1. Introduction
Although the diversity of manufactured nanomaterials (MNMs) is very high, there is currently a continuous development of the technologies used to create new materials with improved properties [
1,
2]. These include titanium dioxide (TiO
2), which is usually utilized in a large variety of consumer products, such as paints, varnishes, plastics [
3,
4], cosmetics, pharmaceuticals [
5], and skin care products [
6]. More recently, due to their unusual physicochemical properties, titanium dioxide nanoparticles (TiO
2 NPs) have begun to be used as photocatalysts in air cleaning [
7], wastewater management [
8], hydrogen production [
9], orthodontic composites [
10,
11], anticorrosion coatings for dental and bone implants, self-cleaning surfaces, and medical instruments [
12].
On surfaces covered with a thin layer of TiO
2, inactivation of microorganisms was observed as a result of advanced oxidation processes (AOP) initiated by ultraviolet radiation (UV) [
13], which represents only 5% of terrestrial sunlight [
14]. Hence, new nanomaterials with enhanced photocatalytic efficiency have been synthesized and tested. TiO
2 nanoparticles were doped with metals (Fe) and non-metals (N) atoms to expand the photocatalytic performance in visible light [
15].
However, the technological progress has brought a continuous need for checks in the safety of these nano-products for the environment and human health. What is known so far, from the nanotoxicological studies, is that the same properties which make MNMs both fascinating and useful are also reasons of serious concerns [
16,
17,
18]. Recent investigations revealed that the lung represents the main target organ following workers’ exposure to NPs via inhalation [
19], and the particles mainly deposit (75%–80%) in the alveolar region where they could interfere with, or within, cells [
20]. Other possible methods of exposure include contact through the skin and ingestion, and the physico-chemical parameters of NPs, such as size and charge, seem to have an important impact on dermal penetration, and also on gastrointestinal absorption [
21].
Different parameters can determine the cytotoxicity of nanomaterials, but the main concern is that they are redox active and due to their small size, some particles can penetrate the cell membranes and have a greater potential to generate oxidative stress [
22]. Several studies, carried out recently both in vitro and in vivo, have suggested that the production of reactive oxygen species (ROS), especially after exposure to visible light or UV, is the main marker of cytotoxicity of TiO
2 nanoparticles and the main responsible mechanism for the inflammatory lung effects induced by inhalation [
23,
24]. At the cellular level, ROS may be generated directly from the particles near, or within, the cell, or indirectly as a result of internalized particles on mitochondrial respiration, as well as due to the antioxidant species depletion [
25]. After all, TiO
2 nanoparticles can cause respiratory toxicity and epithelial inflammation, but compared to other nanoparticles these ones have a low or insignificant cytotoxicity [
26].
It is well known the material properties depend strongly on the raw materials, synthesis method, time, temperature, pressure, etc. In wet methods (like hydrothermal processes) the pH of the solution can trigger the structure, morphology, and photocatalytic properties as well. Taking into account this aspect, in the present study, two types of TiO2 nanoparticles co-doped with 1% atoms of iron and nitrogen (TiO2-1% Fe–N) synthesized under hydrothermal conditions, using two different pH levels were considered. The structure and morphology of these NPs were investigated by X-ray diffraction (XRD), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS), and compared with those of reference material P25 (Degussa) and 1% Fe–N impregnated P25. The photocatalytic properties of these powders, on the degradation of methylene blue (MB), were comparatively revealed by a photocatalytic checker. Further, the new-developed TiO2-1% Fe–N particles were investigated in terms of antimicrobial efficiency and biological interaction with human cells. Cytotoxic effects were assessed in vitro on human dermal (CCD-1070Sk cell line) and pulmonary (MRC-5 cell line) fibroblasts by several parameters, including cell viability, antioxidant response and lipid peroxidation. Further, the antimicrobial activity was tested against Gram-positive fungal (Candida albicans) and bacterial (Staphylococcus aureus, Enterococcus faecalis) strains and Gram-negative (Escherichia coli, Pseudomonas aeruginosa) bacterial strains by quantitative methods according to international standards.
3. Discussion
The present study describes the synthesis, characterization, and biological evaluation of TiO
2 nanoparticles co-doped with N–Fe for an increase of photoactivity in the visible light. The synergistic effect of the N–Fe has been previously explained by the fact that nitrogen causes a band-gap narrowing which allows the absorption of longer wavelengths from visible light region, whereas iron increases the efficiency of charge separation [
40].
Due to its high photoreactivity, TiO
2 has been widely used as a photocatalytic agent, to kill various groups of microorganisms, including bacteria, fungi, and viruses [
41]. The antibacterial effect occurs as an outcome of the ROS interaction with bacterial structural components. ROS especially attack the unsaturated phospholipids from the the cell membrane, lipid peroxidation being considered the lethal mechanism of the photocatalytic process [
42]. What makes photocatalytic sterilization the most effective technique compared to conventional antibacterial treatments is the complete mineralization to CO
2 and H
2O of dead bacteria and the toxins released by them [
43].
There are also scarce reports showing that doping of TiO
2 NPs with N, Fe, or both induces an improvement of their antimicrobial properties. Thus, N-doped TiO
2 exhibited improved antibacterial properties against
E. coli and
B. subtilis under fluorescent light irradiation [
44]. N–Fe co-doped TiO
2 in the form of powders and nanofilms proved to exhibit a good bactericidal activity against
E. coli under visible light irradiation [
45,
46].
The purpose of our study was to obtain biocompatible TiO
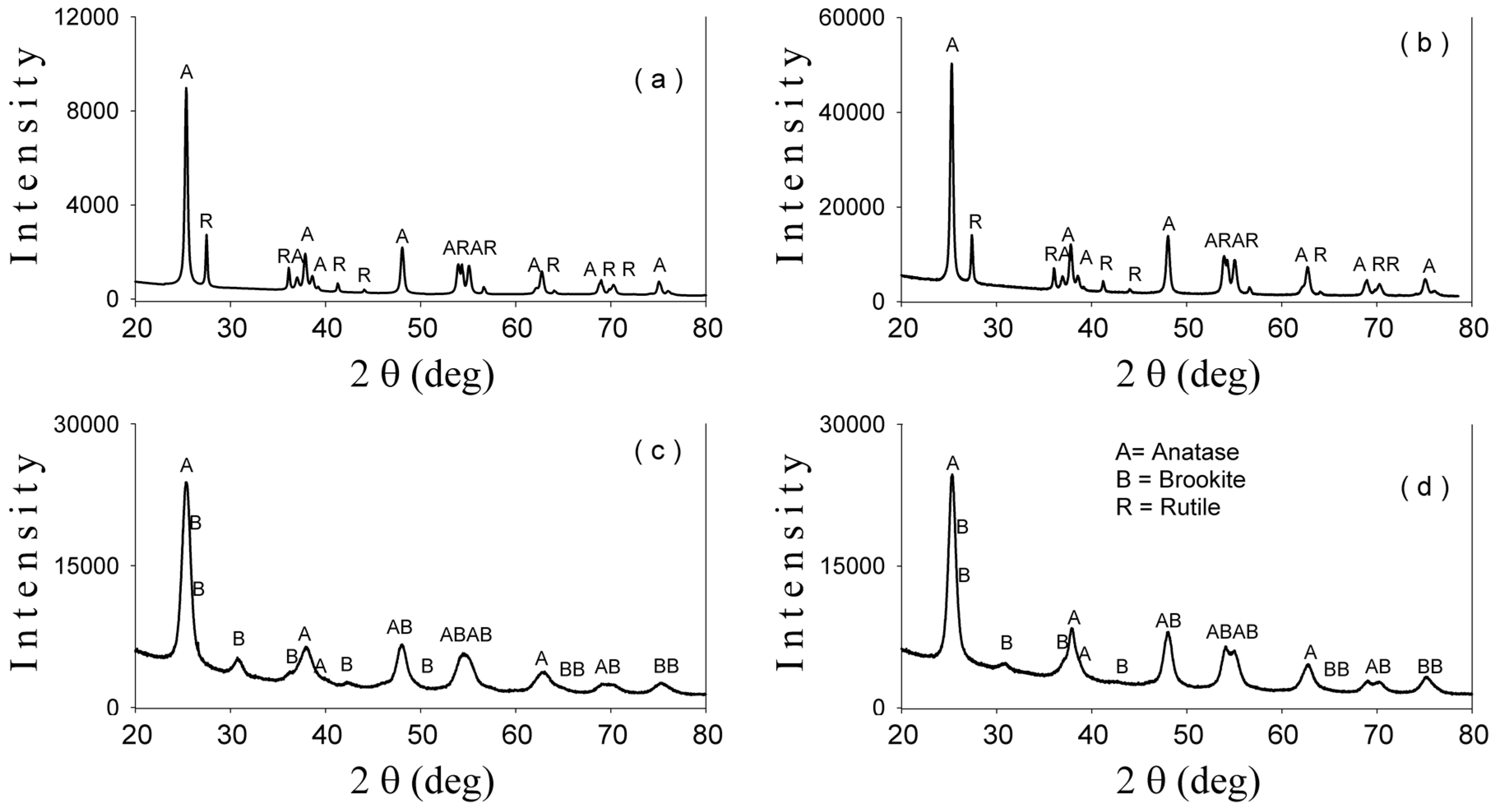
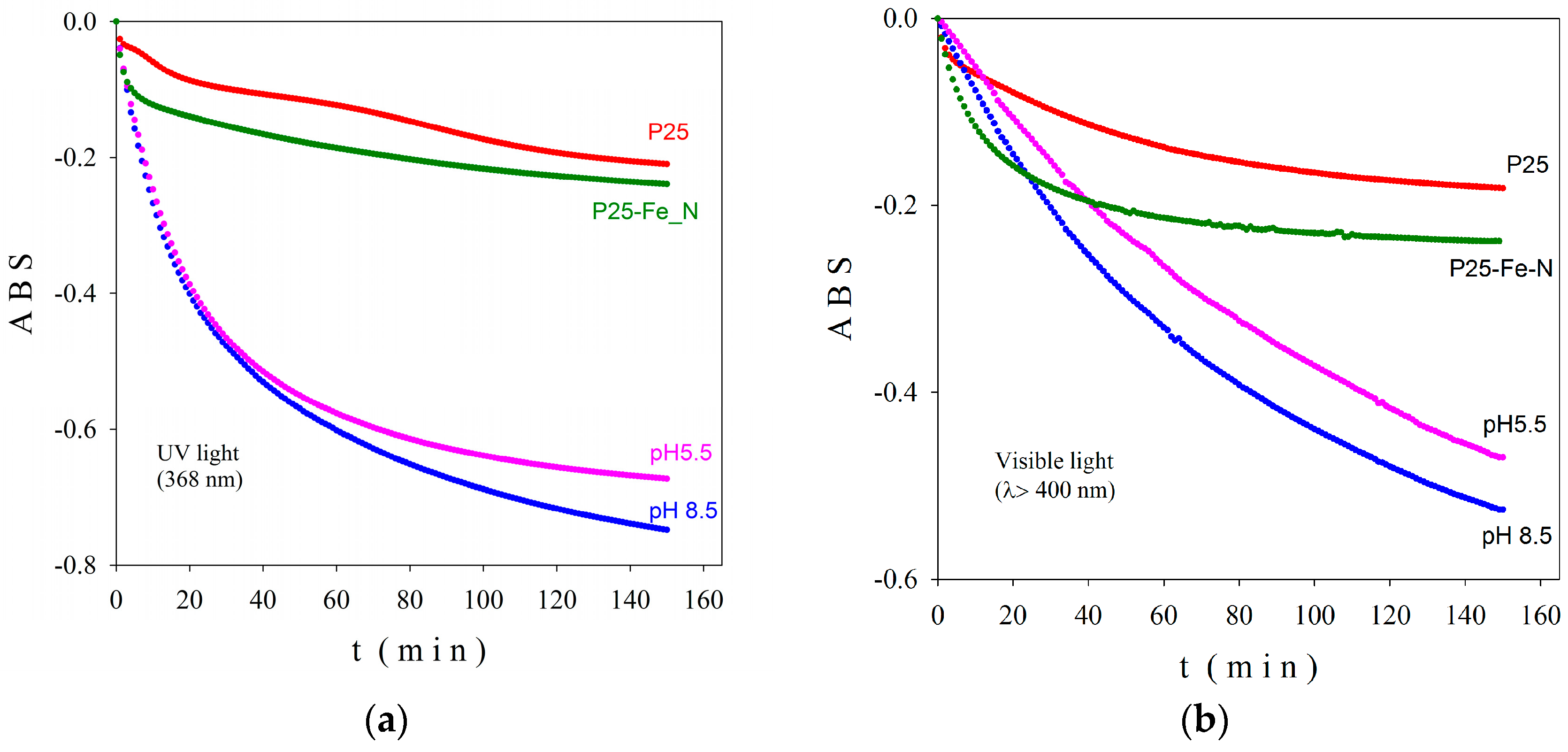
2-based nanoparticles with increased photocatalytic efficiency. The morphological, catalytic, and optical properties of particles depend on their shapes and sizes which are mainly established by the physico-chemical parameters used within the synthesis method. Herein, the pH values during the synthesis obviously change, as expected, with the relative phase content (anatase/brookite), the crystallite and the particle size, as well as the photocatalytic behavior of the powders, as shown by XRD, TEM, and photocatalytic analyses (
Figure 1,
Figure 3 and
Figure 6). Thus, only the hydrothermally-synthesized TiO
2-1% Fe–N NPs (pH 8.5 and 5.5) were selected herein for the biological assessments because the P25 commercial product and P25-based powders did not show significant photocatalytic activity (
Figure 6) and were also previously analyzed in other studies [
47,
48,
49,
50,
51,
52]. Before the biological tests, the complete characterization of co-doped TiO
2 NPs suspensions in cell culture medium revealed their polydispersity and the decrease in stability over time. The surface of TiO
2 nanoparticles dispersed in water is covered by hydroxyl group [
53] and, the ions present in solution are adsorbed on the particle, changing the surface charge and the electric potential between the particle surface and the dispersion solvent [
54].
We investigated the antimicrobial activity of the nano-photocatalysts synthesized in our work on a wide range of microbial strains including bacteria and yeasts, in planktonic and adherent states. The bactericidal activity of the obtained powders was improved under exposure to visible light compared to UV, the highest promising results sustained by the very low minimal inhibitory concentration (MIC) values being obtained against the Gram-negative P. aeruginosa bacterial strain and C. albicans yeast strain. To our best knowledge, the anti-biofilm activity of N–Fe co-doped TiO2 NPs is reported for the first time in this paper. Similar to the activity against planktonic bacteria, the exposure to either visible or UV light improved the efficiency of the photocatalytic NPs against the microbial strains grown in biofilm. The anti-biofilm activity was preserved in many cases at sub-inhibitory concentrations. The MBEC values were much lower compared to MIC values, suggesting the possible usage of these compounds as anti-pathogenic agents, i.e., compounds which do not interfere with the microbial growth, but are able to inhibit the microbial adherence and biofilm development. By this mechanism of action, they could prevent the negative consequences of biofilm formation, paving the way for a large spectrum of applications for the water supply systems, food, cosmetic, and pharmaceutical industries, as well as in different medical fields.
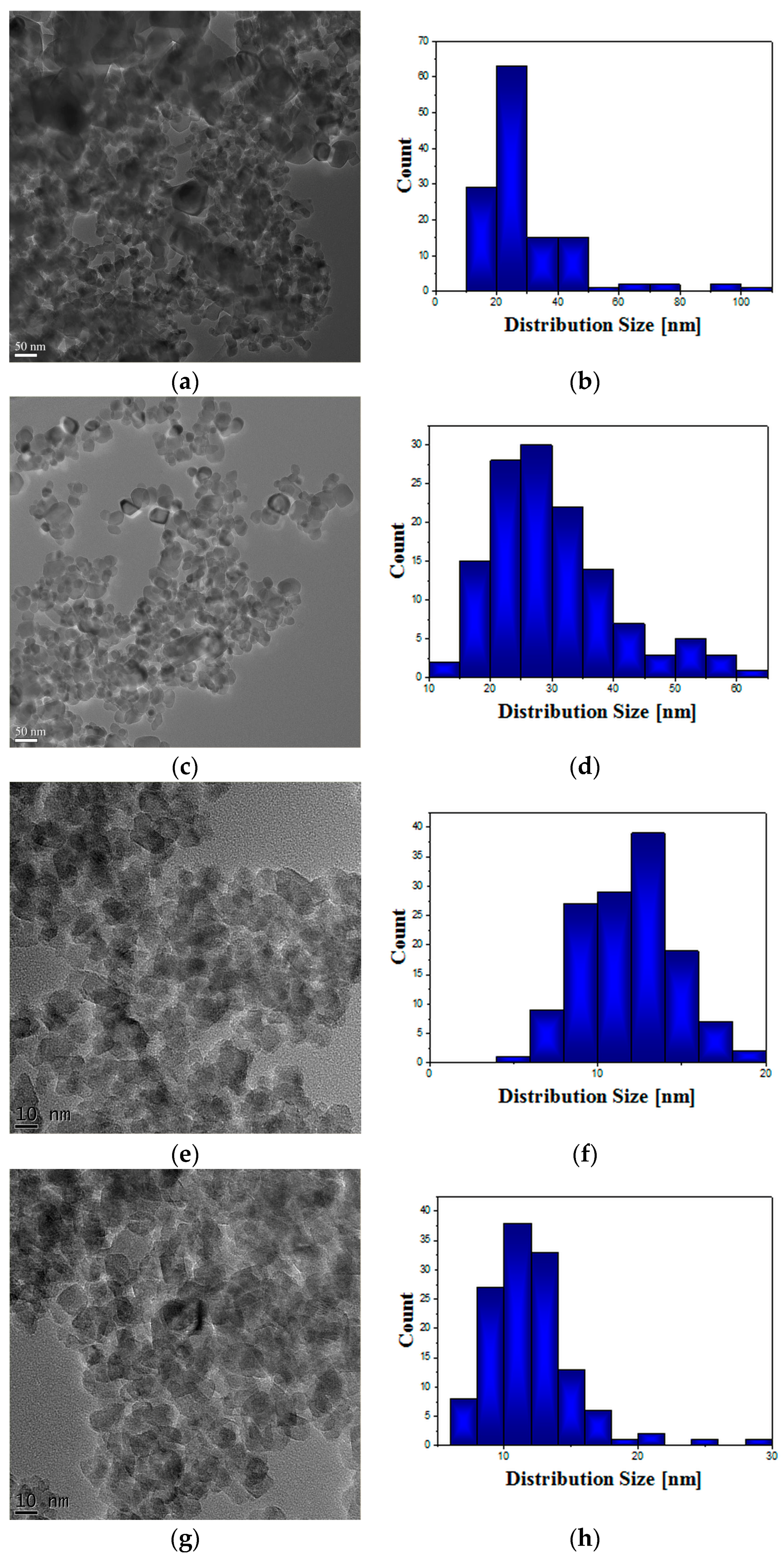
Taking into consideration the enlarged interest for photocatalytic applications of TiO
2 NPs in various fields, it is essential to investigate their biological response initiated after cell exposure. Lately, a number of studies performed on different cell lines revealed that small size TiO
2 NPs (10–20 nm) are more harmful and reactive, even without photoactivation, while normal-sized TiO
2 is biologically inert [
55,
56]. Additionally, the adverse effects of TiO
2 NPs have been investigated in detail at both in vitro and in vivo scales. It was reported that TiO
2 NPs cause cytotoxicity in human bronchial epithelial cells [
22], human lung fibroblasts [
57], and brain microglia [
49]. TiO
2 NPs exposure on aquatic species caused oxidative damage-mediated effects in
Cyprinus carpio [
58],
Oncorhynchus mykiss [
59] and
Daphnia magna [
60]. Thus, our study aimed to fill the current knowledge by investigating the cytotoxic effects induced by new;y-developed TiO
2 NPs in MRC-5 and CCD-1070Sk cells.
Currently, there are several paradigms regarding NPs-mediated toxicity, which include cell death, inflammation, inhibition of cellular proliferation, or DNA damages, but among these the most discussed is oxidative stress [
61]. It occurs as a result of the disproportion between the rate of ROS molecules formation and the cellular antioxidant capacity to neutralize them [
62]. Due to their physicochemical properties, TiO
2 nanoparticles are able to deposit on the cell surface or in cellular organelles where they interact with the structural components [
63].
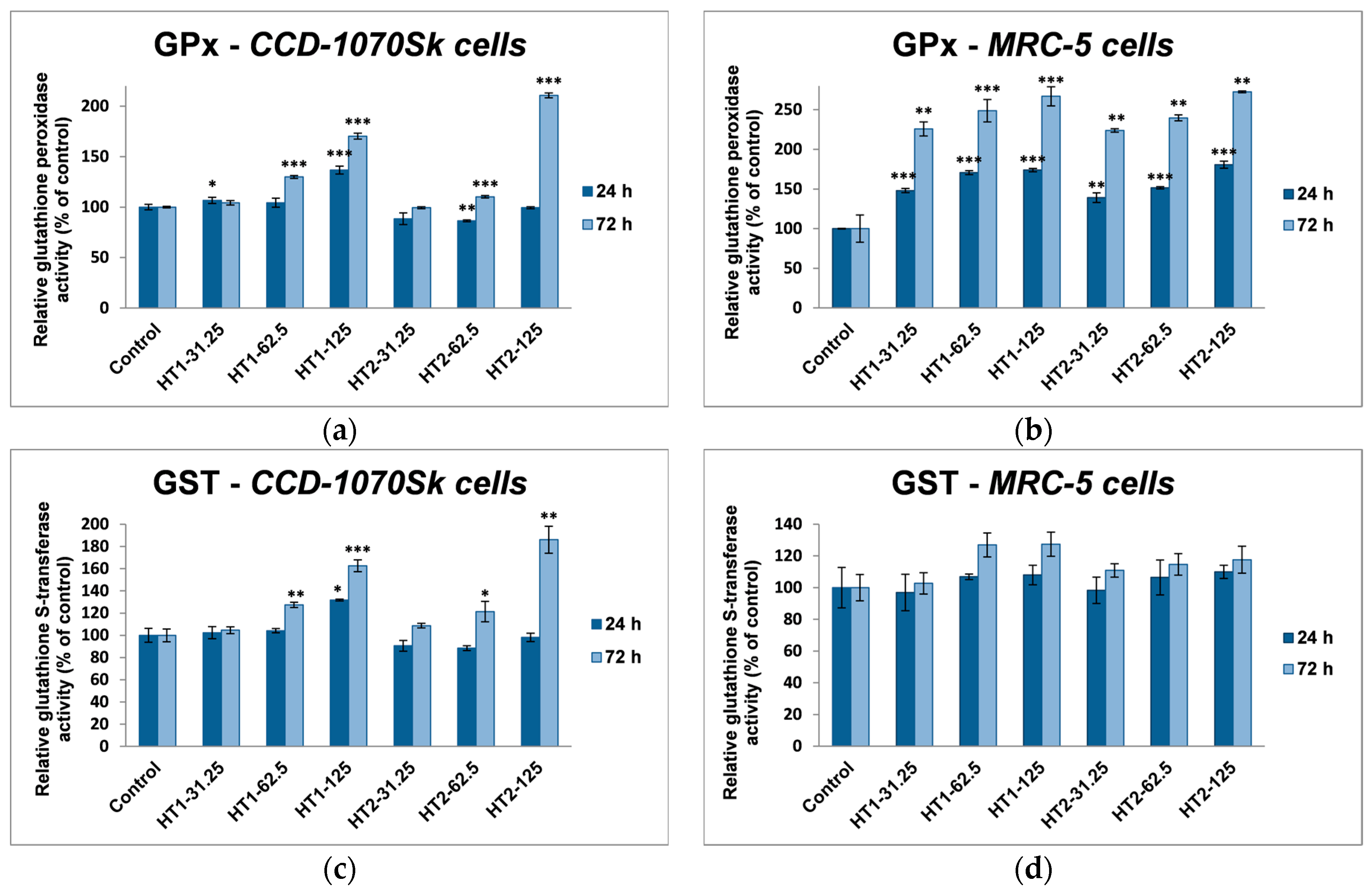
The cells can maintain the ROS level under normal metabolic conditions and prevent oxidative stress by activating enzymatic and non-enzymatic antioxidant systems [
64]. The first line of defense is provided by superoxide dismutase and catalase, which convert ROS into less damaging compounds, followed by glutathione peroxidase and glutathione S-transferase, which detoxify the intermediate products [
65]. However, only the up-regulation of these antioxidant enzymes is not sufficient to neutralize the ROS generation by TiO
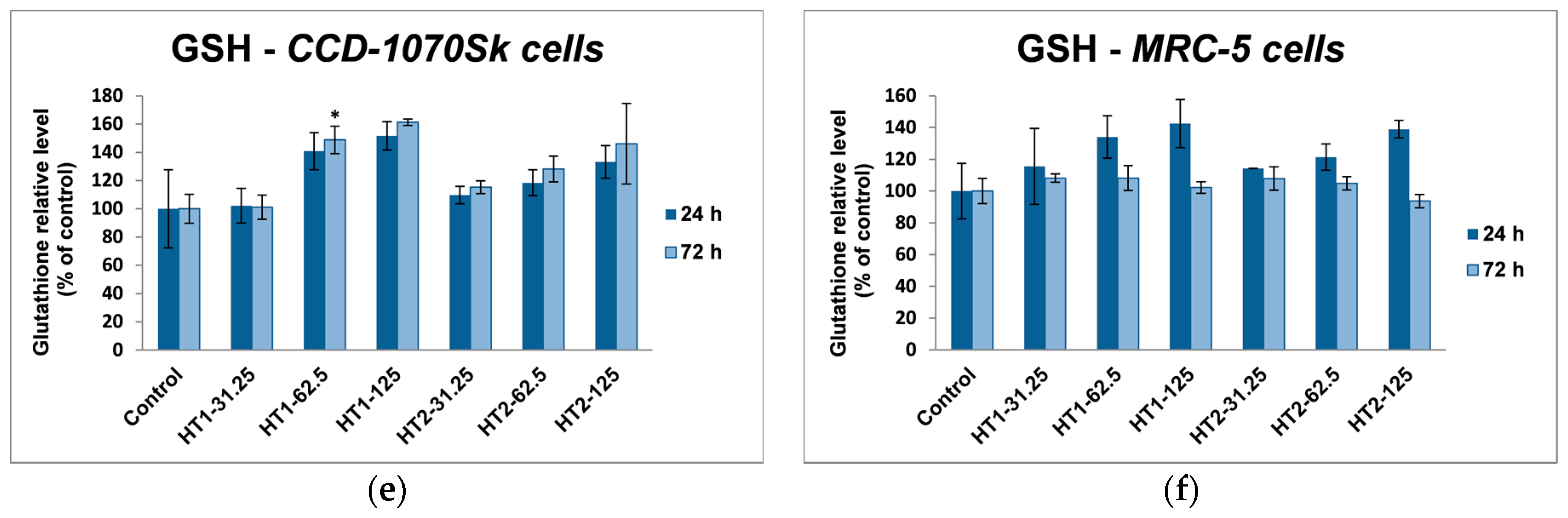
2 NPs in dermal and lung fibroblasts. Glutathione is a tripeptide which acts like a free-radical scavenger and has a key role in these intracellular antioxidant defense processes [
66]. Due to its active thiol group GSH can directly interact with ROS and it acts as a cofactor for GPx and GST enzymes. Moreover, because of its importance in cell defense, GSH is also a vulnerable target in front of different harmful agents, which can directly deplete or inactivate it [
67].
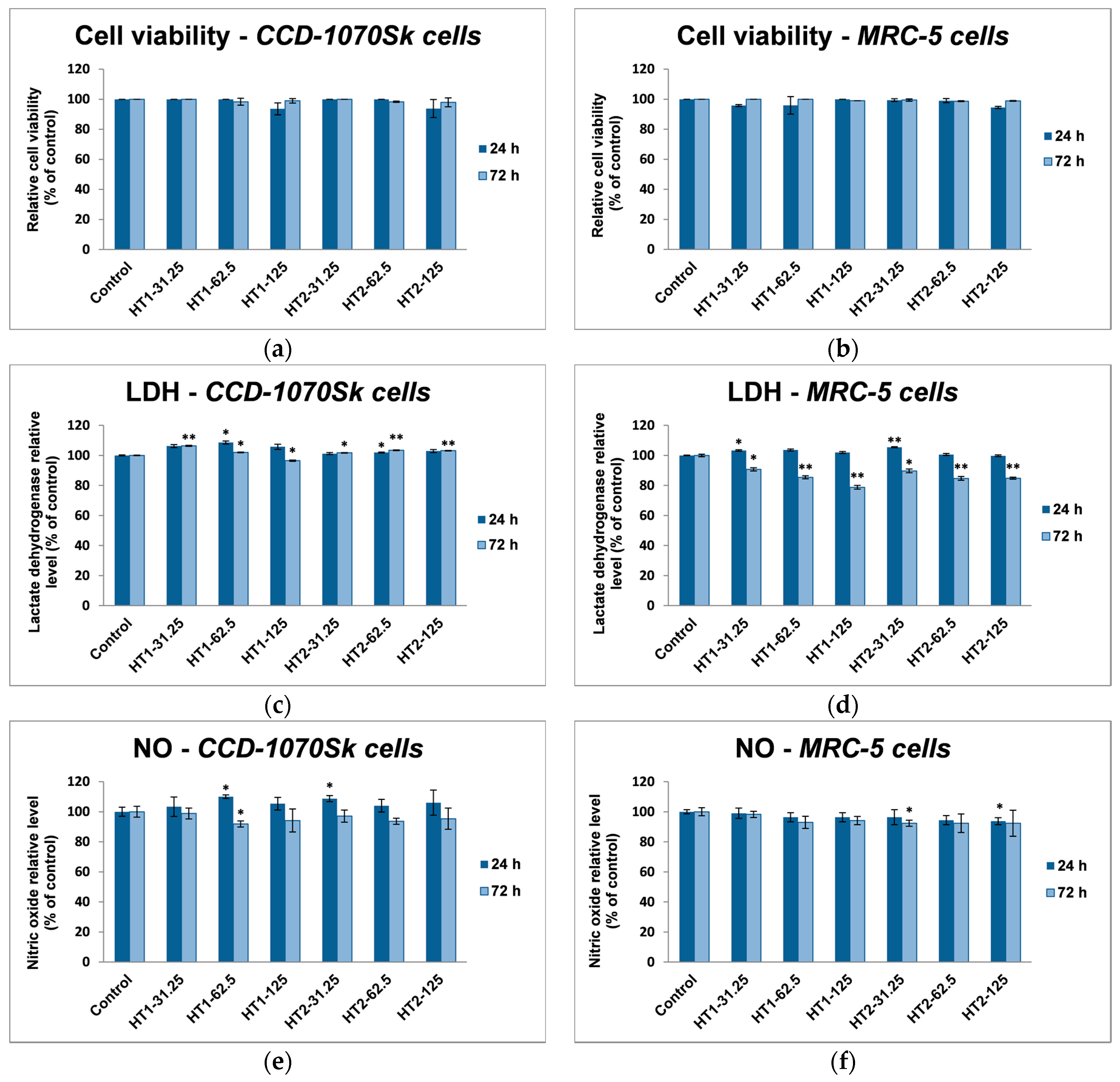
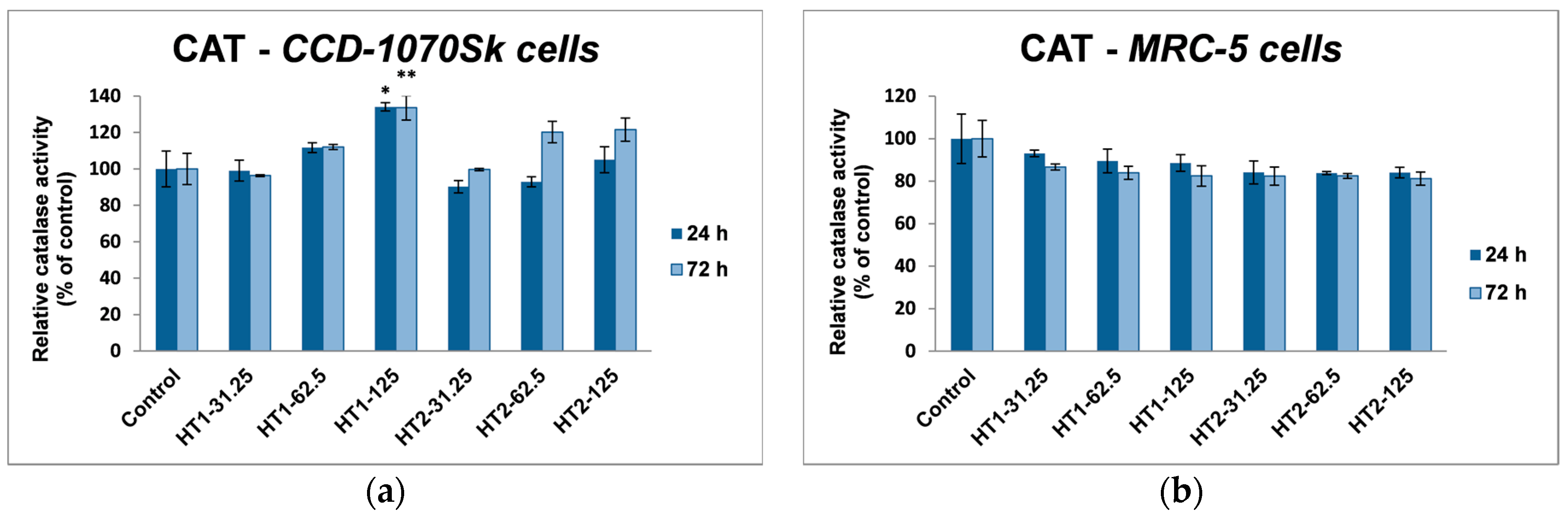
In our experiments, TiO
2-1% Fe–N NPs increased the specific activities of antioxidant enzymes in both cell lines, which proved that they induced oxidative stress and the lung and dermal fibroblasts responded by activating their antioxidant defense system. The CAT activity level decreased only in MRC-5 lung fibroblasts. Additionally, Turkez et al. suggested that a decline of CAT activity could be correlated with an increased genotoxic effect [
68]. Our results could prove that the H
2O
2 amount generated by oxidative stress was low and, as a result, this was decomposed by GPx, which has a lower
KM for H
2O
2 [
69]. GPX and GST activities were significantly elevated in MRC-5 and CCD-1070Sk cells, especially after 72 h, when a slight diminution of the GSH level occurred in MRC-5 cells at the highest concentrations of both TiO
2-1% Fe–N samples. These findings indicated that GPX and GST might have an important influence in counteracting the oxidative stress induced by TiO
2 NPs. Previous reports showed diminished GSH level in different types of cells after TiO
2 NPs exposure [
70,
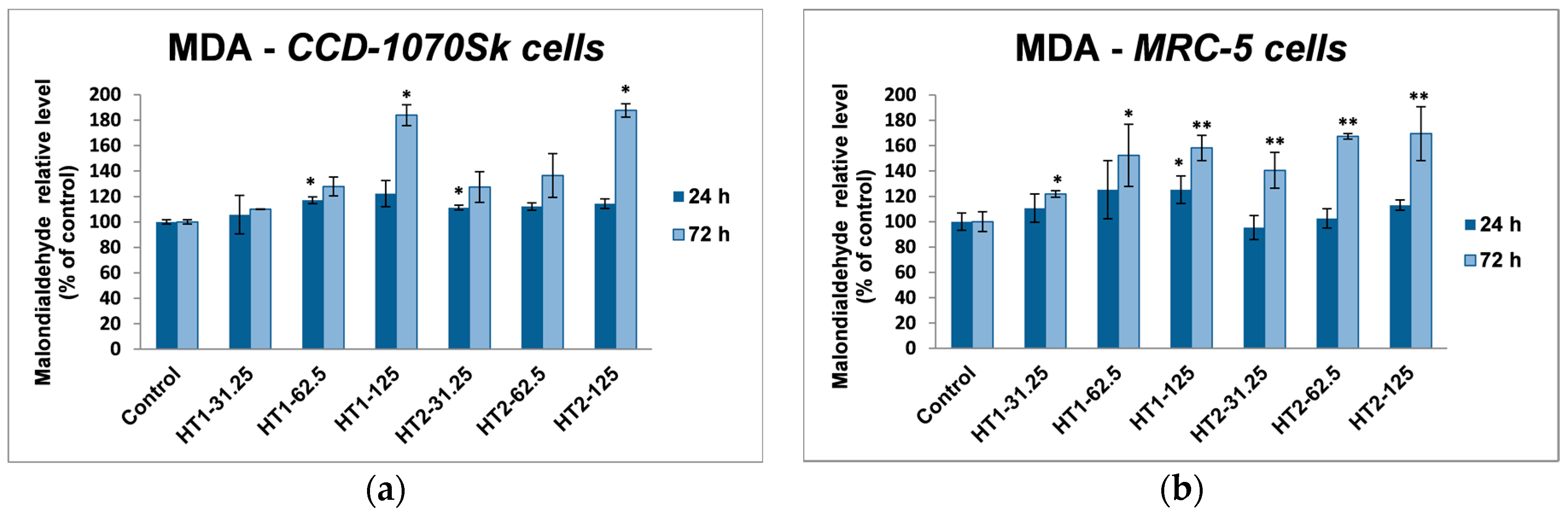
71]. We noticed an insignificant decrease of GSH levels only in MRC-5 cells incubated with nanoparticles for 72 h, which suggested that photocatalysts were well tolerated. Furthermore, an increase in lipid peroxidation level was noticed in both skin and lung cells (
Figure 10). The enhancement of lipids peroxidation by nano TiO
2 was the result of an oxidative attack which was initiated by the reduction of the cellular antioxidant defense mechanism. Similar results were reported in kidney cells exposed to TiO
2 NPs [
72].
4. Materials and Methods
4.1. Synthesis and Characterization of Titanium Dioxide Nanoparticles
P25 powder (Sigma-Aldrich, St. Louis, MO, USA) was impregnated with 1% Fe and N atoms as follows: suitable quantities of TiO2, FeCl3·6H2O, and urea were dispersed/dissolved via mechanical and ultrasonic stirring in distilled water. The resulted mix was heated at 200 °C for two hours in a Teflon-lined autoclave. The resulted powder was rinsed with ultrapure water to eliminate salts (until pH ~6.5), dried, and finally calcined for 2 h at 400 °C.
For the synthesis of TiO2-1% Fe–N particles at pH 5.5 and 8.5 suitable amounts of TiCl3 and FeCl3·6H2O were mixed using energetic stirring in ultrapure water; the pH values were adjusted with 25% NH4OH solution. Then, the obtained Ti(III) precipitates were oxidized in air until the color changed from violet to white. To remove salts, the Ti(IV) and Fe(III) hydroxides that co-precipitated were washed with ultrapure water and dried in air at 105 °C. The nitrogen doping was performed by the hydrothermal treatment (200 °C/2 h) of the resulted co-precipitate in the presence of urea. The dried resulted powder was calcined at 400 °C for 2 h in air.
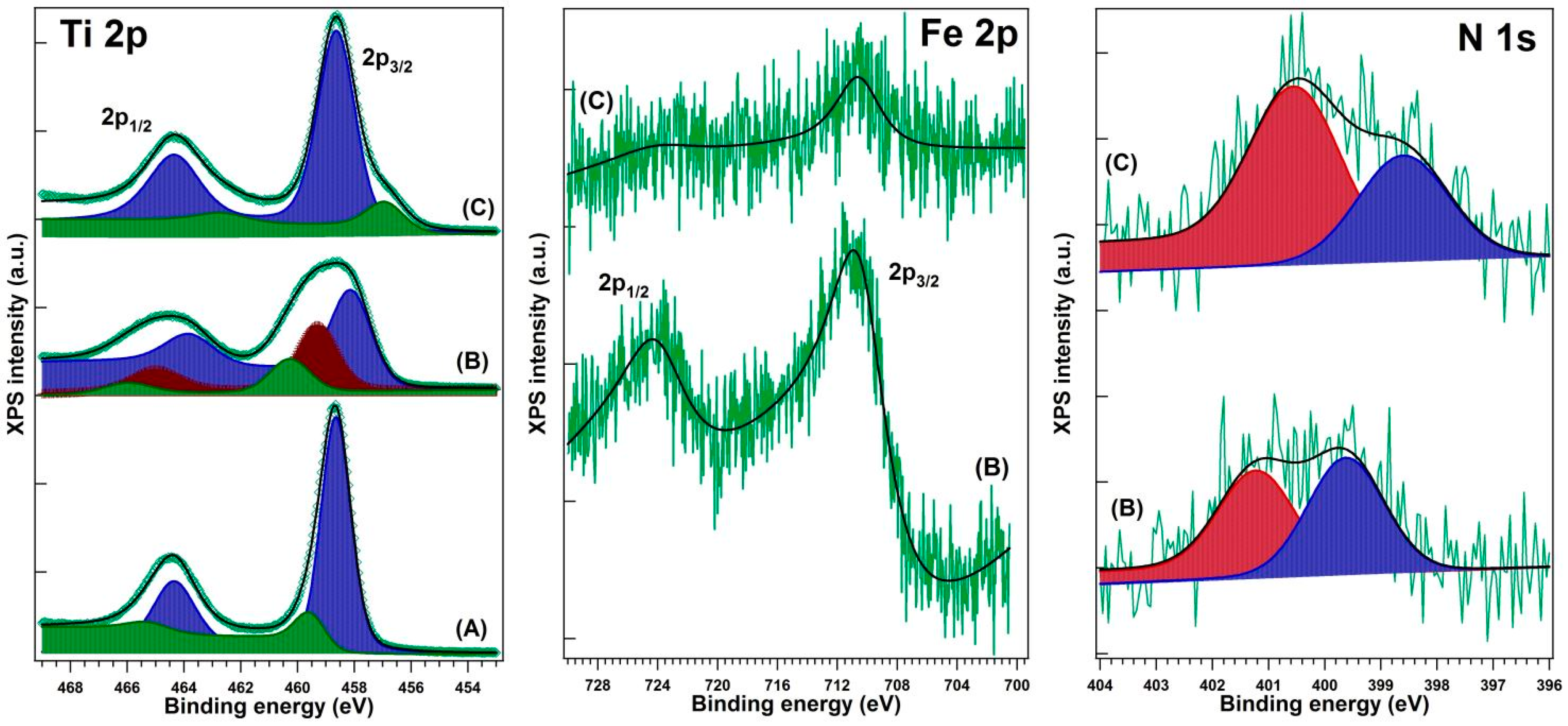
XPS testing was performed in an analysis chamber equipped with a 150 mm hemispherical electron energy analyzer (Phoibos, SPECS Gmbh, Berlin, Germany), a dual anode (Mg/Al Kα) X-ray source, and a monochromatized (Al Kα/Ag Lα) X-ray source, part of a complex surface science cluster (Specs). Monochromatized Al Kα1 (1486.74 eV) radiation was used to extract the electrons from the sample. The analyzer operated in fixed transmission mode with pass energy of 20 eV; the estimated combined (source + analyzer) resolution was about 0.75 ± 0.025 eV. Sample neutralization was achieved using an electron flood gun operating at 1 eV energy and 100 μA electron current. The energy scale was calibrated to the “standard” value of C 1s (284.6 eV) acknowledged by the XPS community. The base pressure during the measurements was about 10−9 Pa.
TEM investigations were performed using a transmission electron microscope (JEOL JEM ARM200F, Tokyo, Japan) operated at 200 kV, on samples obtained by crushing the powders in ethanol, dispersing them by sonication and dropping on lacey carbon grids.
The surface morphology of tested particles was investigated with a scanning electron microscope (SEM; Quanta 200, FEI, Eindhoven, The Netherlands) with a large field detector (LFD) operating at 15 kV. The particles were suspended in absolute ethylic alcohol and sonicated for one hour. A drop of suspension was spread on a thin glass plate and dried under a UV lamp.
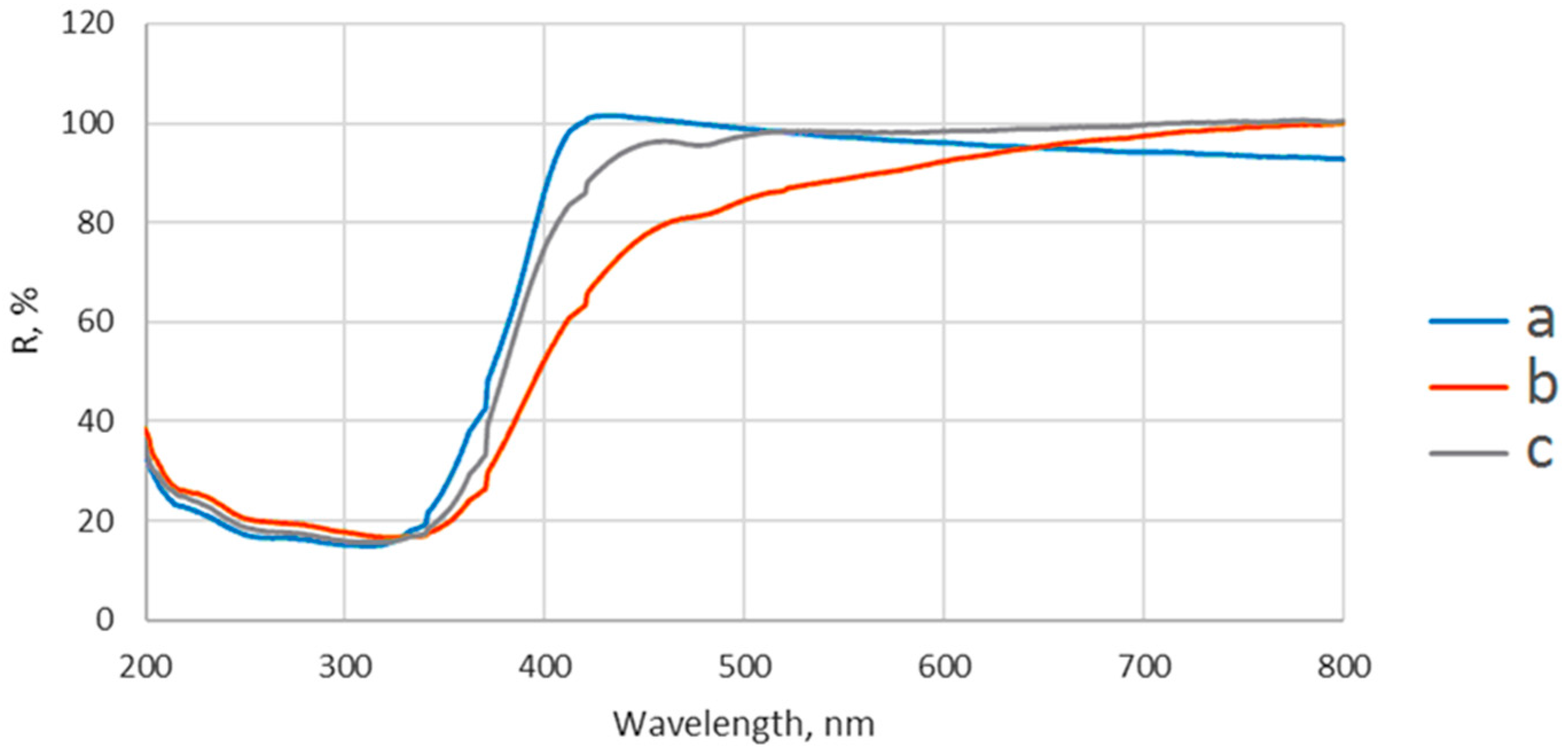
The diffuse reflectance spectra were recorded on Jasco V550 UV-VIS spectrophotometer (Jasco, Inc., Tokyo, Japan), using nanoparticles powder and BaSO4 as a reflectance sample.
In order to study the photocatalytic behavior of co-doped titania nanoscaled powders, we prepared three layered films as follows: The nanopowders were dispersed in distilled water and sonicated in an ultrasonic bath. The final concentration of photocatalytic powder in water was 1 mg/mL. Then, a suitable amount of suspension (calibrated drops) were deposited carefully, to cover the quartz buffer in the horizontal position. The film was dried at 30 °C in the absence of any air current. A new photocatalytic layer (the same number of calibrated drops) was deposited after completely drying. The procedure was three times repeated to obtain a three-layer nanoscaled co-doped TiO2 film. The resulted films were dried in air for 16 h and then cleaned with a 30 W UV (385 nm) lamp for 2 h to remove possible organic contamination. Finally, the films were immersed in methylene blue and dried in the dark at room temperature. We have to mention that within this deposition procedure no additional thermal treatment was necessary, therefore, no structural changes could occur during film’s preparation. In order to reveal the activity in both UV (368 nm) and visible (λ > 400 nm) spectral regions, the obtained films were tested using a PCC-2 (ULVAC RIKO, Chigasaki, Kanagawa, Japan) photocatalytic checker.
In order to assess the hydrodynamic size and zeta potential, the TiO2-based NPs dispersions were prepared at a concentration of 1 mg/mL in ultrapure water and at a concentration of 125 µg/mL in phosphate saline buffer (PBS) or cell culture media, using 5 min ultrasonication and then were analyzed on a Malvern Zetasizer Nano-ZS instrument (Malvern Instruments, Malvern, Worcestershire, UK) by dynamic light scattering (DLS) and laser Doppler velocimetry (LDV) technologies. Three measurements were taken for each sample to determine the particles’ size and zeta potential.
4.2. Antimicrobial Activity Assays
The influence of the obtained powders on the prokaryotic and eukaryotic microbial strains was tested using Gram-negative bacterial strains (
E. coli ATCC 8739,
P. aeruginosa ATCC 27853), Gram-positive bacterial strains (
S. aureus ATCC 6538,
E. faecalis ATCC 29212), and fungal strains (
C. albicans ATCC 10231). For this purpose, microbial suspensions with a density of 1.5 × 10
8 colony forming units (CFU)·mL
−1 equivalent to the 0.5 McFarland’s turbidimetry standard were obtained starting from fresh culture of 15 to 18 h developed on solid media. The tested powders were suspended in dimethyl sulfoxide (DMSO) in order to obtain the stock solution with a concentration of 10 mg·mL
−1. The quantitative assay of the antimicrobial activity was performed by liquid medium micro-dilution method in 96-well plates. In this purpose, two-fold serial dilutions of the nanoparticles’ suspensions, with a concentration range from 1000 to 4 μg·mL
−1 were achieved in a volume of 200 μL of liquid culture medium, irradiated for 30 min in UV (100–290 nm) and VIS light, respectively, using the laminar flow germicidal and illuminating lamps, and then, each well was seeded with 50 μL standardized microbial inoculum. Positive controls (for bacterial culture) were used. After 24 h of incubation at 37 °C, the MIC values were read as the powder concentration found in the last well in which the visible overnight growth of the microbial culture was lower as compared to the positive control, and confirmed by a decreased value of absorbance at 600 nm (Apollo LB 911 ELISA plate reader) [
73].
For the anti-biofilm activity evaluation of the tested nanopowders the microtiter plate method was used. In this purpose, after reading the micro-plates for the establishment of the MIC values, their contents were emptied and, then, the plates were washed three times by phosphate-buffered saline. The microbial cells forming a biofilm adhered on the plastic wells wall resisted to washing and were with fixed cold methanol, which was left to act for 5 min then, colored by crystal violet solution for 15 min and finally resuspended in a 33% acetic acid solution. The density of the microbial biofilm harvested from the plastic wells was measured by reading the optical density at 490 nm for the colored suspensions. The minimal biofilm eradication concentration (MBEC) value was corresponding to the concentration found in the well in which the absorbance values were inferior to those of the positive control [
74].
4.3. Cell Culture
Normal human fibroblasts from skin (CCD-1070Sk cell line, ATCC Cat. No. CRL-2091) and from lung (MRC-5 cell line, ATCC Cat. No. CCL-171) were grown in complete Eagle’s minimum essential medium (MEM; Gibco/Invitrogen, Carlsbad, CA, USA) with the addition of 10% fetal bovine serum (FBS; Gibco/Invitrogen, Carlsbad, CA, USA) at 37 °C in a humidified atmosphere with 5% CO2. The culture medium was replaced every two days with a fresh medium until cells reached 80% confluence, when a 0.25% (w/v) Trypsin 0.53 mM EDTA solution (Sigma-Aldrich) was used to split the cells for future sub-cultivations.
4.4. Culture Treatment Protocol
Two stock suspensions of TiO2 nanoparticles doped with 1% Fe–N atoms synthesized at pH values of 8.5 and 5.5 were prepared in PBS and sterilized by autoclaving at 120 °C for 20 min. The fibroblasts were counted and cultured at a density of 2 × 104 cells/cm2 into 24-well plates (for biocompatibility assessment) or in 75 cm2 culture flasks (for antioxidant enzymes assays, glutathione content, and lipid peroxidation analysis) and allowed to adhere overnight. Afterwards, cells were exposed to various concentrations (31.25, 62.5, and 125 µg/mL) of TiO2-1% Fe–N nanoparticles for 24 and 72 h. Controls represented by untreated cells were used for each test.
4.5. Cell Viability
In order to measure the cell viability, the cells were detached as it was described in
Section 4.3 and counted using Trypan Blue staining. Therefore, 15 μL of cell suspension were mixed with an equal volume of 0.4% (
w/
v) Trypan Blue solution prepared in 0.81% NaCl and 0.06% (
w/
v) dibasic potassium phosphate (Sigma-Aldrich). The cells’ counting was performed using a dual-chamber hemocytometer and a light microscope. Cell viability was given by the following formula:
The cell membrane integrity determined by the LDH amount released in culture medium was assessed using a commercial kit (TOX7, Sigma-Aldrich) according to the manufacturer’s instructions. Thus, 50 μL of culture supernatants were homogenized with 100 μL of mix consisting of equal parts of dye, substrate and cofactor, and incubated for 30 min in dark. The reaction was stopped by adding 15 μL of 1 N HCl, and then the absorbance was read at 490 nm using a GENios Tecan microplate reader (TECAN GENios, Grödig, Austria).
The inflammatory potential determined by the level of NO released in the culture medium was measured using the Griess reagent, which is a stoichiometric solution (v/v) of 0.1% naphthylethylendiamine dihydrochloride and 1% sulphanilamide in 5% H3PO4. Therefore, culture supernatants were mixed with an equal volume of Griess reagent and absorbance was read at 550 nm using a GENios Tecan microplate reader. Finally, NO concentration was calculated by extrapolation on a NaNO2 standard curve.
Cell spreading and actin cytoskeleton morphology were observed via fluorescence microscopy. Firstly, the cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton X-100−2% bovine serum albumin (BSA) for 1 h, following later to mark filamentous actin (F-actin) marked with 20 μg/mL phalloidin conjugated with FITC (Sigma-Aldrich, Munich, Germany). Images were obtained with an inverted fluorescence microscope Olympus IX71 (Olympus, Tokyo, Japan).
4.6. Preparation of Cell Lysates
MRC-5 and CCD-1070Sk cells were collected from culture flasks, washed with PBS and the cell lysates were obtained by sonication (30 s × 3 times) on ice with an ultrasonic processor (Hielscher UP50H, Teltow, Germany). The homogenate was centrifuged at 3000× g for 10 min at 4 °C and the supernatants (total proteic extracts) were collected for biochemical assays.
4.7. Protein Concentration Assay
The protein concentration of the cellular extract was measured according to the method described by Bradford [
75] using the Bradford Reagent (Sigma-Aldrich) and a BSA standard curve.
4.8. Antioxidant Enzymes Assays
The activities of antioxidant enzymes were determined by spectrophotometric methods. Catalase (CAT) (EC 1.11.1.6) activity was measured by monitoring the decrease in absorbance of H
2O
2 at 240 nm, as described in Aebi’s method [
76]. One unit of CAT activity represented the amount of enzyme that catalyzed the conversion of 1 μmol of H
2O
2 in 1 min under standard conditions. Glutathione peroxidase (GPX) (EC 1.11.1.9) activity was assessed according to the method of Beutler [
77] by monitoring at 340 nm a coupled reaction with glutathione reductase that catalyzed NADPH oxidation. One unit of GPx activity was defined as the quantity of enzyme that catalyzes the transformation of 1 μmol of NADPH per minute. Glutathione S-transferase (GST) (EC 2.5.1.18) activity was determined as described in the method of Habig et al. [
78] by measuring the rate of 1-chloro-2,4-dinitrobenzene (CDNB) conjugation with GSH at 340 nm. One unit of GST activity was defined as the amount of enzyme that produced 1 μmol of conjugated product per minute. All data were obtained in physiological conditions at a pH value of 7.4 and room temperature (25 °C) and the final results were calculated as specific enzymatic activities (units/mg of protein) and expressed as a percent of the control levels.
4.9. Glutathione Content
The proteins from cell lysates were precipitated with a 5% sulfosalicylic acid solution (Sigma-Aldrich) (1:1) and removed by centrifugation at 10,000 rpm and 4 °C for 10 min. The GSH content was determined using the commercial glutathione assay kit (Sigma-Aldrich) according to the protocol provided by the manufacturer. Briefly, each sample was incubated with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) for 5 min at room temperature in order to allow the reduction of DTNB into 5-thio-2-nitrobenzoic acid (TNB). The absorbance was recorded at 405 nm using a microplate reader (TECAN GENios, Grödig, Austria). The GSH levels were calculated as nmols/mg protein and the results were expressed relative to control.
4.10. Lipid Peroxidation
Lipid peroxidation expressed by malondialdehyde (MDA) level was assessed using the fluorimetric method described by Dinischiotu et al. [
79]. A volume of 200 µL of cell lysate diluted correspondingly was mixed with 700 µL of 0.1 N HCl and incubated for 20 min at room temperature. Forwards, 900 µL of 0.025 M thiobarbituric acid (TBA) was added and the mixture was incubated for 65 min at 37 °C. Subsequently, relative fluorescence units (RFU) recorded (excitation wavelength = 520 nm; emission wavelength = 549 nm) (FP-750 Spectrofluorometer, Jasco, Tokyo, Japan) were converted to nmols malondialdehyde (MDA) using a 1,1,3,3-tetramethoxypropane standard curve. The MDA concentration was expressed as nmols of MDA/mg protein and all the results were represented relative to control.
4.11. Statistical Analysis
All results were represented as mean value ± standard deviation (SD) of three different experiments. The statistical analysis was performed using comparisons between groups evaluated by Student’s t-test or two-way ANOVA, followed by Bonferroni post hoc test using GraphPad Prism software (version 5; GraphPad Software, Inc., La Jolla, CA, USA), and only a value of p less than 0.05 was considered statistically significant.


 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}