Quinacrine Inhibits ICAM-1 Transcription by Blocking DNA Binding of the NF-κB Subunit p65 and Sensitizes Human Lung Adenocarcinoma A549 Cells to TNF-α and the Fas Ligand


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
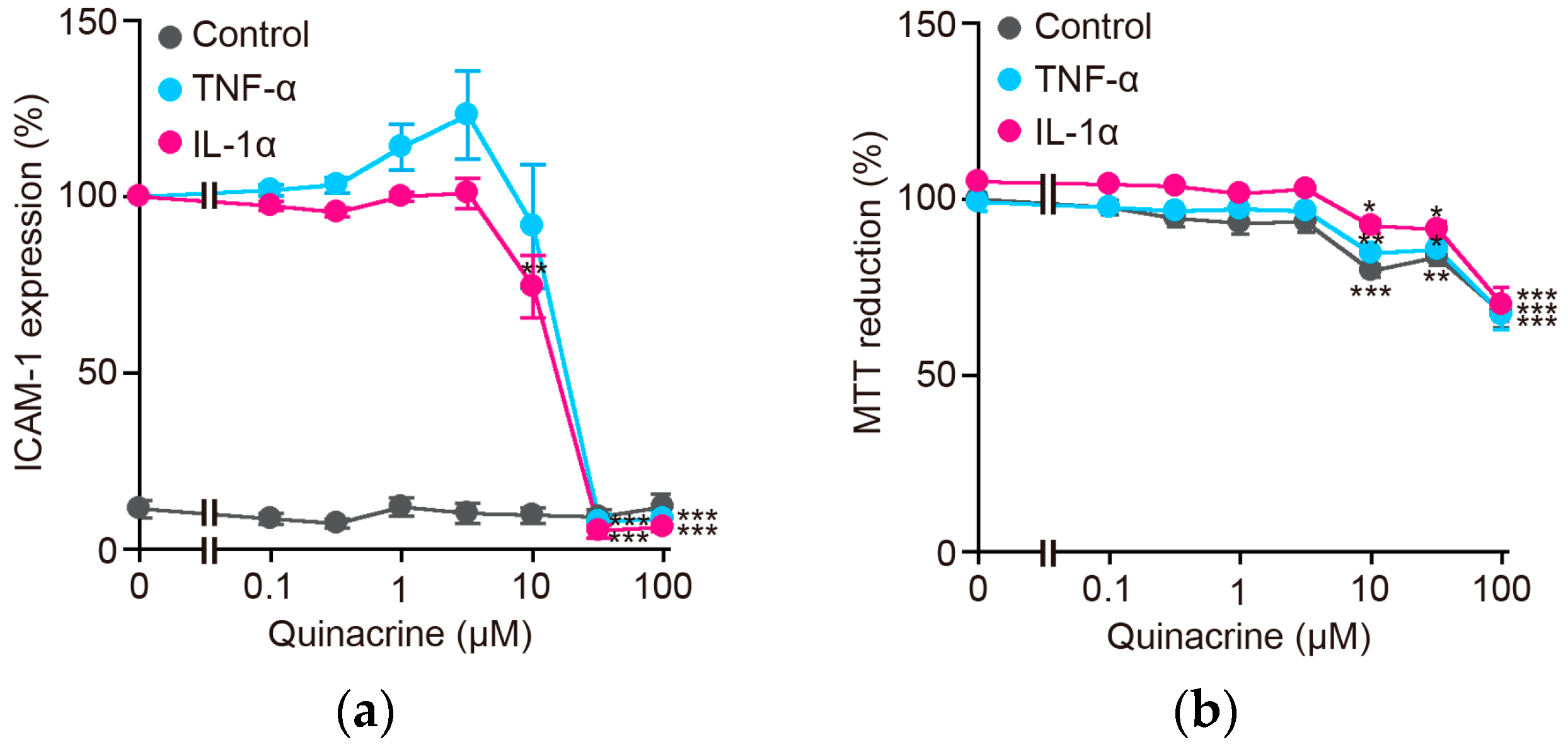
2.1. Quinacrine Inhibits the Expression of the Cell-Surface ICAM-1 Protein Induced by Inflammatory Cytokines
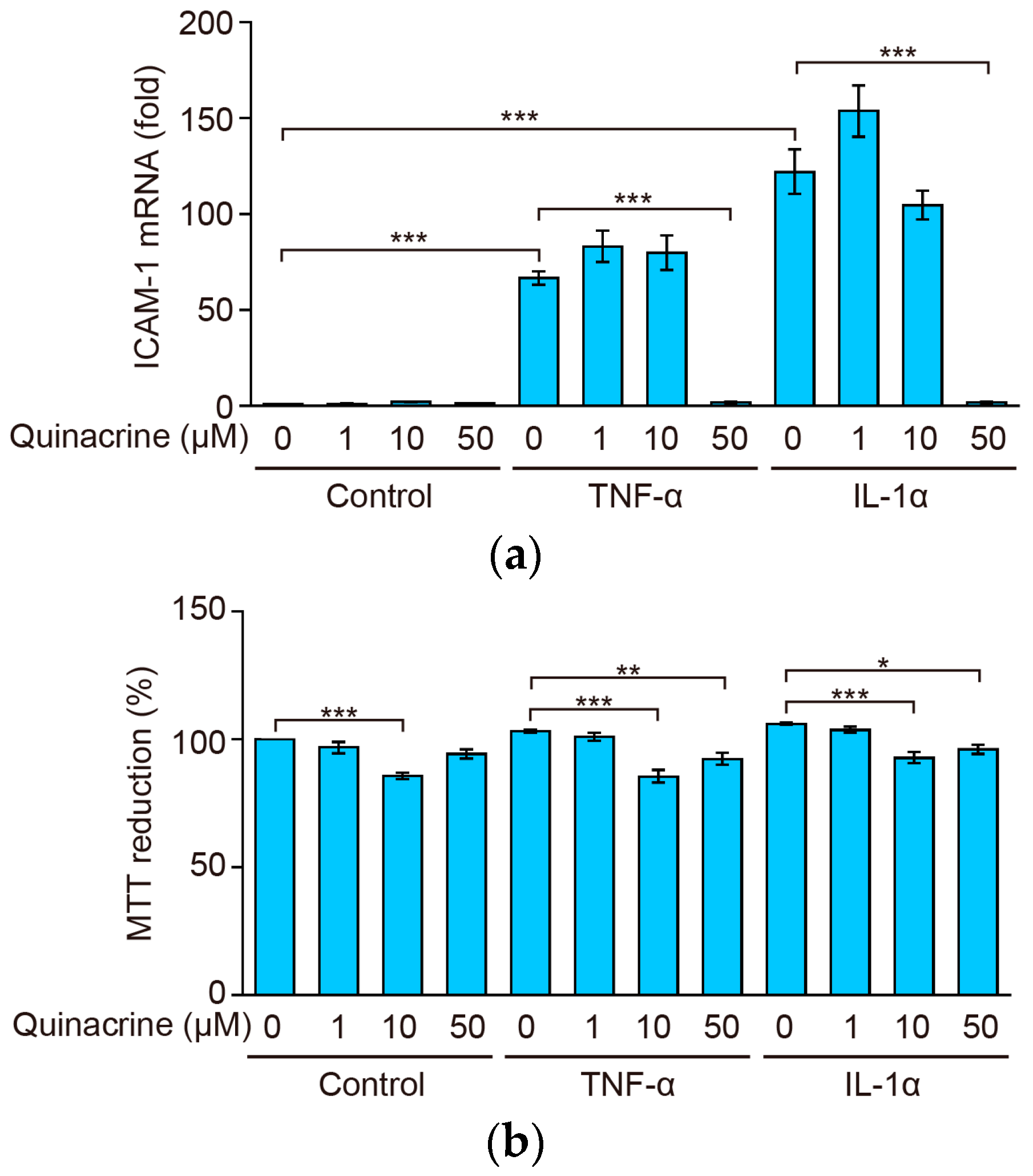
2.2. Quinacrine Inhibits the Expression of ICAM-1 mRNA Induced by Inflammatory Cytokines
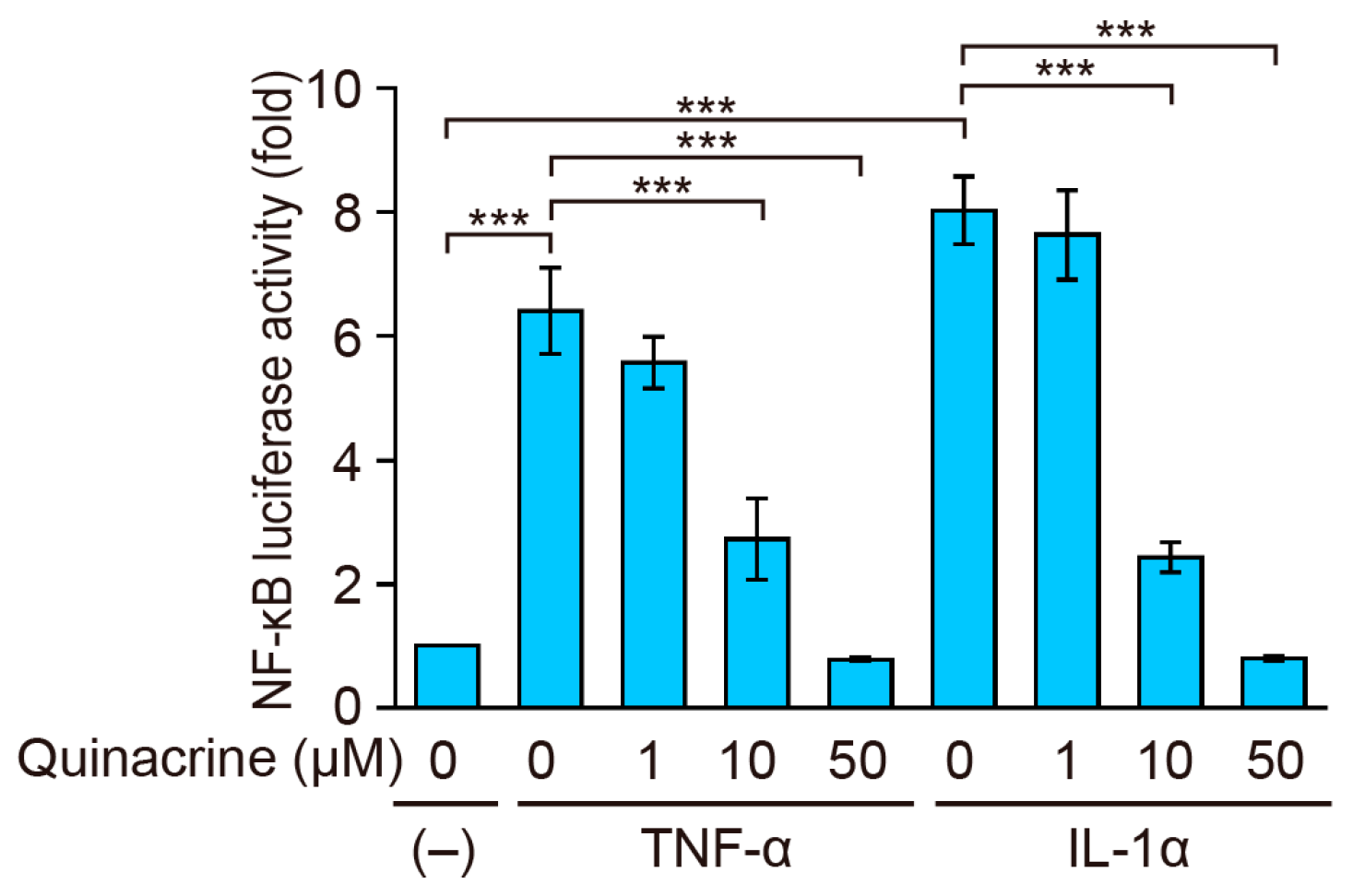
2.3. Quinacrine Inhibits NF-κB-Responsive Luciferase Activity Induced by Inflammatory Cytokines
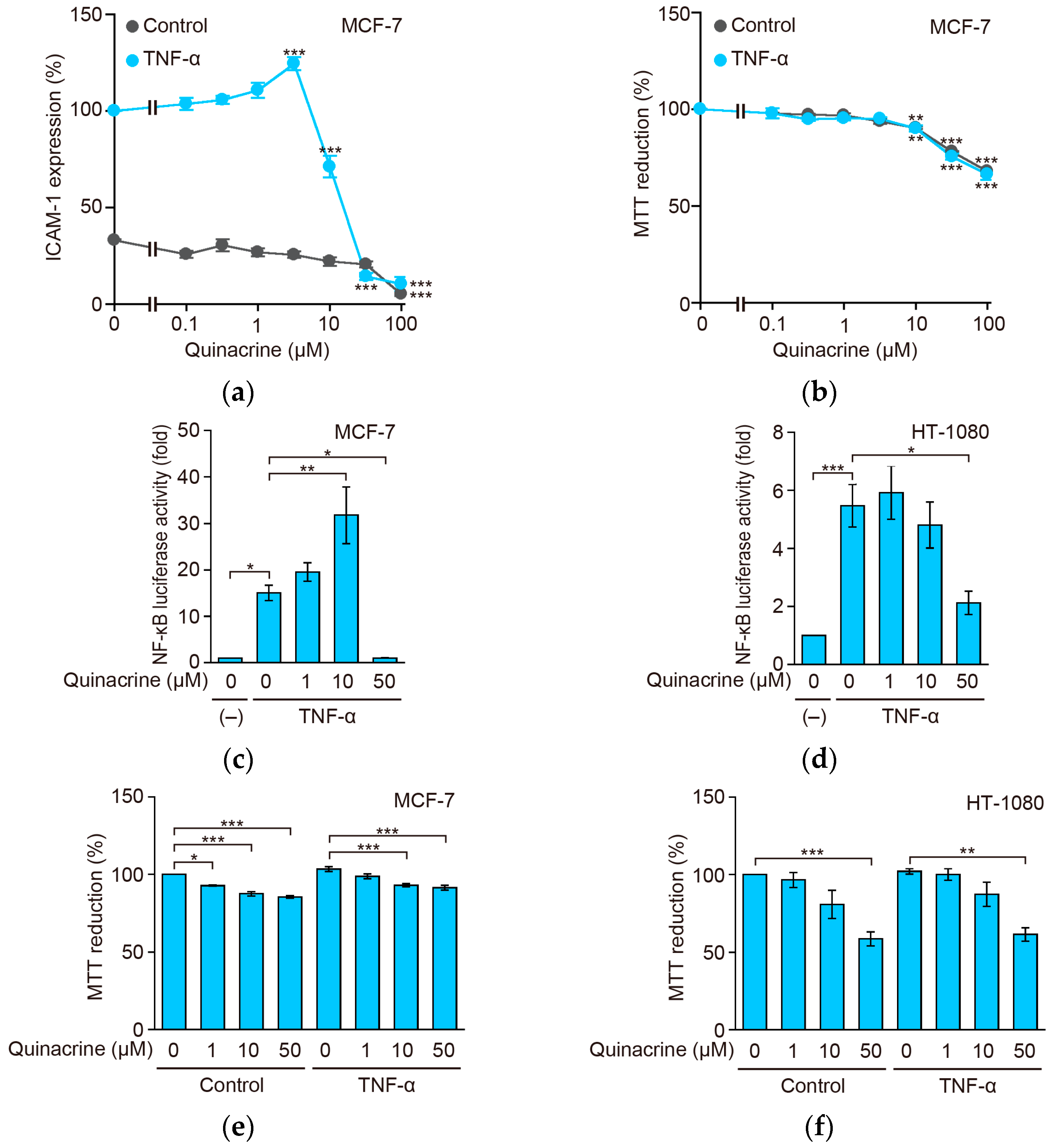
2.4. Quinacrine Inhibits TNF-α-Induced ICAM-1 Protein Expression and NF-κB-Responsive Luciferase Activity in Other Cancer Cell Lines
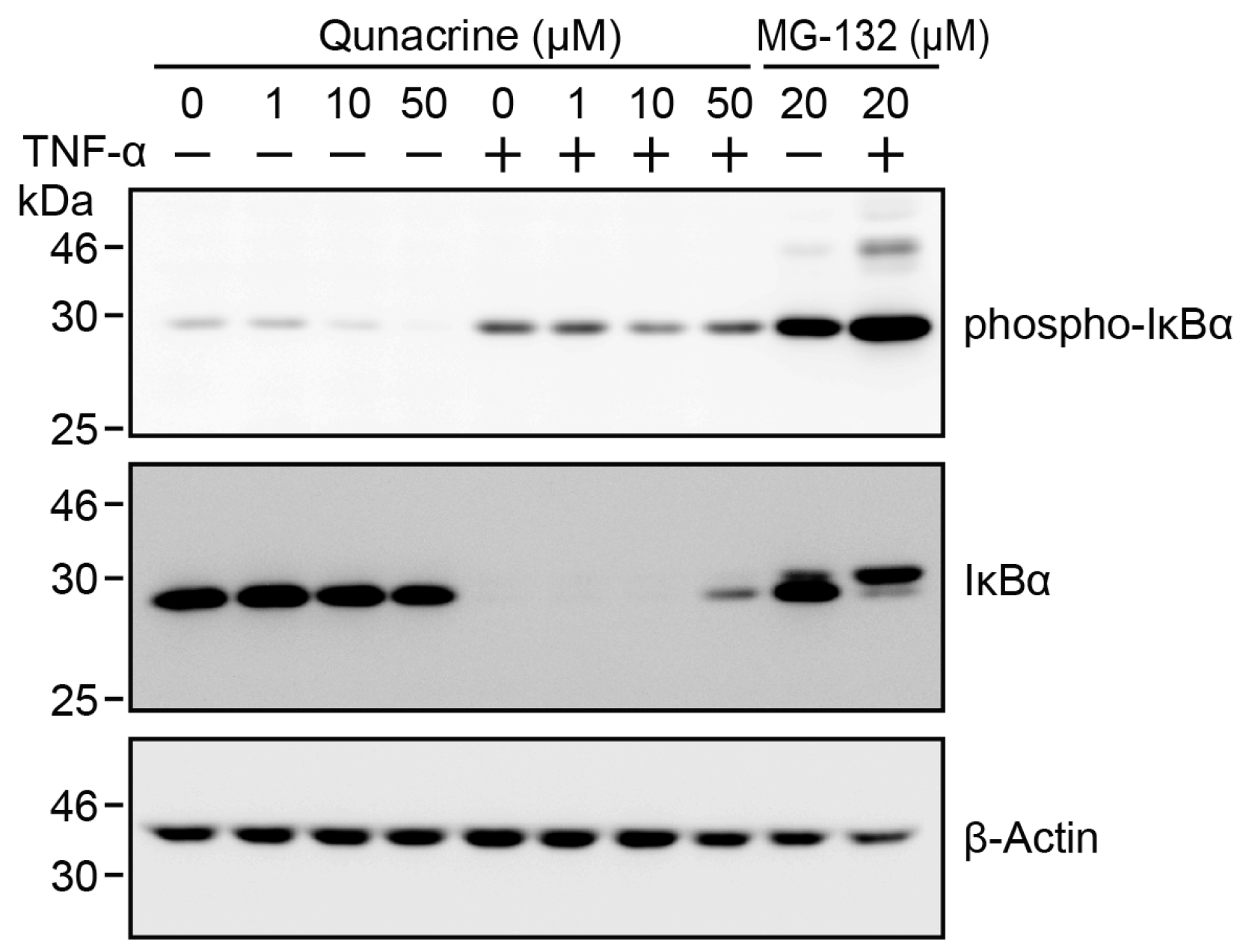
2.5. Quinacrine Does not Inhibit TNF-α-Induced IκBα Degradation
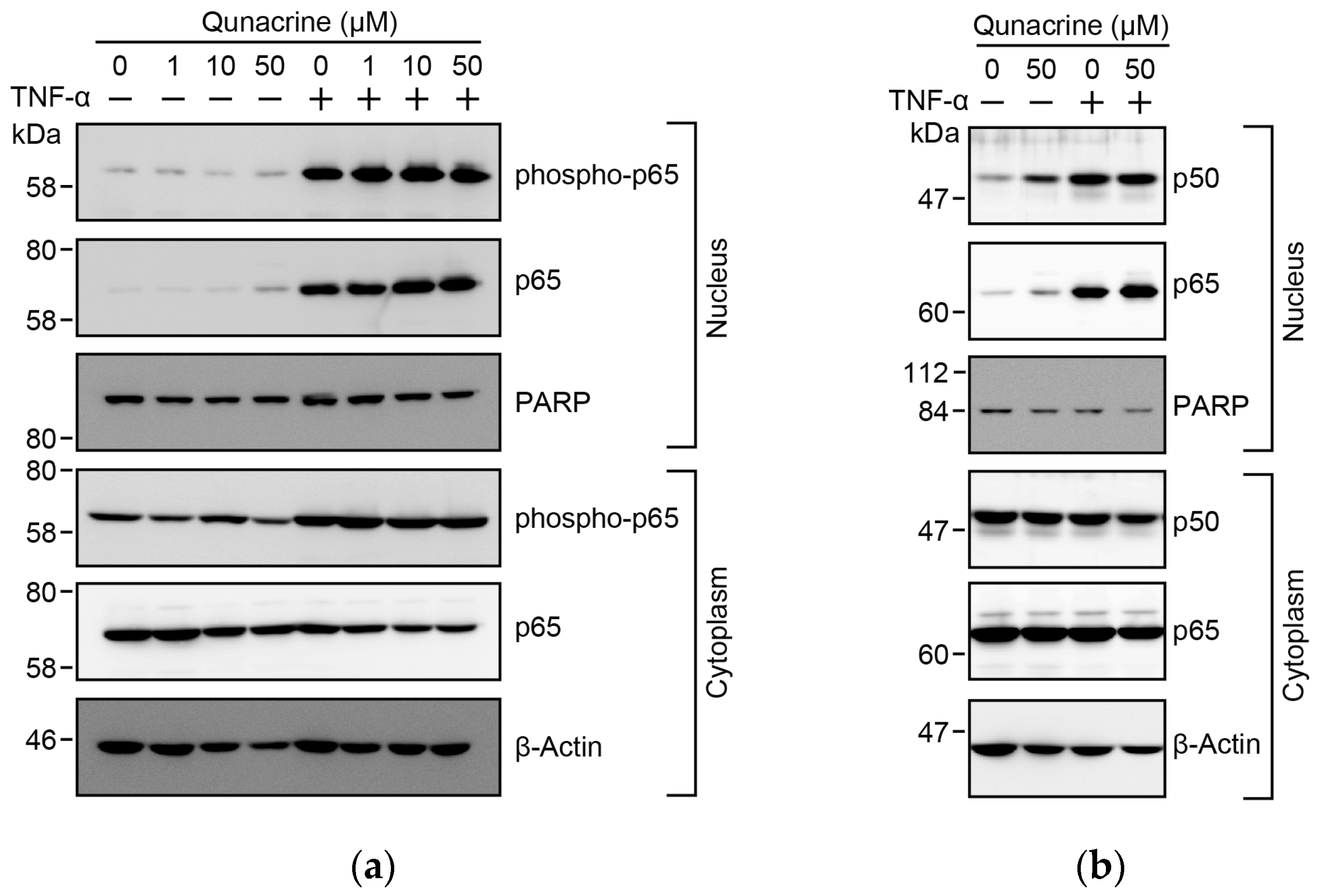
2.6. Quinacrine Does not Prevent TNF-α-Induced S-536 Phosphorylation or the Nuclear Translocation of p65
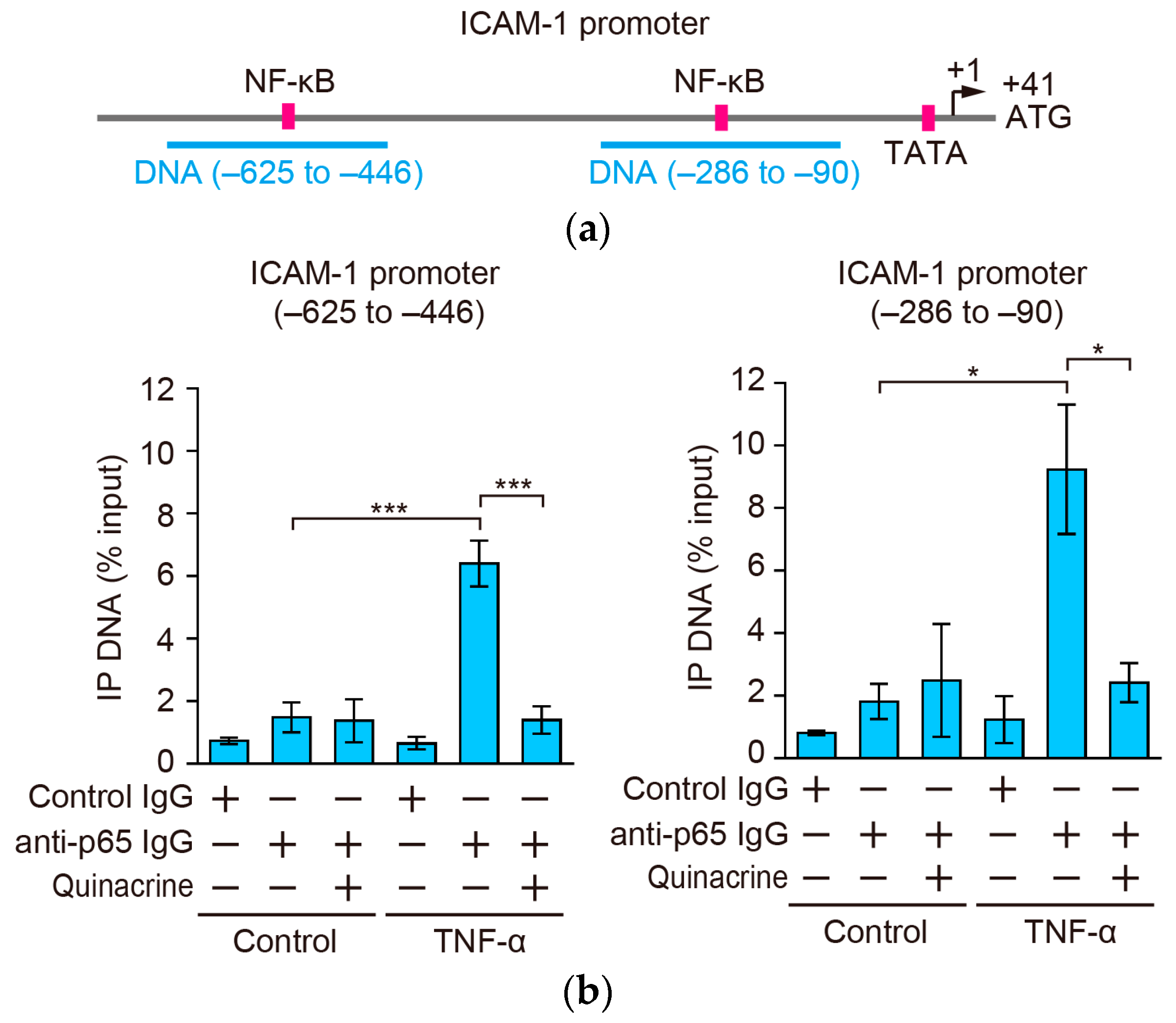
2.7. Quinacrine Inhibits the Binding of p65 to the ICAM-1 Promoter
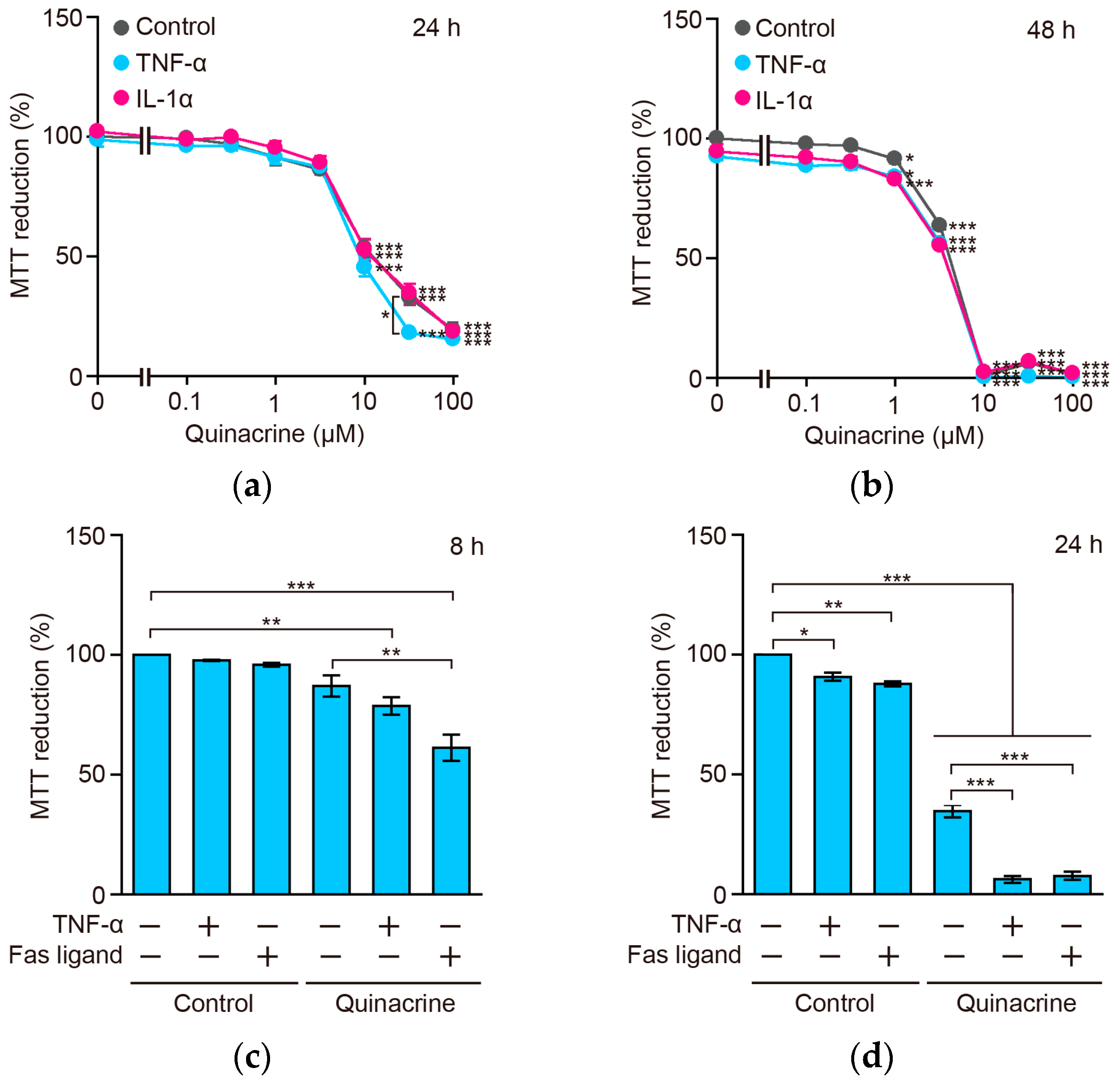
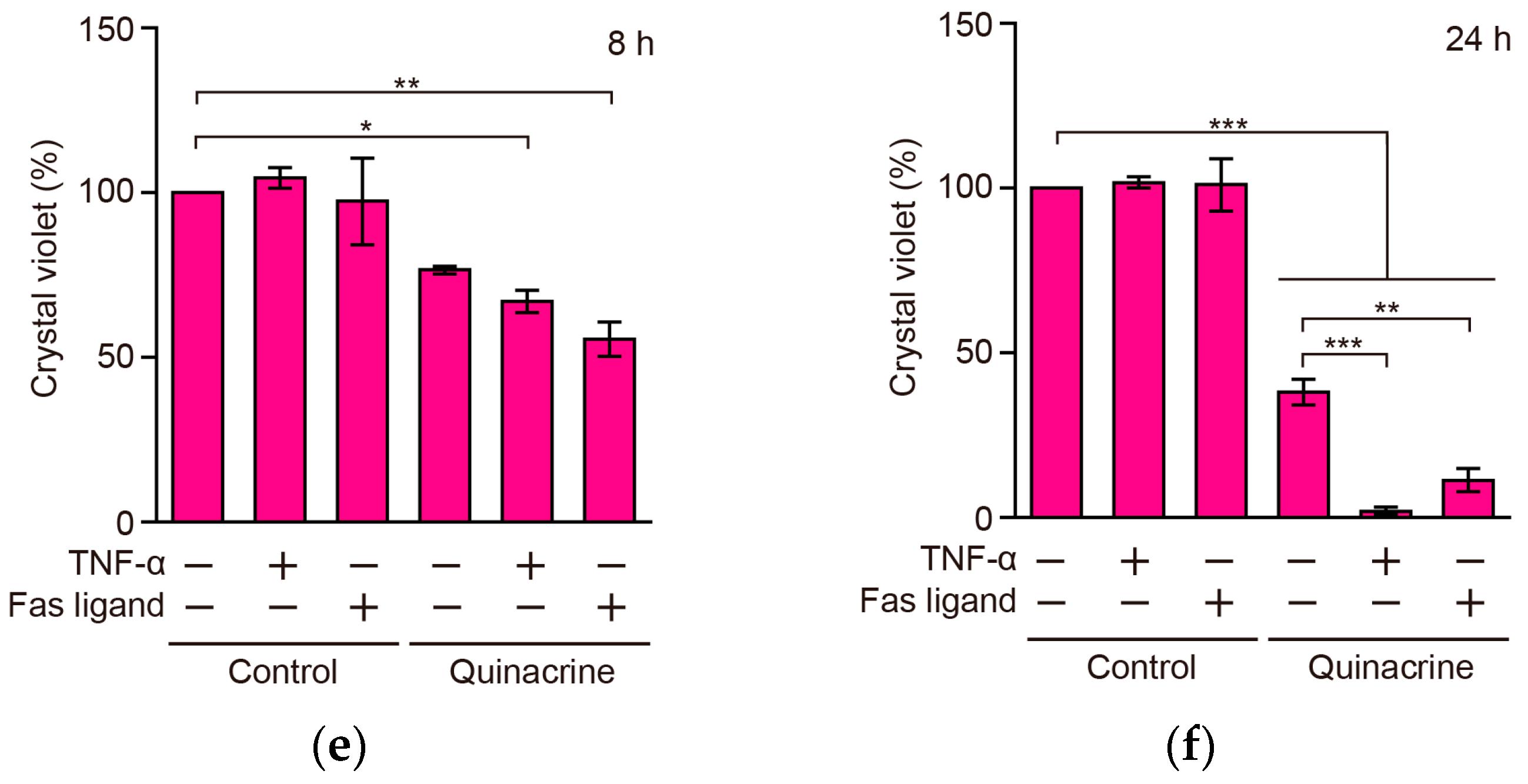
2.8. Quinacrine Sensitizes A549 Cells to TNF-α and the Fas Ligand
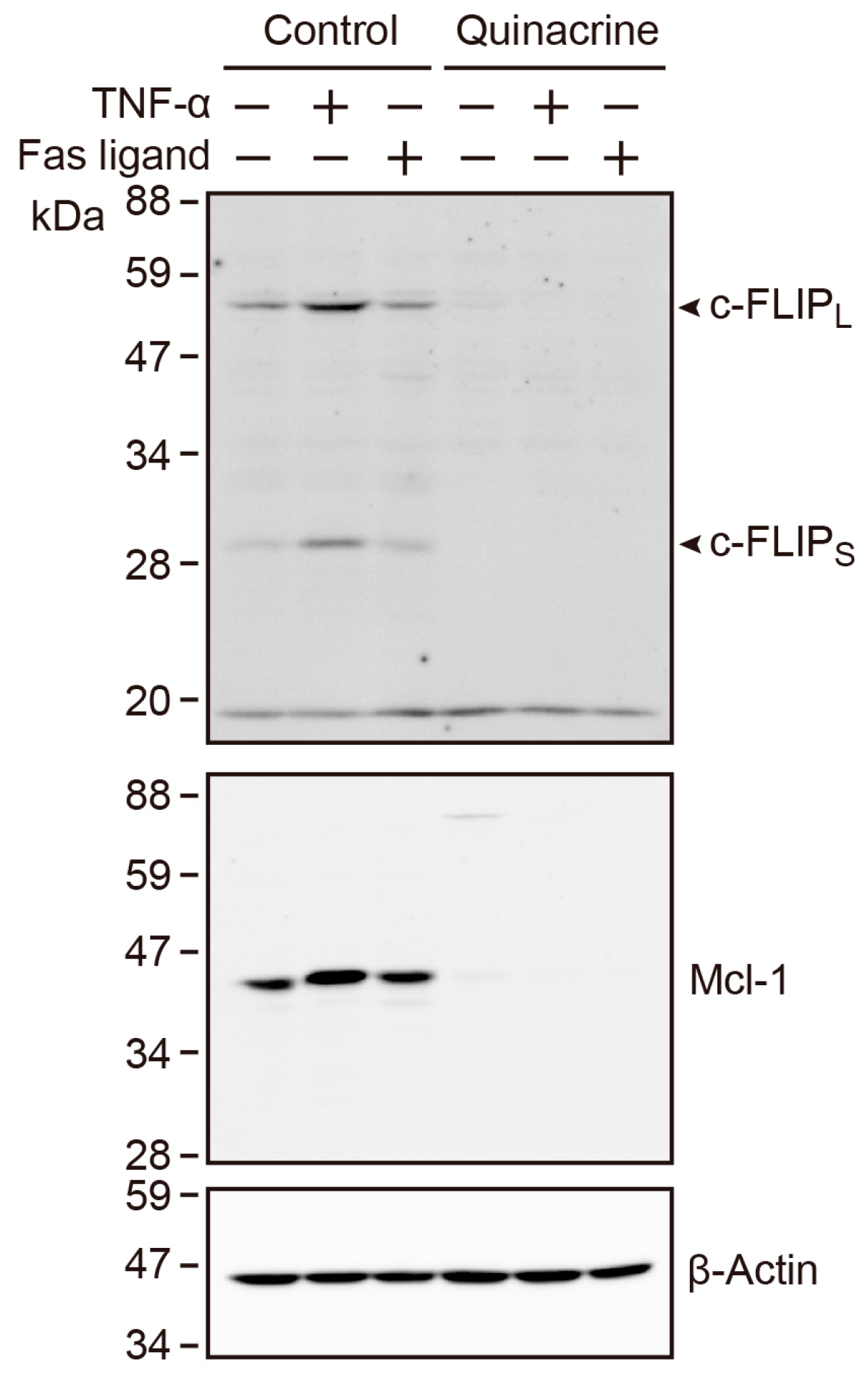
2.9. Quinacrine Down-Regulates the Constitutive and TNF-α-Induced Expression of c-FLIP and Mcl-1
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Reagents
4.3. Antibodies
4.4. ICAM-1 Expression Assay
4.5. Cell Viability Assays
4.6. Assay for ICAM-1 mRNA Expression
4.7. Luciferase Reporter Assay
4.8. Western Blotting
4.9. ChIP Assay
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BSA | bovine serum albumin |
| c-FLIP | cellular FLICE-inhibitory protein |
| ChIP | chromatin immunoprecipitation |
| ICAM-1 | intercellular adhesion molecule-1 |
| IL-1 | interleukin-1 |
| HRP | horseradish peroxidase |
| IκB | inhibitor of nuclear factor κB |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NF-κB | nuclear factor κB |
| PARP | poly(ADP-ribose) polymerase |
| PBS | phosphate-buffered saline |
| TNF | tumor necrosis factor |
| TRAIL | tumor necrosis factor-related apoptosis-inducing ligand |
References
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor promotion via injury- and death-induced inflammation. Immunity 2011, 35, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T. Chemical biology of inflammatory cytokine signaling. J. Antibiot. 2009, 62, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The TLR and IL-1 signalling network at a glance. J. Cell Sci. 2014, 127, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Iwai, K. Roles of linear ubiquitinylation, a crucial regulator of NF-κB and cell death, in the immune system. Immunol. Rev. 2015, 266, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef]
- Ghosh, G.; Wang, V.Y.F.; Huang, D.B.; Fusco, A. NF-κB regulation: Lessons from structures. Immunol. Rev. 2012, 246, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 2006, 25, 6717–6730. [Google Scholar] [CrossRef] [PubMed]
- Christian, F.; Smith, E.L.; Carmody, R.J. The regulation of NF-κB subunits by phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Ehsanian, R.; Van Waes, C.; Feller, S.M. Beyond DNA binding—A review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Commun. Signal. 2011, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Pupe, A.; Degreef, H.; Garmyn, M. Induction of tumor necrosis factor-α by UVB: A role for reactive oxygen intermediates and eicosanoids. Photochem. Photobiol. 2003, 78, 68–74. [Google Scholar] [CrossRef]
- Gurova, K.V.; Hill, J.E.; Guo, C.; Prokvolit, A.; Burdelya, L.G.; Samoylova, E.; Khodyakova, A.V.; Ganapathi, R.; Ganapathi, M.; Tararova, N.D.; et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-κB-dependent mechanism of p53 suppression in tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 17448–17453. [Google Scholar] [CrossRef] [PubMed]
- Gorbachev, A.V.; Gasparian, A.V.; Gurova, K.V.; Gudkov, A.V.; Fairchild, R.L. Quinacrine inhibits the epidermal dendritic cell migration initiating T cell-mediated skin inflammation. Eur. J. Immunol. 2007, 37, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Jani, T.S.; DeVecchio, J.; Mazumdar, T.; Agyeman, A.; Houghton, J.A. Inhibition of NF-κB signaling by quinacrine is cytotoxic to human colon carcinoma cell lines and is synergistic in combination with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) or oxaliplatin. J. Biol. Chem. 2010, 285, 19162–19172. [Google Scholar] [CrossRef] [PubMed]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer compounds that simultaneously suppress NF-κB and activate p53 by targeting FACT. Sci. Transl. Med. 2011, 3, 95ra74. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Stark, G.R. FER tyrosine kinase (FER) overexpression mediates resistance to quinacrine through EGF-dependent activation of NF-κB. Proc. Natl. Acad. Sci. USA 2011, 108, 7968–7973. [Google Scholar] [CrossRef] [PubMed]
- Preet, R.; Mohapatra, P.; Mohanty, S.; Sahu, S.K.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Quinacrine has anticancer activity in breast cancer cells through inhibition of topoisomerase activity. Int. J. Cancer 2012, 130, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Dermawan, J.K.T.; Gurova, K.; Pink, J.; Dowlati, A.; De, S.; Narla, G.; Sharma, N.; Stark, G.R. Quinacrine overcomes resistance to erlotinib by inhibiting FACT, NF-κB, and cell-cycle progression in non-small cell lung cancer. Mol. Cancer Ther. 2014, 13, 2203–2214. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gallant, J.N.; Katz, S.I.; Dolloff, N.G.; Smith, C.D.; Abdulghani, J.; Allen, J.E.; Dicker, D.T.; Hong, B.; Navaraj, A.; et al. Quinacrine sensitizes hepatocellular carcinoma cells to TRAIL and chemotherapeutic agents. Cancer Biol. Ther. 2011, 12, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.N.; Allen, J.E.; Smith, C.D.; Dicker, D.T.; Wang, W.; Dolloff, N.G.; Navaraj, A.; El-Deiry, W.S. Quiancrine synergizes with 5-fluorouracil and other therapies in colorectal cancer. Cancer Biol. Ther. 2011, 12, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A.; Finnegan, A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J. Leukoc. Biol. 1999, 66, 876–888. [Google Scholar] [PubMed]
- Ramos, T.N.; Bullard, D.C.; Barnum, S.R. ICAM-1: Isoforms and phenotypes. J. Immunol. 2014, 192, 4469–4474. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Dye, D.E.; Coombe, D.R. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int. J. Cell Biol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, R.; Fukuhara, S.; Mitsuda, S.; Yokomichi, T.; Kataoka, T. Betulinic acid and oleanolic acid, natural pentacyclic triterpenoids, interfere with N-linked glycan modifications to intercellular adhesion molecule-1, but not its intracellular transport to the cell surface. Eur. J. Pharmacol. 2015, 761, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Yokoigawa, J.; Morimoto, K.; Shiono, Y.; Uesugi, S.; Kimura, K.; Kataoka, T. Allantopyrone A, an α-pyrone metabolite from an endophytic fungus, inhibits the tumor necrosis factor α-induced nuclear factor κB signaling pathway. J. Antibiot. 2015, 68, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Kang, W.; Wang, L.; Sun, B.; Ma, J.; Zheng, C.; Sun, J.; Tian, Z.; Yang, X.; Xiao, W. E2F1 renders prostate cancer cell resistant to ICAM-1 mediated antitumor immunity by NF-κB modulation. Mol. Cancer 2014, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Degitz, K.; Lian-Jie, L.; Caughman, S.W. Cloning and characterization of the 5′-transcriptional regulatory region of the human intercellular adhesion molecule 1 gene. J. Biol. Chem. 1991, 266, 14024–14030. [Google Scholar] [PubMed]
- Ledebur, H.C.; Parks, T.P. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. J. Biol. Chem. 1995, 270, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Ogura, H.; Tsukumo, Y.; Sugimoto, H.; Igarashi, M.; Nagai, K.; Kataoka, T. Ectodomain shedding of TNF receptor 1 induced by protein synthesis inhibitors regulates TNF-α-mediated activation of NF-κB and caspase-8. Exp. Cell Res. 2008, 314, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T. The caspase-8 modulator c-FLIP. Crit. Rev. Immunol. 2005, 25, 31–58. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, I.; Matsuo, K.; Matsushita, Y.; Haruna, Y.; Niwa, M.; Kataoka, T. The C-terminal domain of the long form of cellular FLICE-inhibitory protein (c-FLIPL) inhibits the interaction of the caspase 8 prodomain with the receptor-interacting protein 1 (RIP1) death domain and regulates caspase 8-dependent nuclear factor κB (NF-κB) activation. J. Biol. Chem. 2014, 289, 3876–3887. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Taketani, S.; Osada, H.; Kataoka, T. Cytotrienin A, a translation inhibitor that induces ectodomain shedding of TNF receptor 1 via activation of ERK and p38 MAP kinase. Eur. J. Pharmacol. 2011, 667, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Mitsuda, S.; Yokomichi, T.; Yokoigawa, J.; Kataoka, T. Ursolic acid, a natural pentacyclic triterpenoid, inhibits intracellular trafficking of proteins and induces accumulation of intercellular adhesion molecule-1 linked to high-mannose-type glycans in the endoplasmic reticulum. FEBS Open Bio 2014, 4, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Liu, J.; Ouyang, X. Nucleotide-binding oligomerization domain 1 regulates Porphyromonas gingivalis-induced vascular cell adhesion molecule 1 and intercellular adhesion molecule 1 expression in endothelial cells through NF-κB pathway. J. Periodontal Res. 2015, 50, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lian, F.; Zhu, Y.; Xia, M.; Wang, Q.; Ling, W.; Wang, X.D. Cyanidin-3-O-β-glucoside inhibits LPS-induced expression of inflammatory mediators through decreasing IκBα phosphorylation in THP-1 cells. Inflamm. Res. 2010, 59, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Dohrman, A.; Kataoka, T.; Cuenin, S.; Russell, J.Q.; Tschopp, J.; Budd, R.C. Cellular FLIP (long form) regulates CD8+ T cell activation through caspase-8-dependent NF-κB activation. J. Immunol. 2005, 174, 5270–5278. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.; Kataoka, T. Deoxynivalenol induces ectodomain shedding of TNF receptor 1 and thereby inhibits the TNF-α-induced NF-κB signaling pathway. Eur. J. Pharmacol. 2013, 701, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Thippegowda, P.B.; Hu, G.; Bachmaier, K.; Christman, J.W.; Malik, A.B.; Tiruppathi, C. NF-κB regulates thrombin-induced ICAM-1 gene expression in cooperation with NFAT by binding to the intronic NF-κB site in the ICAM-1 gene. Physiol. Genom. 2009, 38, 42–53. [Google Scholar] [CrossRef] [PubMed]










© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harada, M.; Morimoto, K.; Kondo, T.; Hiramatsu, R.; Okina, Y.; Muko, R.; Matsuda, I.; Kataoka, T. Quinacrine Inhibits ICAM-1 Transcription by Blocking DNA Binding of the NF-κB Subunit p65 and Sensitizes Human Lung Adenocarcinoma A549 Cells to TNF-α and the Fas Ligand. Int. J. Mol. Sci. 2017, 18, 2603. https://doi.org/10.3390/ijms18122603
Harada M, Morimoto K, Kondo T, Hiramatsu R, Okina Y, Muko R, Matsuda I, Kataoka T. Quinacrine Inhibits ICAM-1 Transcription by Blocking DNA Binding of the NF-κB Subunit p65 and Sensitizes Human Lung Adenocarcinoma A549 Cells to TNF-α and the Fas Ligand. International Journal of Molecular Sciences. 2017; 18(12):2603. https://doi.org/10.3390/ijms18122603
Chicago/Turabian StyleHarada, Misuzu, Kyoko Morimoto, Tetsuya Kondo, Reiko Hiramatsu, Yuji Okina, Ryo Muko, Iyo Matsuda, and Takao Kataoka. 2017. "Quinacrine Inhibits ICAM-1 Transcription by Blocking DNA Binding of the NF-κB Subunit p65 and Sensitizes Human Lung Adenocarcinoma A549 Cells to TNF-α and the Fas Ligand" International Journal of Molecular Sciences 18, no. 12: 2603. https://doi.org/10.3390/ijms18122603
APA StyleHarada, M., Morimoto, K., Kondo, T., Hiramatsu, R., Okina, Y., Muko, R., Matsuda, I., & Kataoka, T. (2017). Quinacrine Inhibits ICAM-1 Transcription by Blocking DNA Binding of the NF-κB Subunit p65 and Sensitizes Human Lung Adenocarcinoma A549 Cells to TNF-α and the Fas Ligand. International Journal of Molecular Sciences, 18(12), 2603. https://doi.org/10.3390/ijms18122603