
Acidosis Activates Endoplasmic Reticulum Stress Pathways through GPR4 in Human Vascular Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
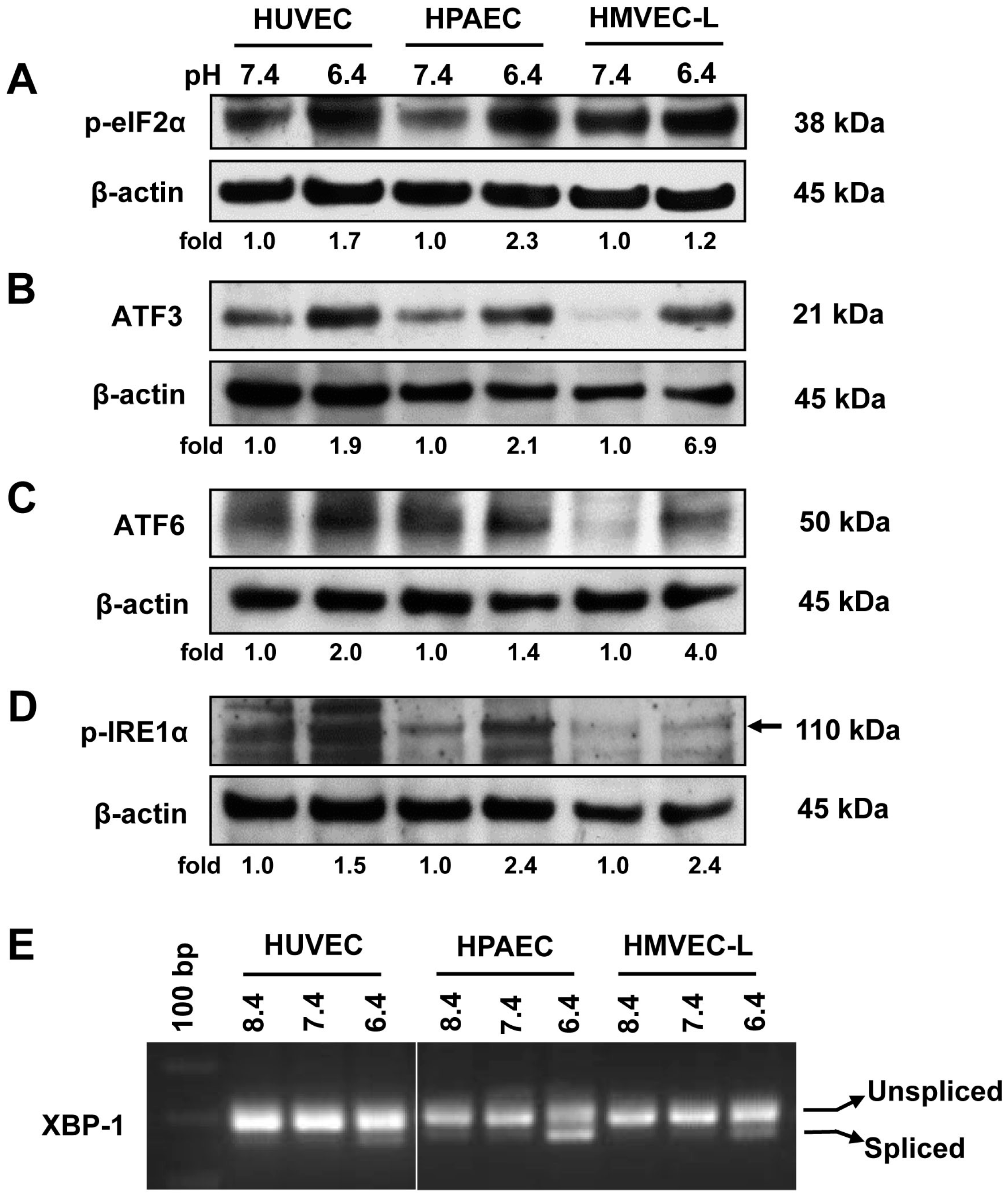
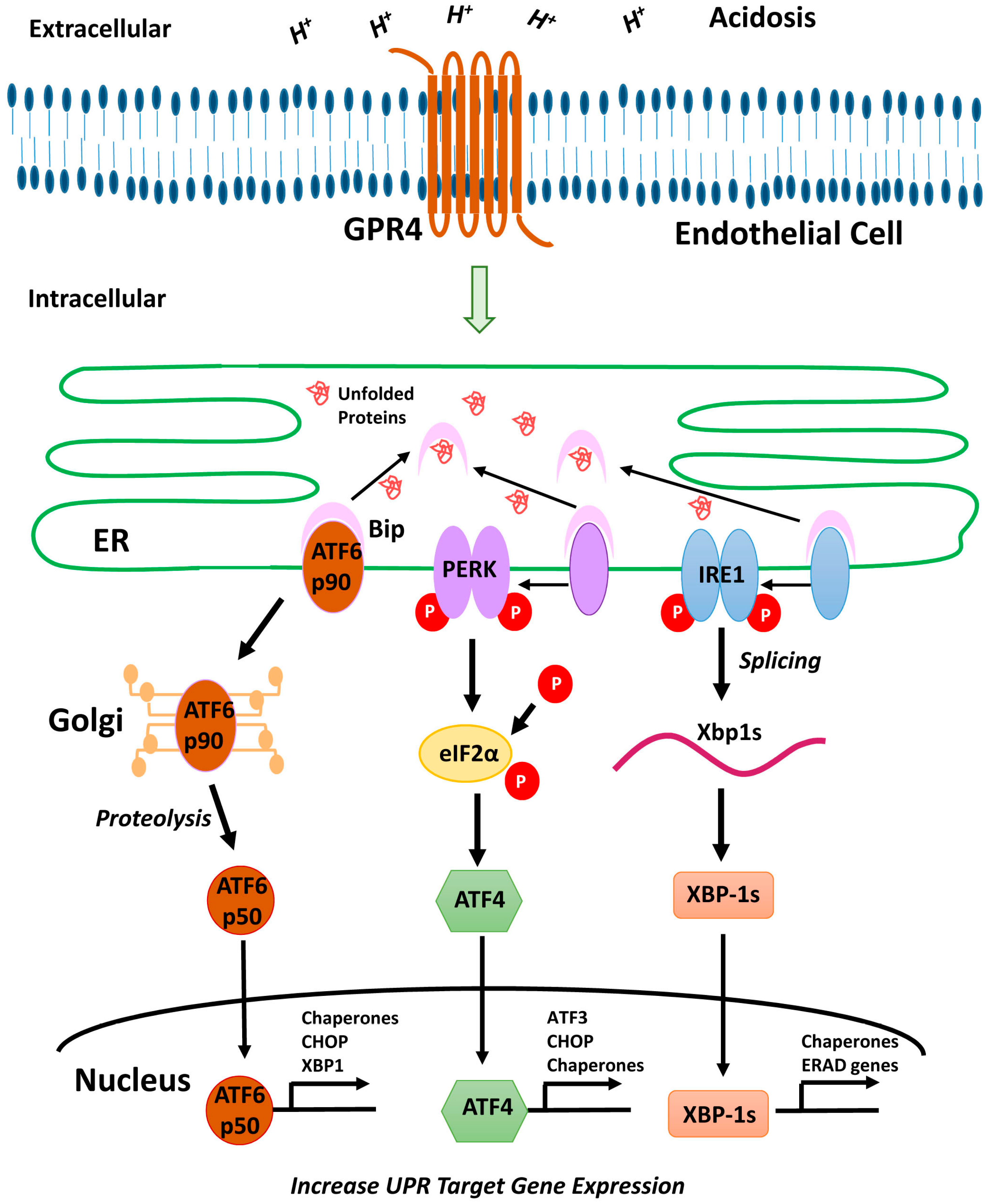
2.1. Acidic pH Activates All Three Arms of the ER Stress/UPR Pathways in Vascular ECs
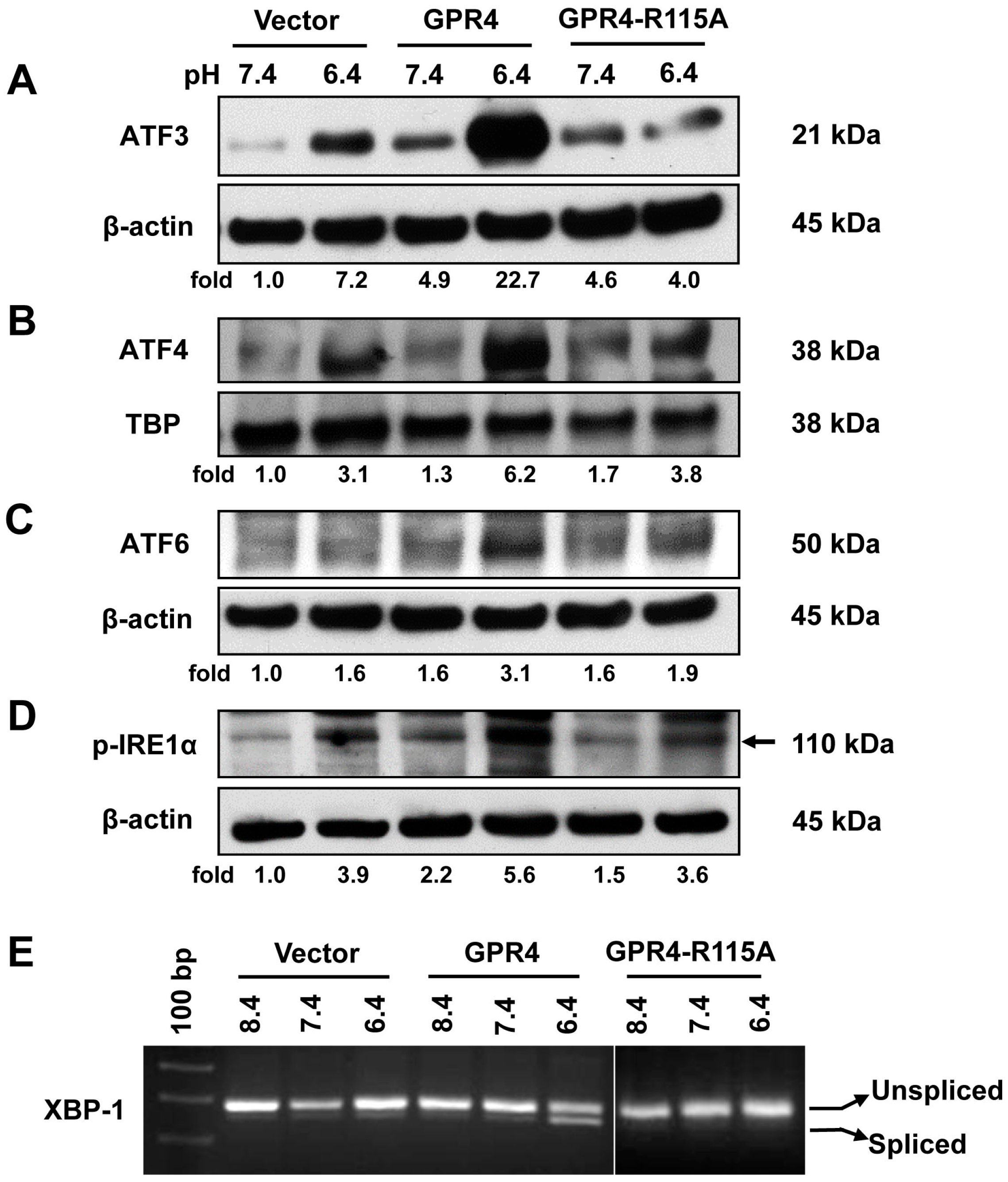
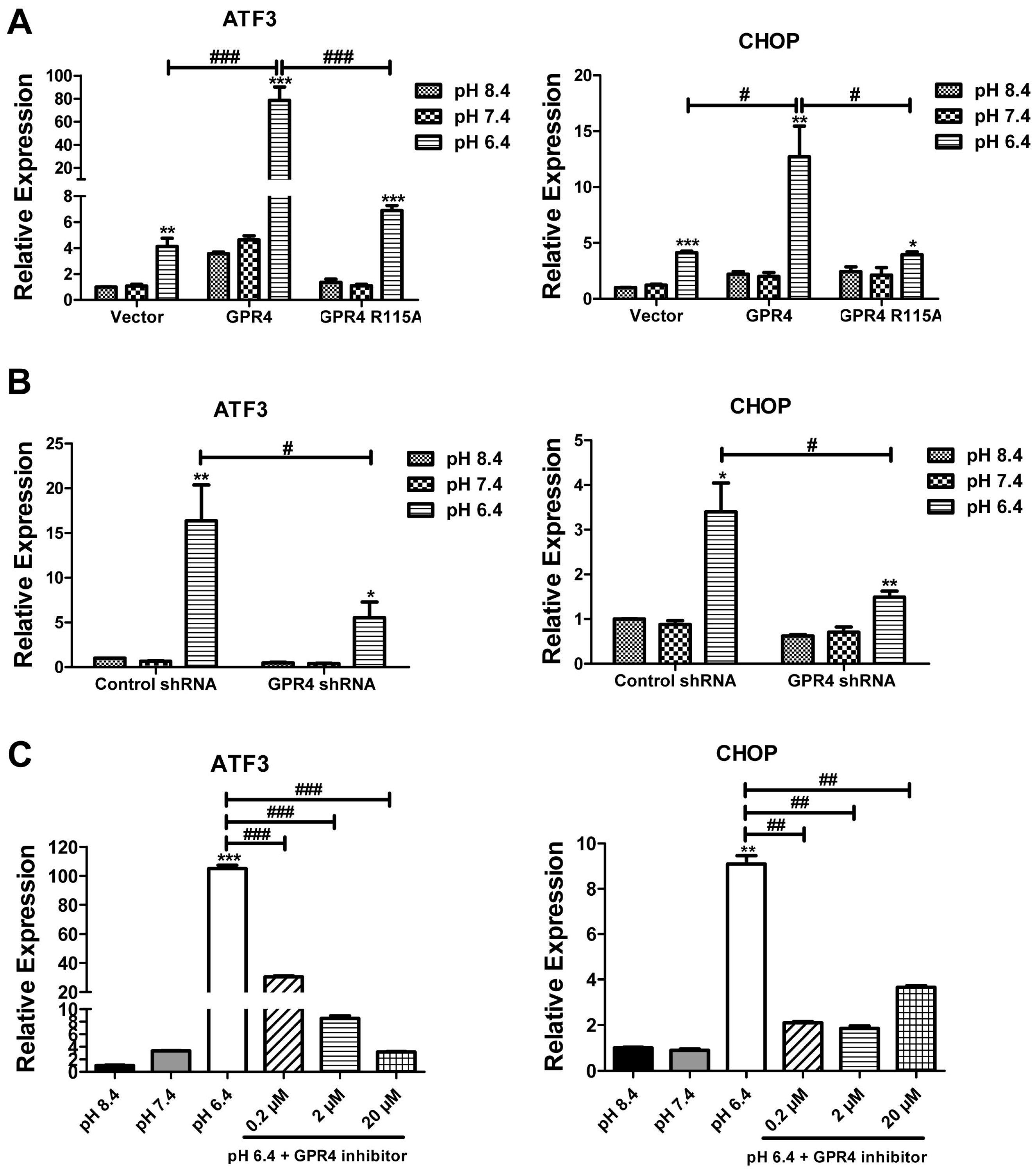
2.2. Overexpression of GPR4, but Not the Signaling Defective GPR4 Mutant, Augments the ER Stress Response Induced by Acidosis in HUVEC
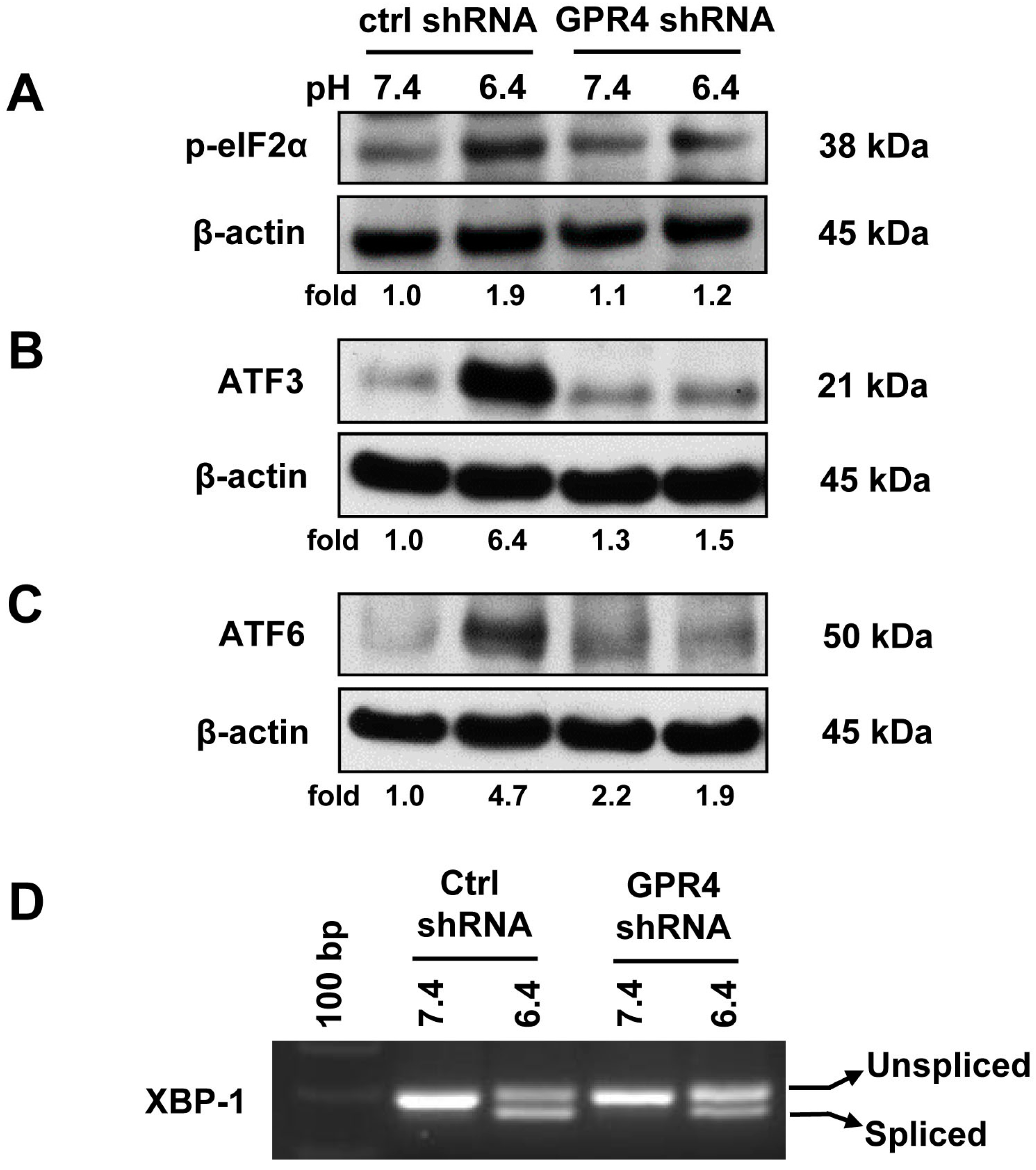
2.3. Knocking Down GPR4 by shRNA Attenuates the ER Stress Response Induced by Acidosis in HUVEC
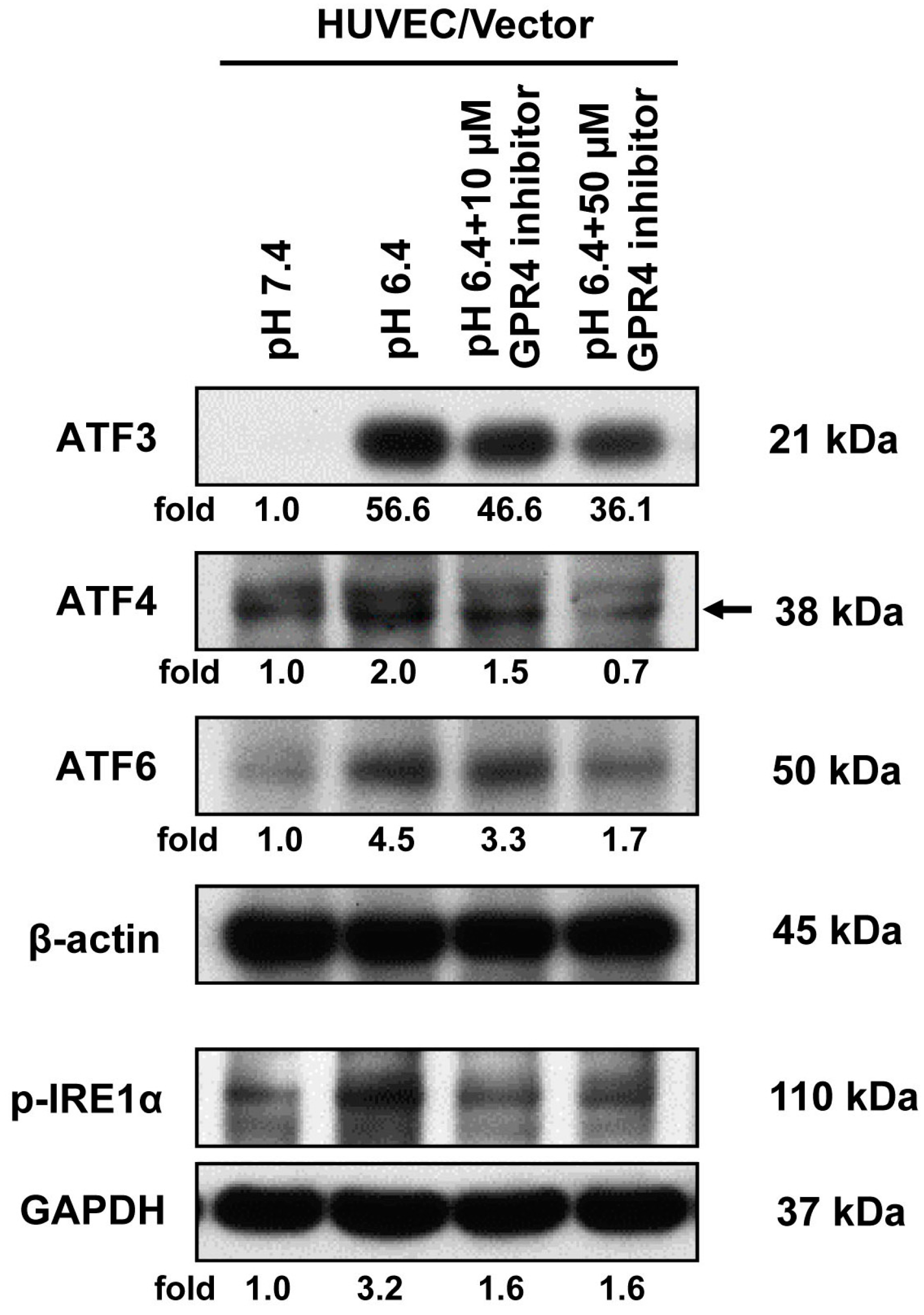
2.4. Blockade of GPR4 Activity by a Small Molecule Inhibitor Diminishes the ER Stress Response Induced by Acidosis in HUVEC
2.5. GPR4 Modulates the mRNA Expression of ER Stress Response Genes Induced by Acidic pH in HUVEC
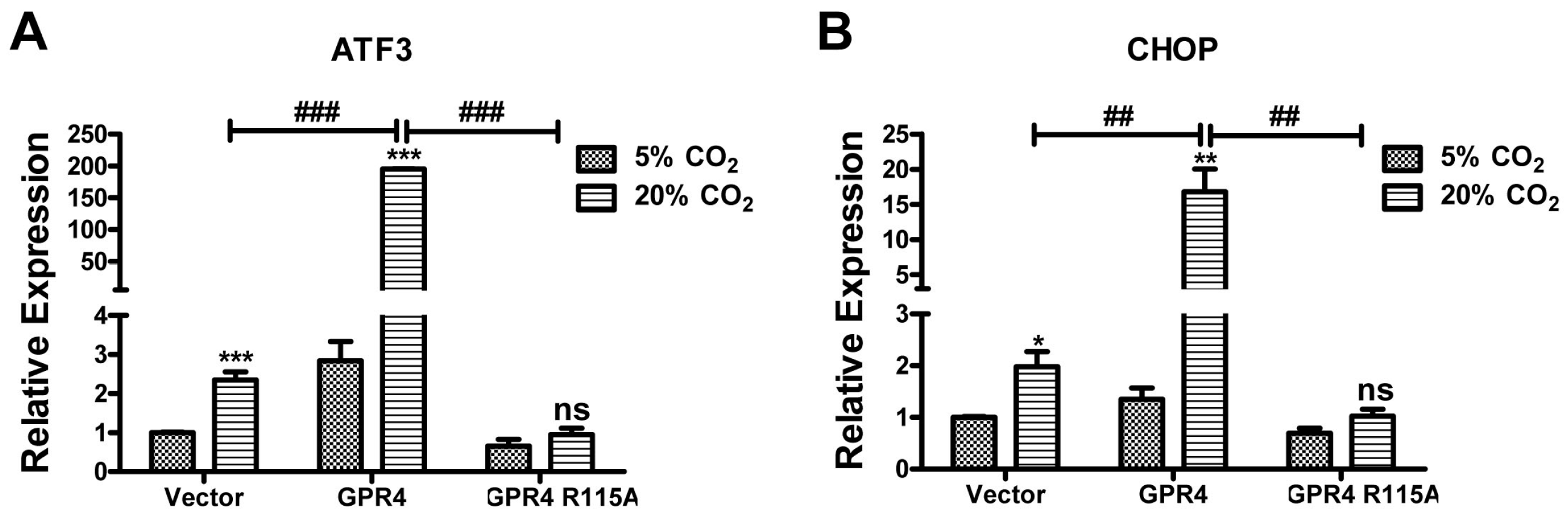
2.6. GPR4 Modulates Hypercapnic Acidosis-Induced ER Stress Gene Expression in HUVEC
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture and Retroviral Transduction
4.3. Isocapnic and Hypercapnic pH Treatment
4.4. Western Blotting
4.5. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.6. Real-Time qRT-PCR
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Chen, A.; Dong, L.; Leffler, N.R.; Asch, A.S.; Witte, O.N.; Yang, L.V. Activation of GPR4 by acidosis increases endothelial cell adhesion through the cAMP/Epac pathway. PLoS ONE 2011, 6, e27586. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Z.; Leffler, N.R.; Asch, A.S.; Chi, J.T.; Yang, L.V. Acidosis activation of the proton-sensing GPR4 receptor stimulates vascular endothelial cell inflammatory responses revealed by transcriptome analysis. PLoS ONE 2013, 8, e61991. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Justus, C.R.; Dong, L.; Yang, L.V. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front. Physiol. 2013, 4, 354. [Google Scholar] [CrossRef] [PubMed]
- Sanderlin, E.J.; Justus, C.R.; Krewson, E.A.; Yang, L.V. Emerging roles for the pH-sensing G protein-coupled receptors in response to acidotic stress. Cell Health Cytoskelet. 2015, 7, 99–109. [Google Scholar]
- Huang, Y.; McNamara, J.O. Ischemic stroke: “Acidotoxicity” is a perpetrator. Cell 2004, 118, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Siesjo, B.K.; Katsura, K.I.; Kristian, T.; Li, P.A.; Siesjo, P. Molecular mechanisms of acidosis-mediated damage. Acta Neurochir. Suppl. 1996, 66, 8–14. [Google Scholar] [PubMed]
- Griffiths, J.R.; McIntyre, D.J.; Howe, F.A.; Stubbs, M. Why are cancers acidic? A carrier-mediated diffusion model for H+ transport in the interstitial fluid. Novartis Found. Symp. 2001, 240, 46–62. [Google Scholar] [PubMed]
- Aoyama, K.; Burns, D.M.; Suh, S.W.; Garnier, P.; Matsumori, Y.; Shiina, H.; Swanson, R.A. Acidosis causes endoplasmic reticulum stress and caspase-12-mediated astrocyte death. J. Cereb. Blood Flow Metab. 2005, 25, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Johno, H.; Ogata, R.; Nakajima, S.; Hiramatsu, N.; Kobayashi, T.; Hara, H.; Kitamura, M. Acidic stress-ER stress axis for blunted activation of NF-κB in mesothelial cells exposed to peritoneal dialysis fluid. Nephrol. Dial. Transplant. 2012, 27, 4053–4060. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Lucas, J.E.; Chen, J.L.; LaMonte, G.; Wu, J.; Wang, M.C.; Koumenis, C.; Chi, J.T. Functional interaction between responses to lactic acidosis and hypoxia regulates genomic transcriptional outputs. Cancer Res. 2012, 72, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Wang, Y.; Alam, G.N.; Ning, Y.; Rados, P.V.; Nor, J.E.; Polverini, P.J. Glucose-regulated protein 78 (Grp78) confers chemoresistance to tumor endothelial cells under acidic stress. PLoS ONE 2014, 9, e101053. [Google Scholar] [CrossRef] [PubMed]
- Cimellaro, A.; Perticone, M.; Fiorentino, T.V.; Sciacqua, A.; Hribal, M.L. Role of endoplasmic reticulum stress in endothelial dysfunction. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Lu, D.; Hai, T.; Harding, H.P.; Wang, X.; Ron, D.; Cavener, D.R.; Wek, R.C. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 2004, 24, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P. Acid-sensitive ion channels and receptors. Handb. Exp. Pharmacol. 2009, 194, 283–332. [Google Scholar]
- Dong, L.; Li, Z.; Yang, L.V. Function and signaling of the pH-sensing G protein-coupled receptors in physiology and diseases. In Molecular Genetics of Dysregulated pH homeostasis; Chi, J.T., Ed.; Springer: New York, NY, USA, 2014; pp. 45–65. [Google Scholar]
- Ludwig, M.G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-sensing G protein-coupled receptors. Nature 2003, 425, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.V.; Radu, C.G.; Roy, M.; Lee, S.; McLaughlin, J.; Teitell, M.A.; Iruela-Arispe, M.L.; Witte, O.N. Vascular abnormalities in mice deficient for the G protein-coupled receptor GPR4 that functions as a pH sensor. Mol. Cell. Biol. 2007, 27, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Tobo, A.; Tobo, M.; Nakakura, T.; Ebara, M.; Tomura, H.; Mogi, C.; Im, D.S.; Murata, N.; Kuwabara, A.; Ito, S.; et al. Characterization of imidazopyridine compounds as negative allosteric modulators of proton-sensing GPR4 in extracellular acidification-induced responses. PLoS ONE 2015, 10, e0129334. [Google Scholar] [CrossRef] [PubMed]
- Sanderlin, E.J.; Leffler, N.R.; Lertpiriyapong, K.; Cai, Q.; Hong, H.; Bakthavatchalu, V.; Fox, J.G.; Oswald, J.Z.; Justus, C.R.; Krewson, E.A.; et al. GPR4 deficiency alleviates intestinal inflammation in a mouse model of acute experimental colitis. Biochim. Biophys. Acta 2016, 1863, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Ito, S.; Watari, K.; Mogi, C.; Arisawa, M.; Okajima, F.; Kurose, H.; Shuto, S. Identification of a potent and selective GPR4 antagonist as a drug lead for the treatment of myocardial infarction. ACS Med. Chem. Lett. 2016, 7, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.N.; Velic, A.; Soliz, J.; Shi, Y.; Li, K.; Wang, S.; Weaver, J.L.; Sen, J.; Abbott, S.B.; Lazarenko, R.M.; et al. Physiology. Regulation of breathing by CO2 requires the proton-activated receptor GPR4 in retrotrapezoid nucleus neurons. Science 2015, 348, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.L.; Pellegrini, P.; di Lernia, G.; Djavaheri-Mergny, M.; Brnjic, S.; Zhang, X.; Hagg, M.; Linder, S.; Fais, S.; Codogno, P.; et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J. Biol. Chem. 2012, 287, 30664–30676. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Rothberg, J.M.; Kumar, V.; Schramm, K.J.; Haller, E.; Proemsey, J.B.; Lloyd, M.C.; Sloane, B.F.; Gillies, R.J. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res. 2012, 72, 3938–3947. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.G.; Zhu, X.M.; Chu, X.P.; Minami, M.; Hey, J.; Wei, W.L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Okito, A.; Nakahama, K.; Akiyama, M.; Ono, T.; Morita, I. Involvement of the G protein-coupled receptor 4 in RANKL expression by osteoblasts in an acidic environment. Biochem. Biophys. Res. Commun. 2015, 458, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Cheah, C.Y.; Seymour, J.F.; Wang, M.L. Mantle cell lymphoma. J. Clin. Oncol. 2016, 34, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef] [PubMed]
- Cawley, K.; Deegan, S.; Samali, A.; Gupta, S. Assays for detecting the unfolded protein response. Methods Enzymol. 2011, 490, 31–51. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, L.; Krewson, E.A.; Yang, L.V. Acidosis Activates Endoplasmic Reticulum Stress Pathways through GPR4 in Human Vascular Endothelial Cells. Int. J. Mol. Sci. 2017, 18, 278. https://doi.org/10.3390/ijms18020278
Dong L, Krewson EA, Yang LV. Acidosis Activates Endoplasmic Reticulum Stress Pathways through GPR4 in Human Vascular Endothelial Cells. International Journal of Molecular Sciences. 2017; 18(2):278. https://doi.org/10.3390/ijms18020278
Chicago/Turabian StyleDong, Lixue, Elizabeth A. Krewson, and Li V. Yang. 2017. "Acidosis Activates Endoplasmic Reticulum Stress Pathways through GPR4 in Human Vascular Endothelial Cells" International Journal of Molecular Sciences 18, no. 2: 278. https://doi.org/10.3390/ijms18020278
APA StyleDong, L., Krewson, E. A., & Yang, L. V. (2017). Acidosis Activates Endoplasmic Reticulum Stress Pathways through GPR4 in Human Vascular Endothelial Cells. International Journal of Molecular Sciences, 18(2), 278. https://doi.org/10.3390/ijms18020278