Aptamers and Glioblastoma: Their Potential Use for Imaging and Therapeutic Applications


Abstract
:1. Introduction
2. Aptamers
3. Glioblastoma Targeting Aptamers
3.1. Aptamers Targeting Tumour Intitiating Cells
3.2. Aptamers Targeting Tenascin-C
3.3. Aptamers Targeting Platelet Derived Growth Factor Receptor β
3.4. Aptamers Targeting Axl
3.5. Aptamers Targeting the Epidermal Growth Factor Receptor
3.6. Targeting Nucleolin with a Guanine-Rich Oligonucleotide
3.7. Glioblastoma Aptamers with Unknown Targets
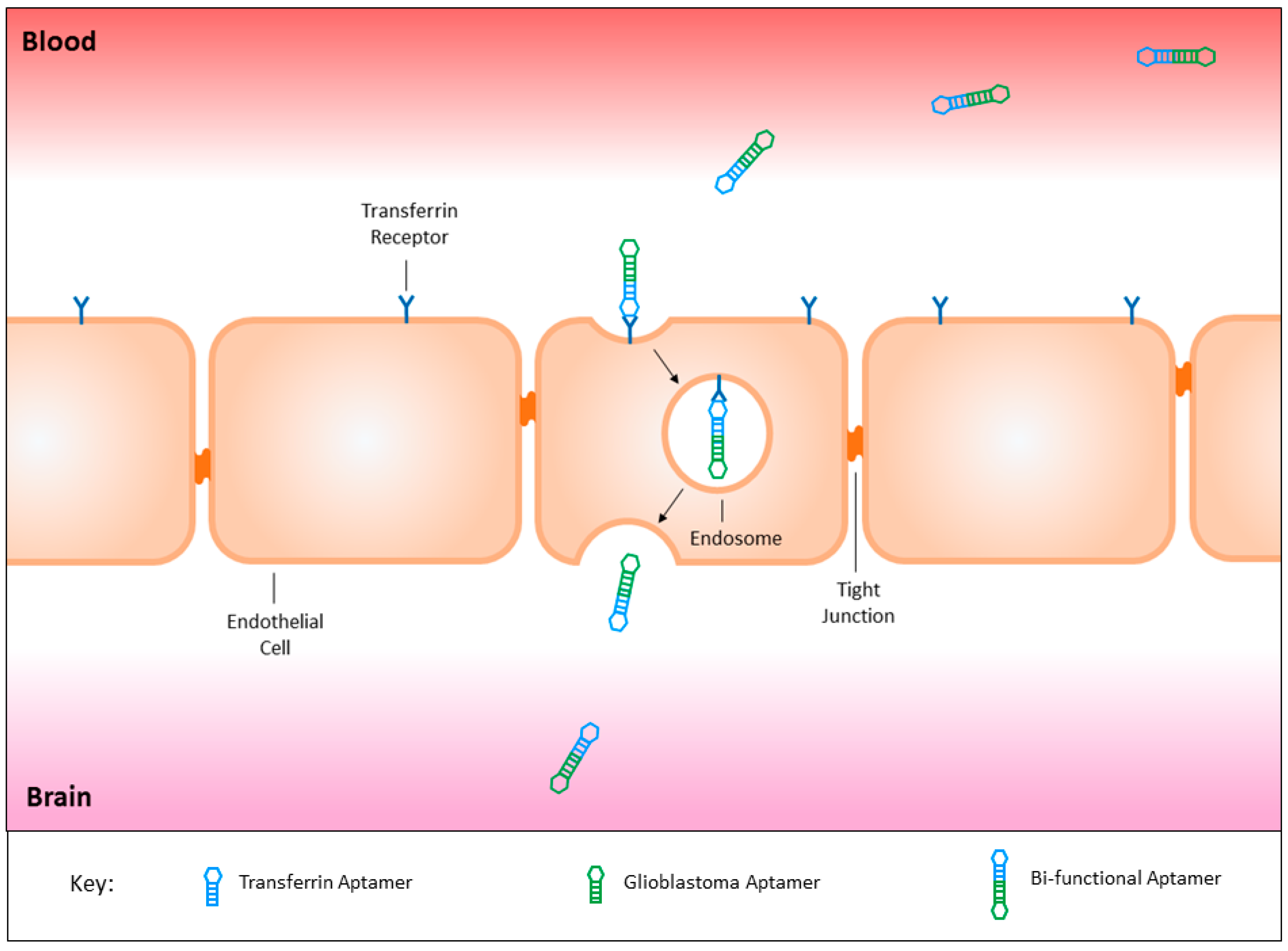
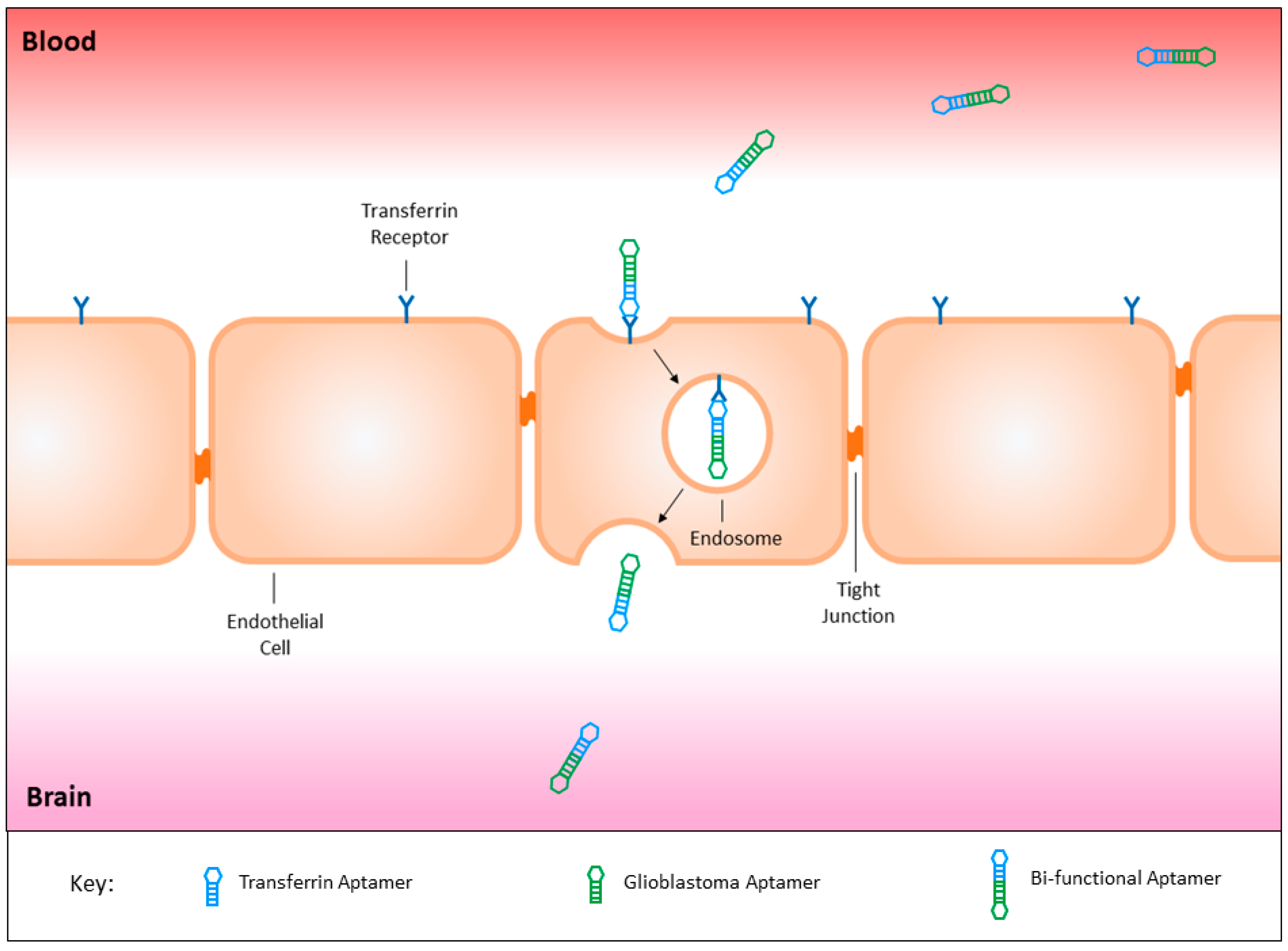
3.8. Circumventing the Blood Brain Barrier
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADCC | Antibody-dependent cellular cytotoxicity |
| ATP | Adenosine triphosphate |
| CDC | Complement-dependent cytotoxicity |
| DNA | Deoxyribose nucleic acid |
| EGFR | Epidermal growth factor receptor |
| EGFRvIII | Epidermal growth factor receptor variant three |
| ERBB2 | Human epidermal growth factor receptor two |
| NF1 | Neurofibromin one |
| NOS | Not otherwise specified |
| PDGFRA | Platelet derived growth factor receptor A |
| PDGFRβ | Platelet derived growth factor receptor β |
| PI3K | Phosphoinositide three-kinase |
| PTEN | Phosphatase and tensin homolog |
| SELEX | Systematic evolution of ligands by exponential enrichment |
| PET | Positron emission tomography |
| RB1 | Retinoblastoma protein |
| RNA | Ribonucleic acid |
| siRNA | Small interfering RNA |
| TIC | Tumour initiating cells |
| TP53 | Tumour suppressor p53 |
| VEGF | Vascular endothelial growth factor |
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. Cbtrus statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2009–2013. Neuro Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Holmen, S.L.; Colman, H. Idh1 and Idh2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Ironside, J.W. Diagnosis and pathogenesis of gliomas. Curr. Diagn. Pathol. 2007, 13, 180–192. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. Who Classification of Tumours of the Central Nervous System, 4th ed.; IARC Press: Lyon, France, 2016. [Google Scholar]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, D.; Figarella-Branger, W.; Cavenee, K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
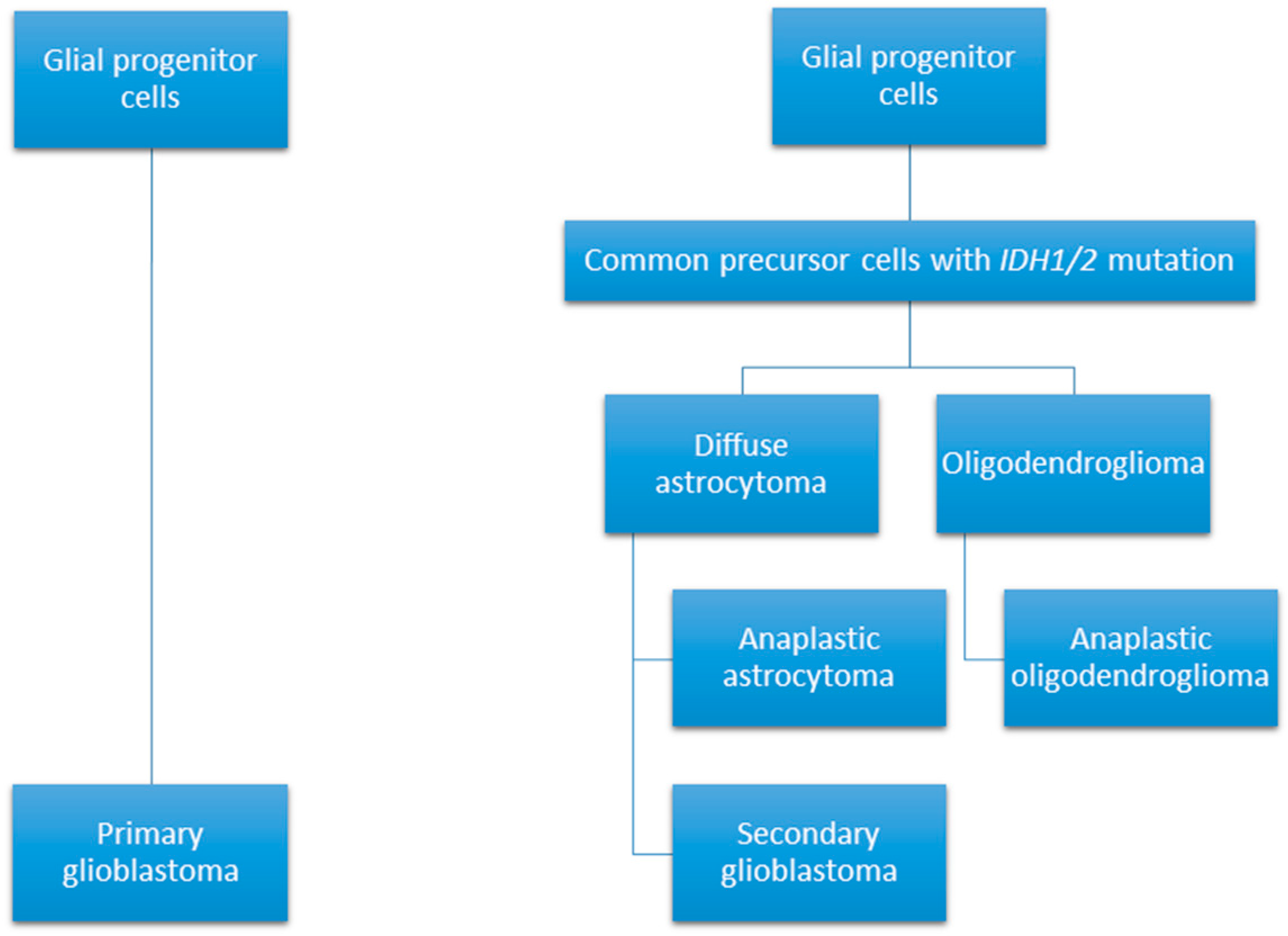
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Radivoyevitch, T.; Maciejewski, J.P.; van Noorden, C.J.F.; Bleeker, F.E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Hou, C.; Chen, H.; Zong, X.; Zong, P. Genetics and epigenetics of glioblastoma: Applications and overall incidence of Idh1 mutation. Front. Oncol. 2016, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and therapeutic biomarkers in glioblastoma: Current status and future perspectives. BioMed Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Eder, K.; Kalman, B. Molecular heterogeneity of glioblastoma and its clinical relevance. Pathol. Oncol. Res. 2014, 20, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Mirimanoff radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Oppenlander, M.E.; Wolf, A.B.; Snyder, L.A.; Bina, R.; Wilson, J.R.; Coons, S.W.; Ashby, L.S.; Brachman, D.; Nakaji, P.; Porter, R.W.; et al. An extent of resection threshold for recurrent glioblastoma and its risk for neurological morbidity. J. Neurosurg. 2014, 120, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, K.; Sokolowska, J.; Szmidt, M.; Sysa, P. Glioblastoma multiforme—An overview. Contemp. Oncol. 2014, 18, 307–312. [Google Scholar]
- Thomas, R.P.; Recht, L.; Nagpal, S. Advances in the management of glioblastoma: The role of temozolomide and mgmt testing. Clin. Pharmacol. 2013, 5, 1–9. [Google Scholar] [PubMed]
- Jakola, A.S.; Gulati, S.; Weber, C.; Unsgard, G.; Solheim, O. Postoperative Deterioration in health related quality of life as predictor for survival in patients with glioblastoma: A prospective study. PLoS ONE 2011, 6, e28592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, A.M. Paul Ehrlich’s passion: The origins of his receptor immunology. Cell. Immunol. 1999, 194, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Levi-Schaffer, F.; Sela, M.; Yarden, Y. Immunotherapy of cancer: From monoclonal to oligoclonal cocktails of anti-cancer antibodies: Iuphar review 18. Br. J. Pharmacol. 2016, 173, 1407–1424. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Reproducibility crisis: Blame it on the antibodies. Nature 2015, 521, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Radom, F.; Jurek, P.M.; Mazurek, M.P.; Otlewski, J.; Jeleń, F. Aptamers: Molecules of great potential. Biotechnol. Adv. 2013, 31, 1260–1274. [Google Scholar] [CrossRef] [PubMed]
- Maggi, E.; Vultaggio, A.; Matucci, A. Acute infusion reactions induced by monoclonal antibody therapy. Expert Rev. Clin. Immunol. 2011, 7, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the Anti-CD28 monoclonal antibody Tgn1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Descotes, J. Immunotoxicity of monoclonal antibodies. mABs 2009, 1, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; van den Bent, M.; Lassman, A.B.; Reardon, D.A.; Scott, A.M. Antibody–drug conjugates in glioblastoma therapy: The right drugs to the right cells. Nat. Rev. Clin. Oncol. 2017, 14, 695. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M. Antibodies to Watch in 2017. mABs 2017, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Sarafraz-Yazdi, E.; Pincus, M.R.; Michl, J. Tumor-targeting peptides and small molecules as anti-cancer agents to overcome drug resistance. Curr. Med. Chem. 2014, 21, 1618–1630. [Google Scholar] [CrossRef] [PubMed]
- Le Joncour, V.; Laakkonen, P. Seek & destroy, use of targeting peptides for cancer detection and drug delivery. Bioorg. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Mousavizadeh, A.; Jabbari, A.; Akrami, M.; Bardania, H. Cell targeting peptides as smart ligands for targeting of therapeutic or diagnostic agents: A systematic review. Colloids Surf. B Biointerfaces 2017, 158, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zong, H.; Ma, C.; Ming, X.; Shang, M.; Li, K.; He, X.; Du, H.; Cao, L. Epidermal growth factor receptor in glioblastoma. Oncol. Lett. 2017, 14, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Faivre, S.; Armand, J.P. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs 2000, 60, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Prados, M.D.; Chang, S.M.; Butowski, N.; DeBoer, R.; Parvataneni, R.; Carliner, H.; Kabuubi, P.; Ayers-Ringler, J.; Rabbitt, J.; Page, M.; et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J. Clin. Oncol. 2009, 27, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: Eortc brain tumor group study. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; Cavallo, G.; Lonardi, S.; Magrini, E.; Tosoni, A.; Grosso, D.; Scopece, L.; Blatt, V.; Urbini, B.; Pession, A.; et al. Gefitinib in patients with progressive high-grade gliomas: A multicentre phase II study by gruppo italiano cooperativo di neuro-oncologia (GICNO). Br. J. Cancer 2007, 96, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Narita, Y. Drug review: Safety and efficacy of bevacizumab for glioblastoma and other brain tumors. Jpn. J. Clin. Oncol. 2013, 43, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Tipping, M.; Eickhoff, J.; Robins, H.I. Clinical outcomes in recurrent glioblastoma with bevacizumab therapy: An analysis of the literature. J. Clin. Neurosci. 2017, 44, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Gabathuler, R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol. Dis. 2010, 37, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.S.; Amin, A.F. Getting into the brain: Approaches to enhance brain drug delivery. CNS Drugs 2009, 23, 35–58. [Google Scholar] [CrossRef] [PubMed]
- Baseri, B.; Choi, J.J.; Tung, Y.S.; Konofagou, E.E. Multi-modality safety assessment of blood-brain barrier opening using focused ultrasound and definity microbubbles: A short-term study. Ultrasound Med. Biol. 2010, 36, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Calias, P.; Banks, W.A.; Begley, D.; Scarpa, M.; Dickson, P. Intrathecal delivery of protein therapeutics to the brain: A critical reassessment. Pharmacol. Ther. 2014, 144, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, S.B.; Kohman, R.E.; Feldman, R.E.; Ramanlal, S.; Han, X. Permeabilization of the blood-brain barrier via mucosal engrafting: Implications for drug delivery to the brain. PLoS ONE 2013, 8, e61694. [Google Scholar]
- Sheikov, N.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.; Hynynen, K. Cellular mechanisms of the blood-brain barrier opening induced by ultrasound in presence of microbubbles. Ultrasound Med. Biol. 2004, 30, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.M.; Bleier, B.S. The blood-brain barrier and nasal drug delivery to the central nervous system. Am. J. Rhinol. Allergy 2015, 29, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Aprili, D.; Bandschapp, O.; Rochlitz, C.; Urwyler, A.; Ruppen, W. Serious complications associated with external intrathecal catheters used in cancer pain patients: A systematic review and meta-analysis. Anesthesiology 2009, 111, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dalwadi, G.; Benson, H.A. Drug delivery across the blood-brain barrier. Curr. Drug Deliv. 2004, 1, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011, 3, 84ra44. [Google Scholar] [CrossRef] [PubMed]
- Pestourie, C.; Tavitian, B.; Duconge, F. Aptamers against extracellular targets for in vivo applications. Biochimie 2005, 87, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.; Henri, J.; Goodman, L.; Xiang, D.; Duan, W.; Shigdar, S. Development of a bifunctional aptamer targeting the transferrin receptor and epithelial cell adhesion molecule (EPCAM) for the treatment of brain cancer metastases. ACS Chem. Neurosci. 2017, 8, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, D.; Schluesener, H.J.; Zhang, Z. Advances in SELEX and application of aptamers in the central nervous system. Biomol. Eng. 2007, 24, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Savory, N.; Abe, K.; Ikebukuro, K. Methods for improving aptamer binding affinity. Molecules 2016, 21, 421. [Google Scholar] [CrossRef] [PubMed]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA aptamers using cell-SELEX. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Bunka, D.H.J.; Stockley, P.G. Aptamers come of age—At last. Nat. Rev. Microbiol. 2006, 4, 588–596. [Google Scholar] [CrossRef] [PubMed]
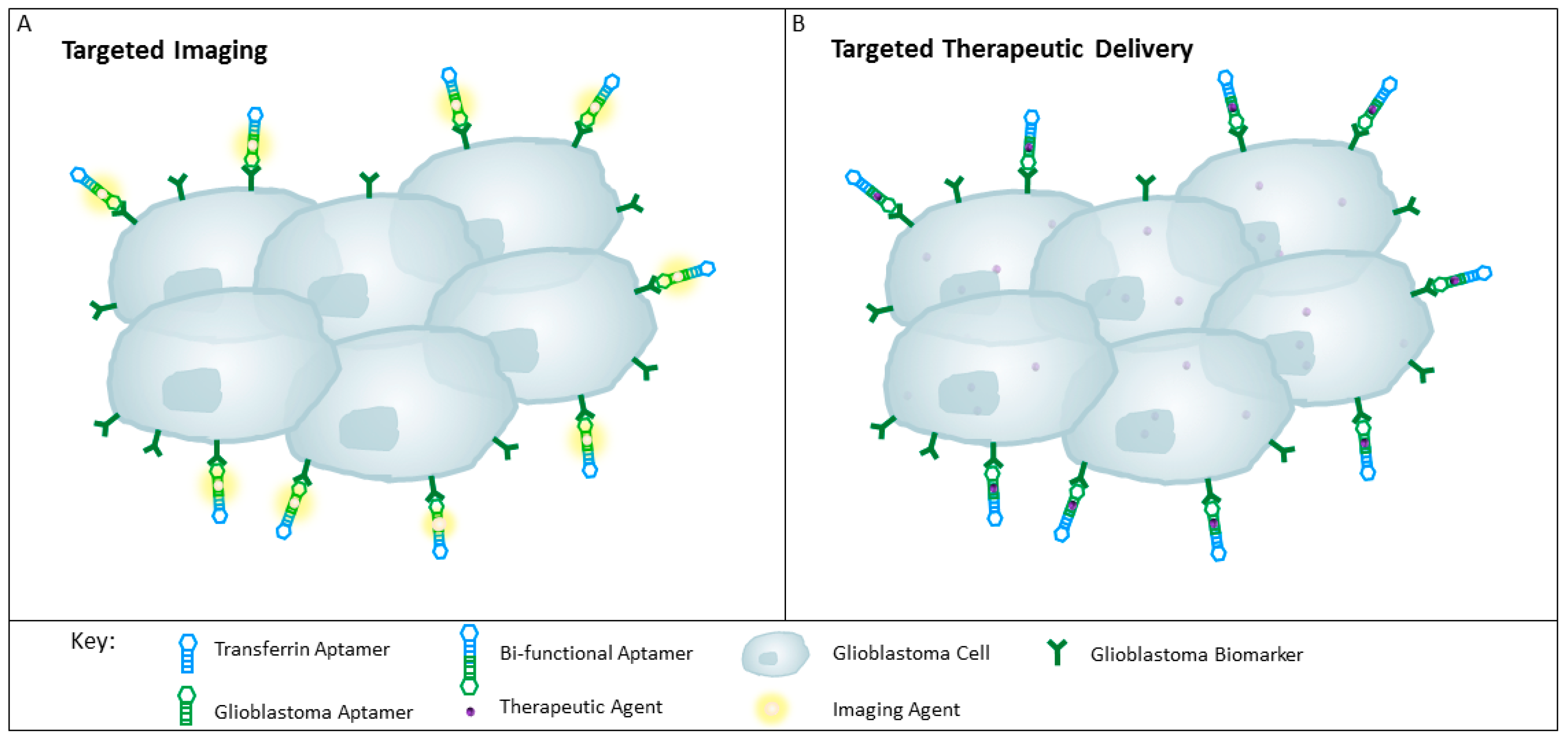
- Hicke, B.J.; Stephens, A.W. Escort aptamers: A delivery service for diagnosis and therapy. J. Clin. Investig. 2000, 106, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, C.A.; Cai, W.; Hong, H. Applications of aptamers in targeted imaging: State of the art. Curr. Top. Med. Chem. 2015, 15, 1138–1152. [Google Scholar] [CrossRef] [PubMed]
- Keshtkar, M.; Shahbazi-Gahrouei, D.; Khoshfetrat, S.M.; Mehrgardi, M.A.; Aghaei, M. Aptamer-conjugated magnetic nanoparticles as targeted magnetic resonance imaging contrast agent for breast cancer. J. Med. Signals Sens. 2016, 6, 243–247. [Google Scholar] [PubMed]
- Hicke, B.J.; Stephens, A.W.; Gould, T.; Chang, Y.F.; Lynott, C.K.; Heil, J.; Borkowski, S.; Hilger, C.S.; Cook, G.; Warren, S.; et al. Tumor targeting by an aptamer. J. Nucl. Med. 2006, 47, 668–678. [Google Scholar] [PubMed]
- Rockey, W.M.; Huang, L.; Kloepping, K.C.; Baumhover, N.J.; Giangrande, P.H.; Schultz, M.K. Synthesis and radiolabeling of chelator-RNA aptamer bioconjugates with copper-64 for targeted molecular imaging. Bioorg. Med. Chem. 2011, 19, 4080–4090. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S. What potential do aptamers hold in therapeutic delivery? Ther. Deliv. 2017, 8, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Chandola, C.; Kalme, S.; Casteleijn, M.G.; Urtti, A.; Neerathilingam, M. Application of aptamers in diagnostics, drug-delivery and imaging. J. Biosci. 2016, 41, 535–561. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Gijs, M.; Aerts, A.; Impens, N.; Baatout, S.; Luxen, A. Aptamers as radiopharmaceuticals for nuclear imaging and therapy. Nucl. Med. Biol. 2016, 43, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wu, Q.; Hamerlik, P.; Hitomi, M.; Sloan, A.E.; Barnett, G.H.; Weil, R.J.; Leahy, P.; Hjelmeland, A.B.; Rich, J.N. Aptamer identification of brain tumor-initiating cells. Cancer Res. 2013, 73, 4923–4936. [Google Scholar] [CrossRef] [PubMed]
- Hicke, B.J.; Marion, C.; Chang, Y.F.; Gould, T.; Lynott, C.K.; Parma, D.; Schmidt, P.G.; Warren, S. Tenascin-C Aptamers are generated using tumor cells and purified protein. J. Biol. Chem. 2001, 276, 48644–48654. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, O.; Yan, X.; Niu, G.; Weiss, I.D.; Ma, Y.; Szajek, L.P.; Shen, B.; Kiesewetter, D.O.; Chen, X. Pet imaging of tenascin-C with a radiolabeled single-stranded DNA aptamer. J. Nucl. Med. 2015, 56, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Deng, J.; Jin, H.; Yang, X.; Fan, X.; Li, L.; Zhao, Y.; Guan, Z.; Wu, Y.; Zhang, L.; et al. Chemical modification improves the stability of the DNA aptamer GBI-10 and its affinity towards tenascin-c. Org. Biomol. Chem. 2017, 15, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Camorani, S.; Esposito, C.L.; Rienzo, A.; Catuogno, S.; Iaboni, M.; Condorelli, G.; de Franciscis, V.; Cerchia, L. Inhibition of receptor signaling and of glioblastoma-derived tumor growth by a novel PDGFRβ aptamer. Mol. Ther. 2014, 22, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.L.; Nuzzo, S.; Kumar, S.A.; Rienzo, A.; Lawrence, C.L.; Pallini, R.; Shaw, L.; Alder, J.E.; Ricci-Vitiani, L.; Catuogno, S.; et al. A combined microrna-based targeted therapeutic approach to eradicate glioblastoma stem-like cells. J. Control Release 2016, 238, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, L.; Esposito, C.L.; Camorani, S.; Rienzo, A.; Stasio, L.; Insabato, L.; Affuso, A.; de Franciscis, V. Targeting Axl with an high-affinity inhibitory aptamer. Mol. Ther. 2012, 20, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Parker, G.J.; Mullamitha, S.; Valle, J.W.; Saunders, M.; Broughton, L.; Lawrance, J.; Carrington, B.; Roberts, C.; Issa, B.; et al. Blockade of platelet-derived growth factor receptor-β by CDP860, a humanized, pegylated DI-FAB’, leads to fluid accumulation and is associated with increased tumor vascularized volume. J. Clin. Oncol. 2005, 23, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Chee, C.E.; Krishnamurthi, S.; Nock, C.J.; Meropol, N.J.; Gibbons, J.; Fu, P.F.; Bokar, J.; Teston, L.; O’Brien, T.; Gudena, V.; et al. Phase II study of dasatinib (BMS-354825) in patients with metastatic adenocarcinoma of the pancreas. Oncologist 2013, 18, 1091–1092. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liang, H.; Tan, Y.; Yuan, C.; Li, S.; Li, X.; Li, G.; Shi, Y.; Zhang, X. Cell-SELEX aptamer for highly specific radionuclide molecular imaging of glioblastoma in vivo. PLoS ONE 2014, 9, e90752. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Shi, Y.S.; Wu, X.D.; Liang, H.Y.; Gao, Y.B.; Li, S.J.; Zhang, X.M.; Wang, F.; Gao, T.M. DNA aptamers that target human glioblastoma multiforme cells overexpressing epidermal growth factor receptor variant iii in vitro. Acta Pharmacol. Sin. 2013, 34, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liang, H.; Tan, Y.; Wu, X.; Li, S.; Shi, Y. A U87-EGFRvIII cell-specific aptamer mediates small interfering RNA delivery. Biomed. Rep. 2014, 2, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Huang, N.; Zhang, X.; Zhou, T.; Tan, Y.; Pi, J.; Pi, L.; Cheng, S.; Zheng, H.; Cheng, Y. Aptamer-conjugated pegylated quantum dots targeting epidermal growth factor receptor variant III for fluorescence imaging of glioma. Int. J. Nanomed. 2017, 12, 3899–3911. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Nguyen, H.H.; Byrom, M.; Ellington, A.D. Inhibition of cell proliferation by an anti-EGFR aptamer. PLoS ONE 2011, 6, e20299. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Liu, Y.; Allen, P.B.; Asghar, W.; Mahmood, M.A.I.; Tan, J.; Duhon, H.; Kim, Y.; Ellington, A.D.; Iqbal, S.M. Capture, isolation and release of cancer cells with aptamer-functionalized glass bead array. Lab Chip 2012, 12, 4693–4701. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Mahmood, M.A.I.; Li, N.; Allen, P.B.; Kim, Y.; Bachoo, R.; Ellington, A.D.; Iqbal, S.M. Nanotextured substrates with immobilized aptamers for cancer cell isolation and cytology. Cancer 2012, 118, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Bayrac, A.T.; Sefah, K.; Parekh, P.; Bayrac, C.; Gulbakan, B.; Oktem, H.A.; Tan, W. In vitro selection of dna aptamers to glioblastoma multiforme. ACS Chem. Neurosci. 2011, 2, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Wang, J.; Zhang, W.; Song, Y.; Li, X.; Zou, Y.; Zhu, M.; Zhu, Z.; Chen, F.; Yang, C.J. Selection of DNA aptamers against glioblastoma cells with high affinity and specificity. PLoS ONE 2012, 7, e42731. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Qiao, L.; Zhou, S.F.; Xiang, D.; Wang, T.; Li, Y.; Lim, L.Y.; Kong, L.; Li, L.; Duan, W. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013, 330, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.L.; Passaro, D.; Longobardo, I.; Condorelli, G.; Marotta, P.; Affuso, A.; de Franciscis, V.; Cerchia, L. A neutralizing RNA aptamer against EGFR causes selective apoptotic cell death. PLoS ONE 2011, 6, e24071. [Google Scholar] [CrossRef] [PubMed]
- Camorani, S.; Crescenzi, E.; Colecchia, D.; Carpentieri, A.; Amoresano, A.; Fedele, M.; Chiariello, M.; Cerchia, L. Aptamer targeting EGFRvIII mutant hampers its constitutive autophosphorylation and affects migration, invasion and proliferation of glioblastoma cells. Oncotarget 2015, 6, 37570–37587. [Google Scholar] [CrossRef] [PubMed]
- Camorani, S.; Crescenzi, E.; Gramanzini, M.; Fedele, M.; Zannetti, A.; Cerchia, L. Aptamer-mediated impairment of EGFR-integrin Αvβ3 complex inhibits vasculogenic mimicry and growth of triple-negative breast cancers. Sci. Rep. 2017, 7, 46659. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Brescia, P.; Ortensi, B.; Fornasari, L.; Levi, D.; Broggi, G.; Pelicci, G. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 2013, 31, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Vallera, D.A. CD133, selectively targeting the root of cancer. Toxins 2016, 8, 165. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.P.; Bourdon, M.A. Tenascin: An extracellular matrix protein prominent in specialized embryonic tissues and tumors. Annu. Rev. Cell Biol. 1989, 5, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Tiitta, O.; Virtanen, I.; Sipponen, P.; Gould, V. Tenascin expression in inflammatory, dysplastic and neoplastic lesions of the human stomach. Virchows Arch. 1994, 425, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Denayer, T.; Stöhr, T.; van Roy, M. Animal models in translational medicine: Validation and prediction. New Horiz. Transl. Med. 2014, 2, 5–11. [Google Scholar] [CrossRef]
- Östman, A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004, 15, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, E.; Wu, Q.; Guryanova, O.; Hitomi, M.; Lathia, J.D.; Serwanski, D.; Sloan, A.E.; Weil, R.J.; Lee, J.; et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev. 2012, 26, 1247–1262. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Shaw, L.; Lawrence, C.; Lea, R.; Alder, J. P50developing a physiologically relevant blood brain barrier model for the study of drug disposition in glioma. Neuro Oncol. 2014, 16, vi8. [Google Scholar] [CrossRef]
- Shieh, Y.-S.; Lai, C.-Y.; Kao, Y.-R.; Shiah, S.-G.; Chu, Y.-W.; Lee, H.-S.; Wu, C.-W. Expression of Axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [PubMed]
- Zhang, Y.X.; Knyazev, P.G.; Cheburkin, Y.V.; Sharma, K.; Knyazev, Y.P.; Orfi, L.; Szabadkai, I.; Daub, H.; Keri, G.; Ullrich, A. Axl is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008, 68, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, M.; Knyazev, P.; Abate, A.; Reschke, M.; Maier, H.; Stefanova, N.; Knyazeva, T.; Barbieri, V.; Reindl, M.; Muigg, A.; et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Ginisty, H.; Sicard, H.; Roger, B.; Bouvet, P. Structure and functions of nucleolin. J. Cell Sci. 1999, 112 Pt 6, 761–772. [Google Scholar] [PubMed]
- Berger, C.M.; Gaume, X.; Bouvet, P. The roles of nucleolin subcellular localization in cancer. Biochimie 2015, 113, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.N.; Castro, J.E.; Motta, M.; Cottam, H.B.; Kyburz, D.; Kipps, T.J.; Corr, M.; Carson, D.A. Selection of oligonucleotide aptamers with enhanced uptake and activation of human leukemia B cells. Hum. Gene Ther. 2003, 14, 849. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A. The experimental use of antisense oligonucleotides: A guide for the perplexed. J. Clin. Investig. 2001, 108, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Yan, Z.; Jin, K.; Pang, Q.; Jiang, T.; Lu, H.; Liu, X.; Pang, Z.; Yu, L.; Jiang, X. Precise glioblastoma targeting by AS1411 aptamer-functionalized poly (l-γ-glutamylglutamine)-paclitaxel nanoconjugates. J. Colloid Interface Sci. 2017, 490, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Bambury, R.M.; van Allen, E.M.; Drabkin, H.A.; Lara, P.N., Jr.; Harzstark, A.L.; Wagle, N.; Figlin, R.A.; Smith, G.W.; Garraway, L.A.; et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Investig. New Drugs 2014, 32, 178–187. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aptamer | Target | SELEX Method | Positive Selection | Negative Selection | Reference(s) |
|---|---|---|---|---|---|
| A3, A4 | Unknown | Differential cell-SELEX | CD133+ TIC | CD133− cells; human neural progenitor cells | [72] |
| TTA1 | Tenascin-C | Crossover-SELEX | U251 cells; human Tenascin-C | No negative selection | [66,73] |
| GBI-10 | Tenascin-C | Cell-SELEX | U251 cells | No negative selection | [74,75] |
| Gint4.T | PDGFRβ | Cell-SELEX | U87MG cells | No negative selection | [76,77] |
| GL21.T | Axl | Differential cell-SELEX | U87MG cells | T98G cells | [77,78,79,80] |
| U2 | EGFRvIII | Differential cell-SELEX | U87MG-EGFRvIII cells | U87MG cells | [81] |
| Aptamer 32 | EGFRvIII | Differential cell-SELEX | U87MG-EGFRvIII cells | U87MG cells | [82,83,84] |
| E07 | EGFR; EGFRvIII | Protein-SELEX | Human EGFR | No negative selection | [85,86,87] |
| GMT 3–9 | Unknown | Differential cell-SELEX | A172 cells | No negative selection | [88] |
| GBM128, GBM131 | Unknown | Differential cell-SELEX | U118-MG cells | SVGp12 cells | [89] |
| Aptamer | Target | SELEX Method | Positive Selection | Negative Selection | Reference(s) |
|---|---|---|---|---|---|
| CD133-A15, CD133-B19 | CD133 | Differential cell-SELEX | HEK293T-CD133 cells | HEK293T cells | [90] |
| CL4 | EGFR; EGFRvIII | Differential cell-SELEX | A549 cells | H460 cells | [91,92,93] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hays, E.M.; Duan, W.; Shigdar, S. Aptamers and Glioblastoma: Their Potential Use for Imaging and Therapeutic Applications. Int. J. Mol. Sci. 2017, 18, 2576. https://doi.org/10.3390/ijms18122576
Hays EM, Duan W, Shigdar S. Aptamers and Glioblastoma: Their Potential Use for Imaging and Therapeutic Applications. International Journal of Molecular Sciences. 2017; 18(12):2576. https://doi.org/10.3390/ijms18122576
Chicago/Turabian StyleHays, Emma M., Wei Duan, and Sarah Shigdar. 2017. "Aptamers and Glioblastoma: Their Potential Use for Imaging and Therapeutic Applications" International Journal of Molecular Sciences 18, no. 12: 2576. https://doi.org/10.3390/ijms18122576
APA StyleHays, E. M., Duan, W., & Shigdar, S. (2017). Aptamers and Glioblastoma: Their Potential Use for Imaging and Therapeutic Applications. International Journal of Molecular Sciences, 18(12), 2576. https://doi.org/10.3390/ijms18122576