ding, R.; Darland, D.C.; Parmacek, M.S.; D’Amore, P.A. Endothelial-mesenchymal interactions in vitro reveal molecular mechanisms of smooth muscle/pericyte differentiation. Stem Cells Dev. 2004, 13, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Liu, Z.; ten Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Boil. 2007, 8, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; Ten Dijke, P. Regulation of endothelial cell plasticity by TGF-beta. Cell Tissue Res. 2012, 347, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, T.; Tamura, K.; Takahashi, K.; Saito, Y.; Akikusa, B.; Oohashi, H.; Kasayuki, N.; Ueda, M.; Morisaki, N. In vivo effect of TGF-beta 1. Enhanced intimal thickening by administration of TGF-beta 1 in rabbit arteries injured with a balloon catheter. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Michon, I.N.; Penning, L.C.; Molenaar, T.J.; van Berkel, T.J.; Biessen, E.A.; Kuiper, J. The effect of TGF-beta receptor binding peptides on smooth muscle cells. Biochem. Biophys. Res. Commun. 2002, 293, 1279–1286. [Google Scholar] [CrossRef]
- Sato, Y.; Okada, F.; Abe, M.; Seguchi, T.; Kuwano, M.; Sato, S.; Furuya, A.; Hanai, N.; Tamaoki, T. The mechanism for the activation of latent TGF-beta during co-culture of endothelial cells and smooth muscle cells: Cell-type specific targeting of latent TGF-beta to smooth muscle cells. J. Cell Biol. 1993, 123, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Lohi, J.; Saarinen, J.; Kovanen, P.T.; Keski-Oja, J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J. Biol. Chem. 1995, 270, 4689–4696. [Google Scholar] [CrossRef] [PubMed]
- Graham, H.K.; Akhtar, R.; Kridiotis, C.; Derby, B.; Kundu, T.; Trafford, A.W.; Sherratt, M.J. Localised micro-mechanical stiffening in the ageing aorta. Mech. Ageing Dev. 2011, 132, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S.; Davies, A.H. Pathogenesis of primary varicose veins. Br. J. Surg. 2009, 96, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, Y.P.; Rudensky, A.Y. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat. Rev. Immunol. 2007, 7, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q. Transforming growth factor-beta1 protects against pulmonary artery endothelial cell apoptosis via ALK5. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L123–L133. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Patel, B.; Harrington, E.O.; Rounds, S. Transforming growth factor-beta1 causes pulmonary microvascular endothelial cell apoptosis via ALK5. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L825–838. [Google Scholar] [CrossRef] [PubMed]
- Toma, I.; McCaffrey, T.A. Transforming growth factor-beta and atherosclerosis: Interwoven atherogenic and atheroprotective aspects. Cell Tissue Res. 2012, 347, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Gojova, A.; Marchiol-Fournigault, C.; Esposito, B.; Kamate, C.; Merval, R.; Fradelizi, D.; Tedgui, A. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ. Res. 2001, 89, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Serrati, S.; Margheri, F.; Pucci, M.; Cantelmo, A.R.; Cammarota, R.; Dotor, J.; Borras-Cuesta, F.; Fibbi, G.; Albini, A.; del Rosso, M. TGFbeta1 antagonistic peptides inhibit TGFbeta1-dependent angiogenesis. Biochem. Pharmacol. 2009, 77, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Ten Dijke, P. Transforming growth factor-beta signaling and tumor angiogenesis. Front. Biosci. 2009, 14, 4848–4861. [Google Scholar] [CrossRef]
- Pepper, M.S. Transforming growth factor-beta: Vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997, 8, 21–43. [Google Scholar] [CrossRef]
- Herpin, A.; Lelong, C.; Favrel, P. Transforming growth factor-beta-related proteins: An ancestral and widespread superfamily of cytokines in metazoans. Dev. Comp. Immunol. 2004, 28, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Abe, M.; Sato, Y.; Miyazono, K.; Harpel, J.; Heldin, C.H.; Rifkin, D.B. Role of the latent TGF-beta binding protein in the activation of latent TGF-beta by co-cultures of endothelial and smooth muscle cells. J. Cell Biol. 1993, 120, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.A.; Al-Musawi, S.L.; Walton, K.L. Prodomains regulate the synthesis, extracellular localisation and activity of TGF-beta superfamily ligands. Growth Factors 2011, 29, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, D.B. Latent transforming growth factor-beta (TGF-beta) binding proteins: Orchestrators of TGF-beta availability. J. Biol. Chem. 2005, 280, 7409–7412. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, J.; Keski-Oja, J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Boil. Cell 2000, 11, 2691–2704. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Kirkbride, K.C.; Ray, B.N.; Blobe, G.C. Cell-surface co-receptors: Emerging roles in signaling and human disease. Trends Biochem. Sci. 2005, 30, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, C.; Lopez-Novoa, J.M.; Quintanilla, M. The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim. Biophys. Acta 2009, 1792, 954–973. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Goumans, M.J.; Ten Dijke, P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yang, X.; Friesel, R.E.; Vary, C.P.; Liaw, L. Mechanisms of TGF-beta-induced differentiation in human vascular smooth muscle cells. J. Vasc. Res. 2011, 48, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-smad signaling pathways of the TGF-beta family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Chen, Y.G. Regulation of TGF-beta signal transduction. Scientifica 2014, 2014, 874065. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-beta structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Stringer, S.E.; Hamilton, A.; Charlton-Menys, V.; Gotting, C.; Muller, B.; Aeschlimann, D.; Alexander, M.Y. Decorin GAG synthesis and TGF-beta signaling mediate Ox-LDL-induced mineralization of human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Chen, Y.G.; Massague, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. The regulation of TGFbeta signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Huang, F.; Chiang, Y.J.; Li, M.; Du, J.; Ding, Y.; Zhang, T.; Lee, H.W.; Jeong, L.S.; Chen, Y.; et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-beta type II receptor. Mol. Cell 2013, 49, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, Q.; Du, J.; Luo, S.; Xia, J.; Chen, Y.G. PICK1 promotes caveolin-dependent degradation of TGF-beta type I receptor. Cell Res. 2012, 22, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhang, L.; Gao, X.; Meng, F.; Wen, J.; Zhou, H.; Meng, A.; Chen, Y.G. The evolutionally conserved activity of Dapper2 in antagonizing TGF-beta signaling. FASEB J. 2007, 21, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Blobe, G.C. The interaction of endoglin with beta-arrestin2 regulates transforming growth factor-beta-mediated ERK activation and migration in endothelial cells. J. Biol. Chem. 2007, 282, 21507–21517. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.B.; Ling, G.H.; Sun, L.; Liu, F.Y. Smad anchor for receptor activation (SARA) in TGF-beta signaling. Front. Biosci. (Elite Ed.) 2010, 2, 857–860. [Google Scholar] [PubMed]
- Dai, F.; Chang, C.; Lin, X.; Dai, P.; Mei, L.; Feng, X.H. Erbin inhibits transforming growth factor beta signaling through a novel Smad-interacting domain. Mol. Cell. Biol. 2007, 27, 6183–6194. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 2009, 19, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Itoh, F. Inhibitory machinery for the TGF-beta family signaling pathway. Growth Factors 2011, 29, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Albig, A.R.; Schiemann, W.P. Fibulin-5 antagonizes vascular endothelial growth factor (VEGF) signaling and angiogenic sprouting by endothelial cells. DNA Cell Biol. 2004, 23, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Schiemann, B.J.; Neil, J.R.; Schiemann, W.P. SPARC inhibits epithelial cell proliferation in part through stimulation of the transforming growth factor-beta-signaling system. Mol. Biol. Cell 2003, 14, 3977–3988. [Google Scholar] [CrossRef] [PubMed]
- Sokol, J.P.; Neil, J.R.; Schiemann, B.J.; Schiemann, W.P. The use of cystatin C to inhibit epithelial-mesenchymal transition and morphological transformation stimulated by transforming growth factor-beta. Breast Cancer Res. BCR 2005, 7, R844–R853. [Google Scholar] [CrossRef] [PubMed]
- Walshe, T.E.; dela Paz, N.G.; D’Amore, P.A. The role of shear-induced transforming growth factor-beta signaling in the endothelium. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2608–2617. [Google Scholar] [CrossRef] [PubMed]
- Wojciak-Stothard, B. Endothelial cell migration under flow. In Cell Migration, 2nd ed.; Wells, C.M., Parsons, M., Eds.; Humana Press: New York, NY, USA, 2011; Volume 769, pp. 137–147. [Google Scholar]
- Birukov, K.G. Cyclic stretch, reactive oxygen species, and vascular remodeling. Antioxid. Redox Signal. 2009, 11, 1651–1667. [Google Scholar] [CrossRef] [PubMed]
- Prandi, F.; Piola, M.; Soncini, M.; Colussi, C.; D’Alessandra, Y.; Penza, E.; Agrifoglio, M.; Vinci, M.C.; Polvani, G.; Gaetano, C.; et al. Adventitial vessel growth and progenitor cells activation in an ex vivo culture system mimicking human saphenous vein wall strain after coronary artery bypass grafting. PLoS ONE 2015, 10, e0117409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.X.; Jiang, J.; Jiang, X.H.; Wang, X.D.; Ji, S.Y.; Han, Y.; Long, D.K.; Shen, B.R.; Yan, Z.Q.; Chien, S.; et al. PDGF-BB and TGF-{beta}1 on cross-talk between endothelial and smooth muscle cells in vascular remodeling induced by low shear stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1908–1913. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, C.J.; Williams, B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells: Role of TGF-beta (1). Hypertension 2000, 36, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yu, P.; Tao, M.; Fernandez, C.; Ifantides, C.; Moloye, O.; Schultz, G.S.; Ozaki, C.K.; Berceli, S.A. TGF-beta- and CTGF-mediated fibroblast recruitment influences early outward vein graft remodeling. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H482–H488. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Y.; Chen, W.D.; Zhu, D.L.; Wu, L.Y.; Zhang, J.; Han, W.Q.; Li, J.D.; Yan, C.; Gao, P.J. The PDE1A-PKCalpha signaling pathway is involved in the upregulation of alpha-smooth muscle actin by TGF-beta1 in adventitial fibroblasts. J. Vasc. Res. 2010, 47, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D.A. Mechanisms of fibrogenesis. Exp. Biol. Med. 2008, 233, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Krieg, T.; Abraham, D.; Lafyatis, R. Fibrosis in connective tissue disease: The role of the myofibroblast and fibroblast-epithelial cell interactions. Arthrit. Res. Ther. 2007, 9 (Suppl. 2), S4. [Google Scholar] [CrossRef]
- Wang, S.; Lincoln, T.M.; Murphy-Ullrich, J.E. Glucose downregulation of PKG-I protein mediates increased thrombospondin1-dependent TGF-{beta} activity in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2010, 298, C1188–C1197. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Suriguga, G.M.; Liu, W.J.; Cui, N.X.; Wang, Y.; Du, X.; Yi, Z.C. High glucose induced endothelial to mesenchymal transition in human umbilical vein endothelial cell. Exp. Mol. Pathol. 2017, 102, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ramos, M.; Calleros, L.; Lopez-Ongil, S.; Raoch, V.; Griera, M.; Rodriguez-Puyol, M.; de Frutos, S.; Rodriguez-Puyol, D. HSP70 increases extracellular matrix production by human vascular smooth muscle through TGF-beta1 up-regulation. Int. J. Biochem. Cell Biol. 2013, 45, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Flanders, K.C. Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 2004, 85, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, W.; Yin, Y.; Li, W. AST IV inhibits H(2)O(2)-induced human umbilical vein endothelial cell apoptosis by suppressing Nox4 expression through the TGF-beta1/Smad2 pathway. Int. J. Mol. Med. 2015, 35, 1667–1674. [Google Scholar] [PubMed]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Higgins, P.J. Integration of non-SMAD and SMAD signaling in TGF-beta1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb. Haemost. 2008, 100, 976–983. [Google Scholar] [PubMed]
- Otsuka, G.; Agah, R.; Frutkin, A.D.; Wight, T.N.; Dichek, D.A. Transforming growth factor beta 1 induces neointima formation through plasminogen activator inhibitor-1-dependent pathways. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Ryer, E.J.; Hom, R.P.; Sakakibara, K.; Nakayama, K.I.; Nakayama, K.; Faries, P.L.; Liu, B.; Kent, K.C. PKCdelta is necessary for Smad3 expression and transforming growth factor beta-induced fibronectin synthesis in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, X.; Sun, B.; Zeng, M.; Xing, C.; Zhao, X.; Yang, J. Role of TGF-beta1 in production of fibronectin in vascular smooth muscle cells cultured under high-phosphate conditions. J. Nephrol. 2013, 26, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Wang, S.; Cai, W.; Sheng, J. Role of TGF-beta1/Smad3 signaling pathway in secretion of type I and III collagen by vascular smooth muscle cells of rats undergoing balloon injury. J. Biomed. Biotechnol. 2012, 2012, 965953. [Google Scholar] [CrossRef] [PubMed]
- Obi, A.T.; Diaz, J.A.; Ballard-Lipka, N.L.; Roelofs, K.J.; Farris, D.M.; Lawrence, D.A.; Wakefield, T.W.; Henke, P.K. Plasminogen activator-1 overexpression decreases experimental postthrombotic vein wall fibrosis by a non-vitronectin-dependent mechanism. J. Thromb. Haemost. JTH 2014, 12, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.F.; Sood, V.; Elfline, M.A.; Luke, C.E.; Dewyer, N.A.; Diaz, J.A.; Myers, D.D.; Wakefield, T.; Henke, P.K. The role of urokinase plasminogen activator and plasmin activator inhibitor-1 on vein wall remodeling in experimental deep vein thrombosis. J. Vasc. Surg. 2012, 56, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, O.; Gonzalez-Santamaria, J.; Lagares, D.; Guinea-Viniegra, J.; Pichol-Thievend, C.; Muller, L.; Rodriguez-Pascual, F. LOXL4 is induced by transforming growth factor beta1 through Smad and JunB/Fra2 and contributes to vascular matrix remodeling. Mol. Cell. Biol. 2013, 33, 2388–2401. [Google Scholar] [CrossRef] [PubMed]
- Rachfal, A.W.; Brigstock, D.R. Structural and functional properties of CCN proteins. Vitam. Horm. 2005, 70, 69–103. [Google Scholar] [PubMed]
- Rodriguez-Vita, J.; Ruiz-Ortega, M.; Ruperez, M.; Esteban, V.; Sanchez-Lopez, E.; Plaza, J.J.; Egido, J. Endothelin-1, via ETA receptor and independently of transforming growth factor-beta, increases the connective tissue growth factor in vascular smooth muscle cells. Circ. Res. 2005, 97, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Shi-Wen, X.; Leask, A.; Abraham, D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008, 19, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.G.; Chen, G.; Zhu, J.Y.; Zhang, W.; Sun, Y.F.; Jia, J.; Zhang, J.; Zhao, Y.F. Downregulation of the transforming growth factor-beta/connective tissue growth factor 2 signalling pathway in venous malformations: Its target potential for sclerotherapy. Br. J. Dermatol. 2014, 171, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Tao, M.; Omalley, K.A.; Wang, D.; Ozaki, C.K.; Berceli, S.A. Established neointimal hyperplasia in vein grafts expands via TGF-beta-mediated progressive fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1200–H1207. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, B.; Pickering, R.J.; Tsorotes, D.; Wang, B.; Bernardi, S.; Kantharidis, P.; Fabris, B.; Zauli, G.; Secchiero, P.; Thomas, M.C. Osteoprotegerin promotes vascular fibrosis via a TGF-beta1 autocrine loop. Atherosclerosis 2011, 218, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Yao, E.H.; Fukuda, N.; Ueno, T.; Matsuda, H.; Nagase, H.; Matsumoto, Y.; Sugiyama, H.; Matsumoto, K. A pyrrole-imidazole polyamide targeting transforming growth factor-beta1 inhibits restenosis and preserves endothelialization in the injured artery. Cardiovasc. Res. 2009, 81, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; Hilaire, C.S.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra234. [Google Scholar] [CrossRef] [PubMed]
- Suwanabol, P.A.; Seedial, S.M.; Zhang, F.; Shi, X.; Si, Y.; Liu, B.; Kent, K.C. TGF-beta and Smad3 modulate PI3K/Akt signaling pathway in vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2211–H2219. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; Hollenbeck, S.T.; Ryer, E.J.; Edlin, R.; Yamanouchi, D.; Kundi, R.; Wang, C.; Liu, B.; Kent, K.C. TGF-beta through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H540–H549. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, C.; Liu, R.; Wei, C.; Duan, J.; Liu, K.; Li, S.; Zou, H.; Zhao, J.; Wang, L.; et al. Effect of TGF-beta1 on the migration and recruitment of mesenchymal stem cells after vascular balloon injury: Involvement of matrix metalloproteinase-14. Sci. Rep. 2016, 6, 21176. [Google Scholar] [CrossRef] [PubMed]
- Badier-Commander, C.; Couvelard, A.; Henin, D.; Verbeuren, T.; Michel, J.B.; Jacob, M.P. Smooth muscle cell modulation and cytokine overproduction in varicose veins. An in situ study. J. Pathol. 2001, 193, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C.; Arnould, T.; Thibaut-Vercruyssen, R.; Bouaziz, N.; Janssens, D.; Remacle, J. Perfused human saphenous veins for the study of the origin of varicose veins: Role of the endothelium and of hypoxia. Int. angiol. J. Int. Union Angiol. 1997, 16, 134–141. [Google Scholar]
- Ghaderian, S.M.; Khodaii, Z. Tissue remodeling investigation in varicose veins. Int. J. Mol. Cell. Med. 2012, 1, 50–61. [Google Scholar]
- Sansilvestri-Morel, P.; Fioretti, F.; Rupin, A.; Senni, K.; Fabiani, J.N.; Godeau, G.; Verbeuren, T.J. Comparison of extracellular matrix in skin and saphenous veins from patients with varicose veins: Does the skin reflect venous matrix changes? Clin. Sci. 2007, 112, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Kowalewski, R.; Malkowski, A.; Gacko, M.; Sobolewski, K. Influence of thrombophlebitis on TGF-beta1 and its signaling pathway in the vein wall. Folia Histochem. Cytobiol. Pol. Acad. Sci. Pol. Histochem. Cytochem. Soc. 2010, 48, 542–548. [Google Scholar]
- Raffetto, J. Chronic Venous insufficiency: Molecular abnormalities and ulcer formation. In The Vein Book, 2nd ed.; Bergan, J.J., Bunke-Paquette, N., Eds.; Oxford University Press: New York, NY, USA, 2014; pp. 58–66. [Google Scholar]
- Sansilvestri-Morel, P.; Rupin, A.; Jullien, N.D.; Lembrez, N.; Mestries-Dubois, P.; Fabiani, J.N.; Verbeuren, T.J. Decreased production of collagen Type III in cultured smooth muscle cells from varicose vein patients is due to a degradation by MMPs: Possible implication of MMP-3. J. Vasc. Res. 2005, 42, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Bujan, J.; Gimeno, M.J.; Jimenez, J.A.; Kielty, C.M.; Mecham, R.P.; Bellon, J.M. Expression of elastic components in healthy and varicose veins. World J. Surg. 2003, 27, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.; Hingorani, A.; Ascher, E. Overexpression of transforming growth factor-beta1 correlates with increased synthesis of nitric oxide synthase in varicose veins. J. Vasc. Surg. 2005, 41, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Serralheiro, P.; Cairrao, E.; Maia, C.J.; Joao, M.; Costa Almeida, C.M.; Verde, I. Effect of TGF-beta1 on MMP/TIMP and TGF-beta1 receptors in great saphenous veins and its significance on chronic venous insufficiency. Phlebology 2017, 32, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Serralheiro, P.; Novais, A.; Cairrão, E.; Maia, C.J.; Costa Almeida, C.M.; Verde, I. Variability of MMP/TIMP and TGF-β1 receptors throughout the clinical progression of chronic venous disease. To be submitted. 2017. [Google Scholar]
- Pappas, P.J.; Lal, B.K.; Padberg, F.T., Jr.; Zickler, R.; Duran, W.N. Pathophysiology of chronic venous insufficiency. In The Vein Book, 2nd ed.; Bergan, J.J., Bunke-Paquette, N., Eds.; Oxford University Press: New York, NY, USA, 2014; pp. 67–78. [Google Scholar]
- Kim, B.C.; Kim, H.T.; Park, S.H.; Cha, J.S.; Yufit, T.; Kim, S.J.; Falanga, V. Fibroblasts from chronic wounds show altered TGF-beta-signaling and decreased TGF-beta Type II receptor expression. J. Cell. Physiol. 2003, 195, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.L.; Patel, A.; Fileta, B.; Chang, A.; Barnes, S.; Flagg, A.; Kidwell, M.; Villavicencio, J.L.; Rich, N.M. Varicose veins possess greater quantities of MMP-1 than normal veins and demonstrate regional variation in MMP-1 and MMP-13. J. Surg. Res. 2002, 106, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Khalil, R.A. Mechanisms of varicose vein formation: Valve dysfunction and wall dilation. Phlebology 2008, 23, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Schonbein, G.W.; Takase, S.; Bergan, J.J. New advances in the understanding of the pathophysiology of chronic venous insufficiency. Angiology 2001, 52 (Suppl. 1), S27–S34. [Google Scholar] [CrossRef] [PubMed]
- Sayer, G.L.; Smith, P.D. Immunocytochemical characterisation of the inflammatory cell infiltrate of varicose veins. Eur. J. Vasc. Endovasc. Surg. 2004, 28, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Korthuis, R.J. Mast Cell Proteases and Inflammation. Drug Discov. Today Dis. Models 2011, 8, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Elsharawy, M.A.; Naim, M.M.; Abdelmaguid, E.M.; Al-Mulhim, A.A. Role of saphenous vein wall in the pathogenesis of primary varicose veins. Interact. Cardiovasc. Thorac. Surg. 2007, 6, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Badier-Commander, C.; Verbeuren, T.; Lebard, C.; Michel, J.B.; Jacob, M.P. Increased TIMP/MMP ratio in varicose veins: A possible explanation for extracellular matrix accumulation. J. Pathol. 2000, 192, 105–112. [Google Scholar] [CrossRef]
- Lal, B.K.; Saito, S.; Pappas, P.J.; Padberg, F.T., Jr.; Cerveira, J.J.; Hobson, R.W., 2nd; Duran, W.N. Altered proliferative responses of dermal fibroblasts to TGF-beta1 may contribute to chronic venous stasis ulcer. J. Vasc. Surg. 2003, 37, 1285–1293. [Google Scholar] [CrossRef]
- Pappas, P.J.; Lal, B.K.; Ohara, N.; Saito, S.; Zapiach, L.; Duran, W.N. Regulation of matrix contraction in chronic venous disease. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Mikula-Pietrasik, J.; Uruski, P.; Aniukiewicz, K.; Sosinska, P.; Krasinski, Z.; Tykarski, A.; Ksiazek, K. Serum from varicose patients induces senescence-related dysfunction of vascular endothelium generating local and systemic proinflammatory conditions. Oxid. Med. Cell. Longev. 2016, 2016, 2069290. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Year | Specimen | TGF-β Expression and/or Activity in Veins |
|---|---|---|---|
| Badier-Commander C. [109] | 2001 | Varicose and non-varicose great saphenous vein segments | Increased total amount of TGF-β1 in varicose segments, but identical amount of active TGF-β1 |
| Bujan J. [116] | 2003 | Varicose and non-varicose great saphenous vein segments | Increased TGF-β expression and decreased elastin synthesis in injured regions of varicose segments |
| Sansilvestri-Morel P. [115] | 2005 | Vascular smooth muscle cells (VSMC) from varicose and non-varicose great saphenous vein segments | Unchanged TGF-β1 levels |
| Jacob T. [117] | 2005 | Varicose and non-varicose great saphenous and tributaries vein segments | Increased TGF-β1 expression in varicose segments |
| Pascual G. [19] | 2007 | Varicose and non-varicose great saphenous vein segments from older subjects | Reduced latent TGF-β1 expression in varicose segments |
| Kowalewski R. [113] | 2010 | Varicose and non-varicose great saphenous vein segments | Unchanged TGF-β1 mRNA levels and decreased latent and active TGF-β1 expression in varicose segments |
| Serralheiro P. [118] | 2017 | Cultured healthy great saphenous veins | TGF-β1 increased mRNA levels of MMP9, MMP12, TIMP1 and TIMP2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serralheiro, P.; Soares, A.; Costa Almeida, C.M.; Verde, I. TGF-?1 in Vascular Wall Pathology: Unraveling Chronic Venous Insufficiency Pathophysiology. Int. J. Mol. Sci. 2017, 18, 2534. https://doi.org/10.3390/ijms18122534
Serralheiro P, Soares A, Costa Almeida CM, Verde I. TGF-?1 in Vascular Wall Pathology: Unraveling Chronic Venous Insufficiency Pathophysiology. International Journal of Molecular Sciences. 2017; 18(12):2534. https://doi.org/10.3390/ijms18122534
Chicago/Turabian StyleSerralheiro, Pedro, Andreia Soares, Carlos M. Costa Almeida, and Ignacio Verde. 2017. "TGF-?1 in Vascular Wall Pathology: Unraveling Chronic Venous Insufficiency Pathophysiology" International Journal of Molecular Sciences 18, no. 12: 2534. https://doi.org/10.3390/ijms18122534
APA StyleSerralheiro, P., Soares, A., Costa Almeida, C. M., & Verde, I. (2017). TGF-?1 in Vascular Wall Pathology: Unraveling Chronic Venous Insufficiency Pathophysiology. International Journal of Molecular Sciences, 18(12), 2534. https://doi.org/10.3390/ijms18122534