
Phosphodiesterase Inhibitors as a Therapeutic Approach to Neuroprotection and Repair

Abstract
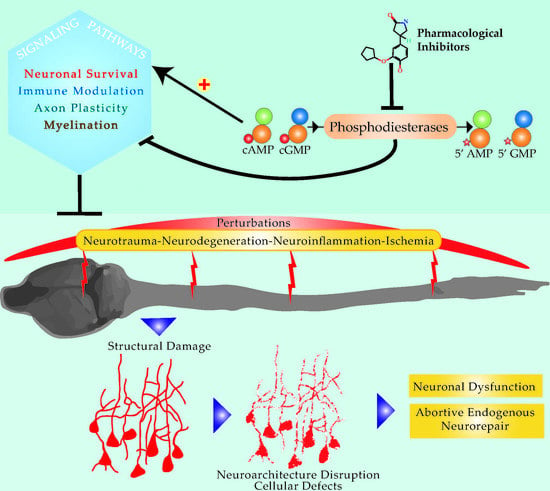
:
1. Introduction
2. Overview of the Cyclic Adenosine Monophosphate Signaling Pathway
2.1. From Membrane to Nucleus
2.2. The Role of Cyclic AMP in the CNS Axon Growth and Cell Survival
3. Overview of Cyclic Nucleotide Phosphodiesterases
3.1. The Phosphodiesterase Family
3.2. The Structure and CNS Distribution of Phosphodiesterases
4. Pathological Alterations to Phosphodiesterases and Cyclic Nucleotides
4.1. Phosphodiesterase and Cyclic Nucleotide Changes Following Neurotrauma
4.2. Phosphodiesterase and Cyclic Nucleotide Alterations in Neurodegenerative Conditions
5. The Utility of PDE Inhibitors for Neuroprotection and Neurorepair
5.1. PDE1
5.2. PDE2
5.3. PDE3
5.4. PDE4
5.5. PDE5
5.6. PDE7
5.7. PDE9
5.8. PDE10
6. Limitations of Currently Available PDE Inhibitors for Therapeutic Use in the CNS
6.1. Synthesis
6.2. Side Effects vs. Potency
7. Clinical Application of PDE Inhibitors in Non-CNS Conditions
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Malpass, K. Multiple sclerosis: Regenerative therapies for MS-hope on the horizon. Nat. Rev. Neurol. 2013, 9, 484. [Google Scholar] [CrossRef] [PubMed]
- Rutland-Brown, W.; Wallace, L.J.D.; Faul, M.D.; Langlois, J.A. Traumatic brain injury hospitalizations among american indians/alaska natives. J. Head Trauma Rehabil. 2005, 20, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, M. Traumatic brain injury: Giving voice to a silent epidemic. Nat. Rev. Neurol. 2013, 9, 186–187. [Google Scholar] [CrossRef] [PubMed]
- New, P.W.; Cripps, R.A.; Bonne Lee, B. Global maps of non-traumatic spinal cord injury epidemiology: Towards a living data repository. Spinal Cord 2014, 52, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.R.; Forgione, N.; Fehlings, M.G. Emerging therapies for acute traumatic spinal cord injury. Can. Med. Assoc. J. 2013, 185, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Tator, C.H.; Fehlings, M.G. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J. Neurosurg. 1991, 75, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Huebner, E.A.; Strittmatter, S.M. Axon regeneration in the peripheral and central nervous systems. Results Probl. Cell Differ. 2009, 48, 339–351. [Google Scholar] [PubMed]
- Ferguson, T.A.; Son, Y.J. Extrinsic and intrinsic determinants of nerve regeneration. J. Tissue Eng. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.Y.; Gordon, T. The cellular and molecular basis of peripheral nerve regeneration. Mol. Neurobiol. 1997, 14, 67–116. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The repair schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.V.; Stevenson, J.A. Peripheral nerve grafts lacking viable schwann cells fail to support central nervous system axonal regeneration. Exp. Brain Res. 1988, 69, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Knott, E.P.; Assi, M.; Pearse, D.D. Cyclic AMP signaling: A molecular determinant of peripheral nerve regeneration. BioMed Res. Int. 2014, 2014, 651625. [Google Scholar] [CrossRef] [PubMed]
- Sansam, K.A. Controversies in the management of traumatic spinal cord injury. Clin. Med. 2006, 6, 202–204. [Google Scholar] [CrossRef]
- Doppenberg, E.M.; Choi, S.C.; Bullock, R. Clinical trials in traumatic brain injury: Lessons for the future. J. Neurosurg. Anesthesiol. 2004, 16, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, T.H.; Murray, F.; Li, X.; Choi, S.S.; Broide, D.H.; Corr, M.; Lee, J.; Webster, N.J.; Insel, P.A.; et al. Cyclic AMP concentrations in dendritic cells induce and regulate Th2 immunity and allergic asthma. Proc. Natl. Acad. Sci. USA 2015, 112, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Mosenden, R.; Tasken, K. Cyclic AMP-mediated immune regulation—Overview of mechanisms of action in T cells. Cell. Signal. 2011, 23, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Schafer, P.H.; Parton, A.; Capone, L.; Cedzik, D.; Brady, H.; Evans, J.F.; Man, H.W.; Muller, G.W.; Stirling, D.I.; Chopra, R. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell. Signal. 2014, 26, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- Heystek, H.C.; Thierry, A.C.; Soulard, P.; Moulon, C. Phosphodiesterase 4 inhibitors reduce human dendritic cell inflammatory cytokine production and Th1-polarizing capacity. Int. Immunol. 2003, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Herve, R.; Schmitz, T.; Evain-Brion, D.; Cabrol, D.; Leroy, M.J.; Mehats, C. The PDE4 inhibitor rolipram prevents NF-κB binding activity and proinflammatory cytokine release in human chorionic cells. J. Immunol. 2008, 181, 2196–2202. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.M.; Gristwood, R.W.; Cooper, N.; Hellewell, P.G. Phosphodiesterase (PDE)4 inhibitors: Anti-inflammatory drugs of the future? Trends Pharmacol. Sci. 1997, 18, 164–171. [Google Scholar] [PubMed]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP: Master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Hannila, S.S.; Filbin, M.T. The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp. Neurol. 2008, 209, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.J.; Shewan, D.A. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol. Cell. Neurosci. 2008, 38, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Pearse, D.D.; Pereira, F.C.; Marcillo, A.E.; Bates, M.L.; Berrocal, Y.A.; Filbin, M.T.; Bunge, M.B. cAMP and schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat. Med. 2004, 10, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Nikulina, E.; Tidwell, J.L.; Dai, H.N.; Bregman, B.S.; Filbin, M.T. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc. Natl. Acad. Sci. USA 2004, 101, 8786–8790. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Yang, H.; Jones, L.L.; Filbin, M.T.; Tuszynski, M.H. Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J. Neurosci. 2004, 24, 6402–6409. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.M.; Oliva, A.A., Jr.; Alonso, O.F.; Pearse, D.D.; Bramlett, H.M.; Dietrich, W.D. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp. Neurol. 2007, 208, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Perez-Torres, S.; Mengod, G. cAMP-specific phosphodiesterases expression in Alzheimer’s disease brains. Int. Congr. Ser. 2003, 1251, 127–138. [Google Scholar] [CrossRef]
- Garcia-Osta, A.; Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Oyarzabal, J.; Franco, R. Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Huang, Y.; Masood, A.; Stolinski, L.R.; Li, Y.; Zhang, L.; Dlaboga, D.; Jin, S.L.; Conti, M.; O’Donnell, J.M. Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4b (PDE4B). Neuropsychopharmacology 2008, 33, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Wild, E.J.; Tabrizi, S.J. Targets for future clinical trials in Huntington’s disease: What’s in the pipeline? Mov. Disord. 2014, 29, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Siuciak, J.A. The role of phosphodiesterases in schizophrenia: Therapeutic implications. CNS Drugs 2008, 22, 983–993. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.M.; Xu, Y. Evidence for global reduction in brain cyclic adenosine monophosphate signaling in depression. Biol. Psychiatry 2012, 72, 524–525. [Google Scholar] [CrossRef] [PubMed]
- Tresguerres, M.; Levin, L.R.; Buck, J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011, 79, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Kleppisch, T. Phosphodiesterases in the central nervous system. Handb. Exp. Pharmacol. 2009, 204, 71–92. [Google Scholar]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. Huntington’s disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Bartolotti, N.; Bennett, D.A.; Lazarov, O. Reduced pcreb in Alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol. Psychiatry 2016, 21, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Kandel, E.R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 2012, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.; Alcolea, D.; Sanchez-Valle, R.; Guardia-Laguarta, C.; Colom-Cadena, M.; Badiola, N.; Suarez-Calvet, M.; Llado, A.; Barrera-Ocampo, A.A.; Sepulveda-Falla, D.; et al. Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 2013, 125, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Vinters, H.V. Emerging concepts in Alzheimer’s disease. Annu. Rev. Pathol. 2015, 10, 291–319. [Google Scholar] [CrossRef] [PubMed]
- Palazidou, E. The neurobiology of depression. Br. Med. Bull. 2012, 101, 127–145. [Google Scholar] [CrossRef] [PubMed]
- Ryder, J.G.; Holtzheimer, P.E. Deep brain stimulation for depression: An update. Curr. Behav. Neurosci. Rep. 2016, 3, 102–108. [Google Scholar] [CrossRef]
- Pearlson, G.D.; Clementz, B.A.; Sweeney, J.A.; Keshavan, M.S.; Tamminga, C.A. Does biology transcend the symptom-based boundaries of psychosis? Psychiatr. Clin. N. Am. 2016, 39, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Huang, Y.; Jin, S.L.; Frith, S.A.; Suvarna, N.; Conti, M.; O’Donnell, J.M. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4d phosphodiesterase enzyme. Neuropsychopharmacology 2002, 27, 587–595. [Google Scholar] [CrossRef]
- Wong, M.L.; Whelan, F.; Deloukas, P.; Whittaker, P.; Delgado, M.; Cantor, R.M.; McCann, S.M.; Licinio, J. Phosphodiesterase genes are associated with susceptibility to major depression and antidepressant treatment response. Proc. Natl. Acad. Sci. USA 2006, 103, 15124–15129. [Google Scholar] [CrossRef] [PubMed]
- Keshavan, M.S.; Nasrallah, H.A.; Tandon, R. Schizophrenia, “just the facts” 6. Moving ahead with the schizophrenia concept: From the elephant to the mouse. Schizophr. Res. 2011, 127, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Jindal, R.D.; Keshavan, M.S. Neurobiology of the early course of schizophrenia. Expert Rev. Neurother. 2008, 8, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr. Res. 2010, 122, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.J.; Chapin, D.S.; Cianfrogna, J.; Corman, M.L.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; Lebel, L.A.; McCarthy, S.A.; Nelson, F.R.; et al. Preclinical characterization of selective phosphodiesterase 10a inhibitors: A new therapeutic approach to the treatment of schizophrenia. J. Pharmacol. Exp. Ther. 2008, 325, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Verhoest, P.R.; Chapin, D.S.; Corman, M.; Fonseca, K.; Harms, J.F.; Hou, X.; Marr, E.S.; Menniti, F.S.; Nelson, F.; O’Connor, R.; et al. Discovery of a novel class of phosphodiesterase 10a inhibitors and identification of clinical candidate 2-[4-(1-methyl-4-pyridin-4-yl-1h-pyrazol-3-yl)-phenoxymethyl]-quinoline (PF-2545920) for the treatment of schizophrenia. J. Med. Chem. 2009, 52, 5188–5196. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, S.A. Earl sutherland (1915–1974) (corrected) and the discovery of cyclic AMP. Perspect. Biol. Med. 2012, 55, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Landry, Y.; Niederhoffer, N.; Sick, E.; Gies, J.P. Heptahelical and other G-protein-coupled receptors (GPCRs) signaling. Curr. Med. Chem. 2006, 13, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Kamenetsky, M.; Middelhaufe, S.; Bank, E.M.; Levin, L.R.; Buck, J.; Steegborn, C. Molecular details of cAMP generation in mammalian cells: A tale of two systems. J. Mol. Biol. 2006, 362, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Sunahara, R.K.; Taussig, R. Isoforms of mammalian adenylyl cyclase: Multiplicities of signaling. Mol. Interv. 2002, 2, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, M.S.; Buck, J.; Levin, L.R. Bicarbonate-regulated soluble adenylyl cyclase. J. Pancreas 2001, 2, 154–158. [Google Scholar]
- Stiles, T.L.; Kapiloff, M.S.; Goldberg, J.L. The role of soluble adenylyl cyclase in neurite outgrowth. Biochim. Biophys. Acta 2014, 1842, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Hanoune, J.; Defer, N. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 145–174. [Google Scholar] [CrossRef] [PubMed]
- Sprang, S.R. G protein mechanisms: Insights from structural analysis. Annu. Rev. Biochem. 1997, 66, 639–678. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.T.; Verheijen, M.H.G.; Cool, R.H.; Nijman, S.M.B.; Wittinghofer, A.; Bos, J.L. Epac is a RAP1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [PubMed]
- Bos, J.L. Epac proteins: Multi-purpose cAMP targets. Trends Biochem. Sci. 2006, 31, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Kopperud, R.; Krakstad, C.; Selheim, F.; Doskeland, S.O. cAMP effector mechanisms. Novel twists for an ‘old’ signaling system. FEBS Lett. 2003, 546, 121–126. [Google Scholar] [CrossRef]
- Bacallao, K.; Monje, P.V. Opposing roles of PKA and Epac in the cAMP-dependent regulation of schwann cell proliferation and differentiation (corrected). PLoS ONE 2013, 8, e82354. [Google Scholar] [CrossRef] [PubMed]
- Montminy, M.R.; Bilezikjian, L.M. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature 1987, 328, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Bonni, A.; Ginty, D.D.; Dudek, H.; Greenberg, M.E. Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol. Cell. Neurosci. 1995, 6, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.B.; Walker, W.H.; Habener, J.F. Cyclic AMP signaling and gene regulation. Annu. Rev. Nutr. 1998, 18, 353–383. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Guan, H.; Ricciardi, R.P. Phosphorylation of serine 337 of NF-κB p50 is critical for DNA binding. J. Biol. Chem. 2003, 278, 45994–45998. [Google Scholar] [CrossRef] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, J.P.; Godlevski, M.M.; Wisely, G.B.; Clay, W.C.; Leesnitzer, L.M.; Ways, J.P.; Gray, J.G. NF-κb regulates IL-1β transcription through a consensus NF-κB binding site and a nonconsensus CRE-like site. J. Immunol. 1994, 153, 712–723. [Google Scholar] [PubMed]
- Verghese, M.W.; McConnell, R.T.; Strickland, A.B.; Gooding, R.C.; Stimpson, S.A.; Yarnall, D.P.; Taylor, J.D.; Furdon, P.J. Differential regulation of human monocyte-derived TNFα and IL-1β by type IV cAMP-phosphodiesterase (cAMP-PDE) inhibitors. J. Pharmacol. Exp. Ther. 1995, 272, 1313–1320. [Google Scholar] [PubMed]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, V.; Zucker, R.S. Enhancement of synaptic transmission by cyclic AMP modulation of presynaptic Ih channels. Nat. Neurosci. 2000, 3, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, T.; Fujioka, A.; Duman, R.S. Activation of cAMP signaling facilitates the morphological maturation of newborn neurons in adult hippocampus. J. Neurosci. 2004, 24, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Titus, D.J.; Furones, C.; Kang, Y.; Atkins, C.M. Age-dependent alterations in cAMP signaling contribute to synaptic plasticity deficits following traumatic brain injury. Neuroscience 2013, 231, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Bregman, B.S.; Broude, E.; McAtee, M.; Kelley, M.S. Transplants and neurotrophic factors prevent atrophy of mature CNS neurons after spinal cord injury. Exp. Neurol. 1998, 149, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Mckeon, R.J.; Schreiber, R.C.; Rudge, J.S.; Silver, J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J. Neurosci. 1991, 11, 3398–3411. [Google Scholar] [PubMed]
- Mukhopadhyay, G.; Doherty, P.; Walsh, F.S.; Crocker, P.R.; Filbin, M.T. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 1994, 13, 757–767. [Google Scholar] [CrossRef]
- Cai, D.; Shen, Y.; de Bellard, M.; Tang, S.; Filbin, M.T. Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron 1999, 22, 89–101. [Google Scholar] [CrossRef]
- Gao, Y.; Nikulina, E.; Mellado, W.; Filbin, M.T. Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J. Neurosci. 2003, 23, 11770–11777. [Google Scholar] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the GP130/JAK/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Gao, Y.; Bryson, J.B.; Hou, J.; Chaudhry, N.; Siddiq, M.; Martinez, J.; Spencer, T.; Carmel, J.; Hart, R.B.; et al. The cytokine interleukin-6 is sufficient but not necessary to mimic the peripheral conditioning lesion effect on axonal growth. J. Neurosci. 2006, 26, 5565–5573. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Deng, K.; Hou, J.; Bryson, J.B.; Barco, A.; Nikulina, E.; Spencer, T.; Mellado, W.; Kandel, E.R.; Filbin, M.T. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 2004, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Deng, K.; Mellado, W.; Lee, J.; Ratan, R.R.; Filbin, M.T. Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 2002, 35, 711–719. [Google Scholar] [CrossRef]
- Lange, P.S.; Langley, B.; Lu, P.; Ratan, R.R. Novel roles for arginase in cell survival, regeneration, and translation in the central nervous system. J. Nutr. 2004, 134, 2812S–2819S. [Google Scholar] [PubMed]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Iorgulescu, J.B.; Patel, S.P.; Louro, J.; Andrade, C.M.; Sanchez, A.R.; Pearse, D.D. Acute putrescine supplementation with schwann cell implantation improves sensory and serotonergic axon growth and functional recovery in spinal cord injured rats. Neural Plast. 2015, 2015, 186385. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.; He, H.; Qiu, J.; Lorber, B.; Bryson, J.B.; Filbin, M.T. Increased synthesis of spermidine as a result of upregulation of arginase I promotes axonal regeneration in culture and in vivo. J. Neurosci. 2009, 29, 9545–9552. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.C.; Fitzpatrick, E.A.; Maley, M.E.; Gellin, G.L.; Tsuei, B.J.; Arden, W.A.; Boulanger, B.R.; Kearney, P.A.; Ochoa, J.B. β Adrenoceptor regulation of macrophage arginase activity. Surgery 2000, 127, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Corraliza, I.M.; Soler, G.; Eichmann, K.; Modolell, M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem. Biophys. Res. Commun. 1995, 206, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Csoka, B.; Selmeczy, Z.; Koscso, B.; Nemeth, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, K.E.; Shandilya, H.; Kepka-Lenhart, D.; Poljakovic, M.; Ghosh, A.; Morris, S.M., Jr. Shaping the murine macrophage phenotype: IL-4 and cyclic AMP synergistically activate the arginase I promoter. J. Immunol. 2013, 191, 2290–2298. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.C.; Campana, A.; Lange, P.S.; Lee, H.H.; Banerjee, K.; Bryson, J.B.; Mahishi, L.; Alam, S.; Giger, R.J.; Barnes, S.; et al. A large-scale chemical screen for regulators of the arginase 1 promoter identifies the soy isoflavone daidzeinas a clinically approved small molecule that can promote neuronal protection or regeneration via a cAMP-independent pathway. J. Neurosci. 2010, 30, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Stout, J.M.; Knapp, A.N.; Banz, W.J.; Wallace, D.G.; Cheatwood, J.L. Subcutaneous daidzein administration enhances recovery of skilled ladder rung walking performance following stroke in rats. Behav. Brain Res. 2013, 256, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Epac: A new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell Biol. 2003, 4, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Roscioni, S.S.; Elzinga, C.R.; Schmidt, M. Epac: Effectors and biological functions. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 377, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Zambon, A.C.; Zhang, L.; Minovitsky, S.; Kanter, J.R.; Prabhakar, S.; Salomonis, N.; Vranizan, K.; Dubchak, I.; Conklin, B.R.; Insel, P.A. Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc. Natl. Acad. Sci. USA 2005, 102, 8561–8566. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Sand, C.; Jakobs, K.H.; Michel, M.C.; Weernink, P.A. Epac and the cardiovascular system. Curr. Opin. Pharmacol. 2007, 7, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kim, C.; Cheng, C.Y.; Brown, S.H.; Wu, J.; Kannan, N. Signaling through cAMP and cAMP-dependent protein kinase: Diverse strategies for drug design. Biochim. Biophys. Acta 2008, 1784, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Ponsioen, B.; Gloerich, M.; Ritsma, L.; Rehmann, H.; Bos, J.L.; Jalink, K. Direct spatial control of Epac1 by cyclic AMP. Mol. Cell. Biol. 2009, 29, 2521–2531. [Google Scholar] [CrossRef] [PubMed]
- Hochbaum, D.; Tanos, T.; Ribeiro-Neto, F.; Altschuler, D.; Coso, O.A. Activation of JNK by Epac is independent of its activity as a rap guanine nucleotide exchanger. J. Biol. Chem. 2003, 278, 33738–33746. [Google Scholar] [CrossRef] [PubMed]
- Lopez De Jesus, M.; Stope, M.B.; Oude Weernink, P.A.; Mahlke, Y.; Borgermann, C.; Ananaba, V.N.; Rimmbach, C.; Rosskopf, D.; Michel, M.C.; Jakobs, K.H.; et al. Cyclic AMP-dependent and Epac-mediated activation of R-Ras by G protein-coupled receptors leads to phospholipase D stimulation. J. Biol. Chem. 2006, 281, 21837–21847. [Google Scholar] [CrossRef] [PubMed]
- Ster, J.; de Bock, F.; Guerineau, N.C.; Janossy, A.; Barrere-Lemaire, S.; Bos, J.L.; Bockaert, J.; Fagni, L. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 2519–2524. [Google Scholar] [CrossRef] [PubMed]
- Nijholt, I.M.; Dolga, A.M.; Ostroveanu, A.; Luiten, P.G.; Schmidt, M.; Eisel, U.L. Neuronal AKAP150 coordinates PKA and Epac-mediated PKB/Akt phosphorylation. Cell. Signal. 2008, 20, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Ha, S.H.; Lee, M.N.; Huston, E.; Kim, D.H.; Jang, S.K.; Suh, P.G.; Houslay, M.D.; Ryu, S.H. Cyclic AMP controls mTOR through regulation of the dynamic interaction between Rheb and phosphodiesterase 4D. Mol. Cell. Biol. 2010, 30, 5406–5420. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Llancao, P.; Henriquez, D.R.; Wilson, C.; Bodaleo, F.; Boddeke, E.W.; Lezoualc’h, F.; Schmidt, M.; Gonzalez-Billault, C. Exchange protein directly activated by cAMP (Epac) regulates neuronal polarization through Rap1B. J. Neurosci. 2015, 35, 11315–11329. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.E.; Selheim, F.; de Rooij, J.; Dremier, S.; Schwede, F.; Dao, K.K.; Martinez, A.; Maenhaut, C.; Bos, J.L.; Genieser, H.G.; et al. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC12 cell neurite extension. J. Biol. Chem. 2003, 278, 35394–35402. [Google Scholar] [CrossRef] [PubMed]
- Kiermayer, S.; Biondi, R.M.; Imig, J.; Plotz, G.; Haupenthal, J.; Zeuzem, S.; Piiper, A. Epac activation converts cAMP from a proliferative into a differentiation signal in PC12 cells. Mol. Biol. Cell 2005, 16, 5639–5648. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Hsu, K.S. Presynaptic mechanism underlying cAMP-induced synaptic potentiation in medial prefrontal cortex pyramidal neurons. Mol. Pharmacol. 2006, 69, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Gekel, I.; Neher, E. Application of an Epac activator enhances neurotransmitter release at excitatory central synapses. J. Neurosci. 2008, 28, 7991–8002. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Zhang, L.; Zhu, J.J.; Schwede, F.; Thomas, S.A. Epac signaling is required for hippocampus-dependent memory retrieval. Proc. Natl. Acad. Sci. USA 2008, 105, 11993–11997. [Google Scholar] [CrossRef] [PubMed]
- Ster, J.; de Bock, F.; Bertaso, F.; Abitbol, K.; Daniel, H.; Bockaert, J.; Fagni, L. Epac mediates PACAP-dependent long-term depression in the hippocampus. J. Physiol. 2009, 587, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, J.N.; Banko, J.L.; Peters, M.M.; Klann, E.; Weeber, E.J.; Nguyen, P.V. Activation of exchange protein activated by cyclic-AMP enhances long-lasting synaptic potentiation in the hippocampus. Learn. Mem. 2008, 15, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Dao, K.K.; Teigen, K.; Kopperud, R.; Hodneland, E.; Schwede, F.; Christensen, A.E.; Martinez, A.; Doskeland, S.O. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J. Biol. Chem. 2006, 281, 21500–21511. [Google Scholar] [CrossRef] [PubMed]
- Gloerich, M.; Bos, J.L. Epac: Defining a new mechanism for cAMP action. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 355–375. [Google Scholar] [CrossRef] [PubMed]
- Parnell, E.; Palmer, T.M.; Yarwood, S.J. The future of Epac-targeted therapies: Agonism vs. antagonism. Trends Pharmacol. Sci. 2015, 36, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; So, K.F. Involvement of cAMP in neuronal survival and axonal regeneration. Anat. Sci. Int. 2004, 79, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Franke, A.; Wilkinson, G.A.; Kruttgen, A.; Hu, M.; Munro, E.; Hanson, M.G., Jr.; Reichardt, L.F.; Barres, B.A. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 1998, 21, 681–693. [Google Scholar] [CrossRef]
- Brunet, A.; Datta, S.R.; Greenberg, M.E. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 2001, 11, 297–305. [Google Scholar] [CrossRef]
- Doronzo, G.; Viretto, M.; Russo, I.; Mattiello, L.; di Martino, L.; Cavalot, F.; Anfossi, G.; Trovati, M. Nitric oxide activates PI3-K and MAPK signalling pathways in human and rat vascular smooth muscle cells: Influence of insulin resistance and oxidative stress. Atherosclerosis 2011, 216, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.J.; Morisset, J.; Vachon, P.H.; Reed, J.C.; Lainé, J.; Rivard, N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J. Cell. Biochem. 2000, 79, 355–369. [Google Scholar] [CrossRef]
- Jin, K.; Mao, X.O.; Zhu, Y.; Greenberg, D.A. MEK and ERK protect hypoxic cortical neurons via phosphorylation of bad. J. Neurochem. 2002, 80, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Butcher, R.W.; Sutherland, E.W. Adenosine 3′,5′-phosphate in biological materials. I. Purification and properties of cyclic 3′,5′-nucleotide phosphodiesterase and use of this enzyme to characterize adenosine 3′,5′-phosphate in human urine. J. Biol. Chem. 1962, 237, 1244–1250. [Google Scholar] [PubMed]
- Manganiello, V.C.; Murata, T.; Taira, M.; Belfrage, P.; Degerman, E. Diversity in cyclic nucleotide phosphodiesterase isoenzyme families. Arch. Biochem. Biophys. 1995, 322, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Moorthy, B.S.; Xin Xiang, L.; Xin Shan, L.; Bharatham, K.; Tulsian, N.K.; Mihalek, I.; Anand, G.S. Active site coupling in PDE:PKA complexes promotes resetting of mammalian cAMP signaling. Biophys. J. 2014, 107, 1426–1440. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, B.S.; Gao, Y.; Anand, G.S. Phosphodiesterases catalyze hydrolysis of c cAMP-bound to regulatory subunit of protein kinase A and mediate signal termination. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Houslay, M.D.; Conti, M. Phosphodiesterase inhibitors: Factors that influence potency, selectivity, and action. In Phosphodiesterases as Drug Targets; Springer Nature: Berlin, Germany, 2011; pp. 47–84. [Google Scholar]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Sette, C.; Conti, M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J. Biol. Chem. 1996, 271, 16526–16534. [Google Scholar] [PubMed]
- Houslay, M.D.; Sullivan, M.; Bolger, G.B. The multienzyme PDE4 cyclic adenosine monophosphate–specific phosphodiesterase family: Intracellular targeting, regulation, and selective inhibition by compounds exerting anti-inflammatory and antidepressant actions. In Advances in Pharmacology; Elsevier BV: Amsterdam, The Netherlands, 1998; pp. 225–342. [Google Scholar]
- MacKenzie, S.J.; Baillie, G.S.; McPhee, I.; Bolger, G.B.; Houslay, M.D. ERK2 mitogen-activated protein kinase binding, phosphorylation, and regulation of the PDE4D cAMP-specific phosphodiesterases. The involvement of COOH-terminal docking sites and NH2-terminal UCR regions. J. Biol. Chem. 2000, 275, 16609–16617. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; MacKenzie, S.J.; McPhee, I.; Houslay, M.D. Sub-family selective actions in the ability of ERK2 map kinase to phosphorylate and regulate the activity of PDE4 cyclic AMP-specific phosphodiesterases. Br. J. Pharmacol. 2000, 131, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A. Cyclic AMP, protein kinase A, and phosphodiesterases: Proceedings of an international workshop. Horm. Metab. Res. 2012, 44, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.Y.; Card, G.L.; Suzuki, Y.; Artis, D.R.; Fong, D.; Gillette, S.; Hsieh, D.; Neiman, J.; West, B.L.; Zhang, C.; et al. A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases. Mol. Cell 2004, 15, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 2010, 35, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterases (PDE) and peptide motifs. Curr. Pharm. Des. 2010, 16, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Nagakura, A.; Niimura, M.; Takeo, S. Effects of a phosphodiesterase IV inhibitor rolipram on microsphere embolism-induced defects in memory function and cerebral cyclic AMP signal transduction system in rats. Br. J. Pharmacol. 2002, 135, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Vitolo, O.V.; Trinchese, F.; Liu, S.; Shelanski, M.; Arancio, O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Investig. 2004, 114, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Cheng, Y.F.; Lin, H.B.; Wang, C.; Xu, J.P.; Zhang, H.T. Prevention of cerebral ischemia-induced memory deficits by inhibition of phosphodiesterase-4 in rats. Metab. Brain Dis. 2011, 26, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Cheng, Y.F.; Huang, Y.; Conti, M.; Wilson, S.P.; O’Donnell, J.M.; Zhang, H.T. Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J. Neurosci. 2011, 31, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Wiescholleck, V.; Manahan-Vaughan, D. PDE4 inhibition enhances hippocampal synaptic plasticity in vivo and rescues MK801-induced impairment of long-term potentiation and object recognition memory in an animal model of psychosis. Transl. Psychiatry 2012, 2, e89. [Google Scholar] [CrossRef] [PubMed]
- Titus, D.J.; Oliva, A.A.; Wilson, N.M.; Atkins, C.M. Phosphodiesterase inhibitors as therapeutics for traumatic brain injury. Curr. Pharm. Des. 2014, 21, 332–342. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Sharma, R.K.; Soderling, T.R. Regulation of Ca2+/calmodulin-dependent cyclic nucleotide phosphodiesterase by the autophosphorylated form of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1989, 264, 10884–10887. [Google Scholar] [PubMed]
- Florio, V.A.; Sonnenburg, W.K.; Johnson, R.; Kwak, K.S.; Jensen, G.S.; Walsh, K.A.; Beavo, J.A. Phosphorylation of the 61-kDa calmodulin-stimulated cyclic nucleotide phosphodiesterase at serine 120 reduces its affinity for calmodulin. Biochemistry 1994, 33, 8948–8954. [Google Scholar] [CrossRef] [PubMed]
- Shakur, Y.; Holst, L.S.; Landstrom, T.R.; Movsesian, M.; Degerman, E.; Manganiello, V. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog. Nucleic Acid Res. Mol. Biol. 2001, 66, 241–277. [Google Scholar] [PubMed]
- Colbran, J.L.; Francis, S.H.; Leach, A.B.; Thomas, M.K.; Jiang, H.; McAllister, L.M.; Corbin, J.D. A phenylalanine in peptide substrates provides for selectivity between cGMP- and cAMP-dependent protein kinases. J. Biol. Chem. 1992, 267, 9589–9594. [Google Scholar] [PubMed]
- Gross-Langenhoff, M.; Hofbauer, K.; Weber, J.; Schultz, A.; Schultz, J.E. cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J. Biol. Chem. 2006, 281, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, V.A.; Desai, M.; Dua, S.; Yamazaki, M.; Amin, R.H.; Yousif, K.K.; Kinumi, T.; Ohashi, M.; Komori, N.; Matsumoto, H.; et al. Residues within the polycationic region of cGMP phosphodiesterase γ subunit crucial for the interaction with transducin α subunit. Identification by endogenous adp-ribosylation and site-directed mutagenesis. J. Biol. Chem. 1997, 272, 15856–15864. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, V.A.; Yamazaki, M.; Hayashi, F.; Yamazaki, A. Suppression of GTP/T α-dependent activation of cGMP phosphodiesterase by adp-ribosylation by its γ subunit in amphibian rod photoreceptor membranes. Biochemistry 1999, 38, 7755–7763. [Google Scholar] [CrossRef] [PubMed]
- Granovsky, A.E.; Natochin, M.; Artemyev, N.O. The γ subunit of rod cGMP-phosphodiesterase blocks the enzyme catalytic site. J. Biol. Chem. 1997, 272, 11686–11689. [Google Scholar] [CrossRef] [PubMed]
- Morin, F.; Vannier, B.; Houdart, F.; Regnacq, M.; Berges, T.; Voisin, P. A proline-rich domain in the γ subunit of phosphodiesterase 6 mediates interaction with SH3-containing proteins. Mol. Vis. 2003, 9, 449–459. [Google Scholar] [PubMed]
- Houslay, M.D.; Adams, D.R. PDE4 cAMP phosphodiesterases: Modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem. J. 2003, 370, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Conti, M. Dimerization of the type 4 cAMP-specific phosphodiesterases is mediated by the upstream conserved regions (UCRS). J. Biol. Chem. 2002, 277, 40212–40221. [Google Scholar] [CrossRef] [PubMed]
- Bolger, G.; Michaeli, T.; Martins, T.; St John, T.; Steiner, B.; Rodgers, L.; Riggs, M.; Wigler, M.; Ferguson, K. A family of human phosphodiesterases homologous to the dunce learning and memory gene product of drosophila melanogaster are potential targets for antidepressant drugs. Mol. Cell. Biol. 1993, 13, 6558–6571. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Wilkinson, I.R.; McCallum, J.F.; Engels, P.; Houslay, M.D. cAMP-specific phosphodiesterase HSPDE4D3 mutants which mimic activation and changes in rolipram inhibition triggered by protein kinase a phosphorylation of Ser-54: Generation of a molecular model. Biochem. J. 1998, 333, 139–149. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.J.; Baillie, G.S.; McPhee, I.; MacKenzie, C.; Seamons, R.; McSorley, T.; Millen, J.; Beard, M.B.; van Heeke, G.; Houslay, M.D. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in upstream conserved region 1 (UCR1). Br. J. Pharmacol. 2002, 136, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Baillie, G.S.; MacKenzie, S.J.; Yarwood, S.J.; Houslay, M.D. The map kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999, 18, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Sonati, P.; Rubin, C.; Michaeli, T. PDE7A1, a cAMP-specific phosphodiesterase, inhibits cAMP-dependent protein kinase by a direct interaction with C. J. Biol. Chem. 2006, 281, 15050–15057. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kotera, J.; Omori, K. Transcriptional activation of phosphodiesterase 7B1 by dopamine D1 receptor stimulation through the cyclic AMP/cyclic AMP-dependent protein kinase/cyclic AMP-response element binding protein pathway in primary striatal neurons. J. Neurochem. 2004, 89, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, P.; Egan, R.W.; Billah, M.M. Human phosphodiesterase 8A splice variants: Cloning, gene organization, and tissue distribution. Gene 2001, 280, 183–194. [Google Scholar] [CrossRef]
- Gamanuma, M.; Yuasa, K.; Sasaki, T.; Sakurai, N.; Kotera, J.; Omori, K. Comparison of enzymatic characterization and gene organization of cyclic nucleotide phosphodiesterase 8 family in humans. Cell. Signal. 2003, 15, 565–574. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, H.T.; O’Donnell, J.M. Phosphodiesterases in the central nervous system: Implications in mood and cognitive disorders. Handb. Exp. Pharmacol. 2011, 447–485. [Google Scholar]
- Dietrich, W.D.; Chatzipanteli, K.; Vitarbo, E.; Wada, K.; Kinoshita, K. The role of inflammatory processes in the pathophysiology and treatment of brain and spinal cord trauma. In Mechanisms of Secondary Brain Damage from Trauma and Ischemia; Springer Nature: Berlin, Germany, 2004; pp. 69–74. [Google Scholar]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Velumian, A.A.; Fehlings, M.G. The role of excitotoxicity in secondary mechanisms of spinal cord injury: A review with an emphasis on the implications for white matter degeneration. J. Neurotrauma 2004, 21, 754–774. [Google Scholar] [CrossRef] [PubMed]
- Sharif, S.F.; Hariri, R.J.; Chang, V.A.; Barie, P.S.; Wang, R.S.; Ghajar, J.B. Human astrocyte production of tumour necrosis factor-α, interleukin-1β, and interleukin-6 following exposure to lipopolysaccharide endotoxin. Neurol. Res. 1993, 15, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Breder, C.D.; Tsujimoto, M.; Terano, Y.; Scott, D.W.; Saper, C.B. Distribution and characterization of tumor necrosis factor-α-like immunoreactivity in the murine central nervous system. J. Comp. Neurol. 1993, 337, 543–567. [Google Scholar] [CrossRef] [PubMed]
- Lucin, K.M.; Wyss-Coray, T. Immune activation in brain aging and neurodegeneration: Too much or too little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Rice, T.; Larsen, J.; Rivest, S.; Yong, V.W. Characterization of the early neuroinflammation after spinal cord injury in mice. J. Neuropathol. Exp. Neurol. 2007, 66, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Garcia-Castillo, D.; Aguirre, V.; Golshani, R.; Atkins, C.M.; Bramlett, H.M.; Dietrich, W.D.; Pearse, D.D. Proinflammatory cytokine regulation of cyclic AMP-phosphodiesterase 4 signaling in microglia in vitro and following CNS injury. Glia 2012, 60, 1839–1859. [Google Scholar] [CrossRef] [PubMed]
- Vitarbo, E.A.; Chatzipanteli, K.; Kinoshita, K.; Truettner, J.S.; Alonso, O.F.; Dietrich, W.D. Tumor necrosis factor α expression and protein levels after fluid percussion injury in rats: The effect of injury severity and brain temperature. Neurosurgery 2004, 55, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Shohami, E.; Bass, R.; Wallach, D.; Yamin, A.; Gallily, R. Inhibition of tumor necrosis factor α (TNFα) activity in rat brain is associated with cerebroprotection after closed head injury. J. Cereb. Blood Flow Metab. 1996, 16, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Young, P.R.; Barone, F.C.; Feuerstein, G.Z.; Smith, D.H.; McIntosh, T.K. Experimental brain injury induces differential expression of tumor necrosis factor-α mRNA in the CNS. Brain Res. Mol. Brain Res. 1996, 36, 287–291. [Google Scholar] [CrossRef]
- Taupin, V.; Toulmond, S.; Serrano, A.; Benavides, J.; Zavala, F. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J. Neuroimmunol. 1993, 42, 177–185. [Google Scholar] [CrossRef]
- Kinoshita, K.; Chatzipanteli, K.; Vitarbo, E.; Truettner, J.S.; Alonso, O.F.; Dietrich, W.D. Interleukin-1β messenger ribonucleic acid and protein levels after fluid-percussion brain injury in rats: Importance of injury severity and brain temperature. Neurosurgery 2002, 51, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Katayama, Y.; Kawamata, T.; Tsubokawa, T. Excitatory amino acid release from contused brain tissue into surrounding brain areas. In Brain Edema IX; Springer Nature: Berlin, Germany, 1994; pp. 524–527. [Google Scholar]
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L., Jr.; Baron, P.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature 1995, 374, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.L.; Hicks, R.R.; Mahesh, J.; Billings, B.B.; Kotwall, G.J. Local neutrophil influx following lateral fluid-percussion brain injury in rats is associated with accumulation of complement activation fragments of the third component (C3) of the complement system. J. Neuroimmunol. 2000, 105, 20–30. [Google Scholar] [CrossRef]
- Hu, S.; Peterson, P.K.; Chao, C.C. Cytokine-mediated neuronal apoptosis. Neurochem. Int. 1997, 30, 427–431. [Google Scholar] [CrossRef]
- Hicks, R.; Soares, H.; Smith, D.; McIntosh, T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996, 91, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Tymianski, M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol. Neurobiol. 2001, 24, 107–129. [Google Scholar] [CrossRef]
- Ghosh, M.; Aguirre, V.; Wai, K.; Felfly, H.; Dietrich, W.D.; Pearse, D.D. The interplay between cyclic AMP, MAPK, and NF-κb pathways in response to proinflammatory signals in microglia. BioMed Res. Int. 2015, 2015, 308461. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From monocytes to M1/M2 macrophages: Phenotypical vs. Functional differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, 1619, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2A phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflamm. 2016, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.W.; Williams, A.J.; Liao, Z.; Yao, C.; Tortella, F.C.; Dave, J.R. Broad spectrum neuroprotection profile of phosphodiesterase inhibitors as related to modulation of cell-cycle elements and caspase-3 activation. Neurosci. Lett. 2007, 418, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.R.; Usechak, P.; Anwer, M.S. cAMP inhibits bile acid-induced apoptosis by blocking caspase activation and cytochrome c release. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G727–G738. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K.; Karmakar, S.; Nowak, M.W.; Banik, N.L. Inhibition of calpain and caspase-3 prevented apoptosis and preserved electrophysiological properties of voltage-gated and ligand-gated ion channels in rat primary cortical neurons exposed to glutamate. Neuroscience 2006, 139, 577–595. [Google Scholar] [CrossRef] [PubMed]
- Mckerracher, L.; David, S.; Jackson, D.L.; Kottis, V.; Dunn, R.J.; Braun, P.E. Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 1994, 13, 805–811. [Google Scholar] [CrossRef]
- Kottis, V.; Thibault, P.; Mikol, D.; Xiao, Z.C.; Zhang, R.; Dergham, P.; Braun, P.E. Oligodendrocyte-myelin glycoprotein (OMGP) is an inhibitor of neurite outgrowth. J. Neurochem. 2002, 82, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- GrandPre, T.; Nakamura, F.; Vartanian, T.; Strittmatter, S.M. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 2000, 403, 439–444. [Google Scholar] [PubMed]
- Chen, M.S.; Huber, A.B.; van der Haar, M.E.; Frank, M.; Schnell, L.; Spillmann, A.A.; Christ, F.; Schwab, M.E. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody in-1. Nature 2000, 403, 434–439. [Google Scholar] [PubMed]
- Wang, K.C.; Kim, J.A.; Sivasankaran, R.; Segal, R.; He, Z. P75 interacts with the nogo receptor as a co-receptor for Nogo, MAG and OMGP. Nature 2002, 420, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Prinjha, R.; Moore, S.E.; Vinson, M.; Blake, S.; Morrow, R.; Christie, G.; Michlovich, D.; Simmons, D.L.; Walsh, F.S. Neurobiology—Inhibitor of neurite outgrowth in humans. Nature 2000, 403, 383–384. [Google Scholar] [PubMed]
- Fournier, A.E.; GrandPre, T.; Strittmatter, S.M. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 2001, 409, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Raiker, S.J.; Venkatesh, K.; Geary, R.; Robak, L.A.; Zhang, Y.; Yeh, H.H.; Shrager, P.; Giger, R.J. Synaptic function for the Nogo-66 receptor NgR1: Regulation of dendritic spine morphology and activity-dependent synaptic strength. J. Neurosci. 2008, 28, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Hayashi, A.; Baba, J.; Sawa, A. Rolipram, a phosphodiesterase-4-selective inhibitor, promotes the survival of cultured rat dopaminergic neurons. Jpn. J. Pharmacol. 1997, 75, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Forgione, N.; Fehlings, M.G. Rho-ROCK inhibition in the treatment of spinal cord injury. World Neurosurg. 2014, 82, e535–e539. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Fournier, A.; Selles-Navarro, I.; Dergham, P.; Sebok, A.; Leclerc, N.; Tigyi, G.; McKerracher, L. Inactivation of Rho signaling pathway promotes CNS axon regeneration. J. Neurosci. 1999, 19, 7537–7547. [Google Scholar] [PubMed]
- Lang, P.; Gesbert, F.; Delespine-Carmagnat, M.; Stancou, R.; Pouchelet, M.; Bertoglio, J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996, 15, 510–519. [Google Scholar] [PubMed]
- Dong, J.M.; Leung, T.; Manser, E.; Lim, L. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase Rokα. J. Biol. Chem. 1998, 273, 22554–22562. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.; Filbin, M.T. A role for cAMP in regeneration of the adult mammalian CNS. J. Anat. 2004, 204, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Irisarri, E.; Markerink-Van Ittersum, M.; Mengod, G.; de Vente, J. Expression of the cGMP-specific phosphodiesterases 2 and 9 in normal and Alzheimer’s disease human brains. Eur. J. Neurosci. 2007, 25, 3332–3338. [Google Scholar] [CrossRef] [PubMed]
- Domek-Lopacinska, K.U.; Strosznajder, J.B. Cyclic GMP and nitric oxide synthase in aging and Alzheimer’s disease. Mol. Neurobiol. 2010, 41, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Heckman, P.R.; Wouters, C.; Prickaerts, J. Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer’s disease: A translational overview. Curr. Pharm. Des. 2015, 21, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, Z.; Pan, J.; Zhang, H.-T.; O’Donnell, J.M. Memory enhancement induced by PDE2 knockdown in an Alzheimer’s disease model of mice. FASEB J. 2016, 30, 707.6. [Google Scholar]
- Ugarte, A.; Gil-Bea, F.; Garcia-Barroso, C.; Cedazo-Minguez, A.; Ramirez, M.J.; Franco, R.; Garcia-Osta, A.; Oyarzabal, J.; Cuadrado-Tejedor, M. Decreased levels of guanosine 3′,5′-monophosphate (cGMP) in cerebrospinal fluid (CSF) are associated with cognitive decline and amyloid pathology in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2015, 41, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Perez-Torres, S.; Cortes, R.; Tolnay, M.; Probst, A.; Palacios, J.M.; Mengod, G. Alterations on phosphodiesterase type 7 and 8 isozyme mRNA expression in Alzheimer’s disease brains examined by in situ hybridization. Exp. Neurol. 2003, 182, 322–334. [Google Scholar] [CrossRef]
- McLachlan, C.S.; Chen, M.L.; Lynex, C.N.; Goh, D.L.; Brenner, S.; Tay, S.K. Changes in PDE4D isoforms in the hippocampus of a patient with advanced Alzheimer disease. Arch. Neurol. 2007, 64, 456–457. [Google Scholar] [CrossRef] [PubMed]
- Foroud, T.; Siemers, E.; Kleindorfer, D.; Bill, D.J.; Hodes, M.E.; Norton, J.A.; Conneally, P.M.; Christian, J.C. Cognitive scores in carriers of Huntington’s disease gene compared to noncarriers. Ann. Neurol. 1995, 37, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, A.D.; Sahakian, B.J.; Hodges, J.R.; Rosser, A.E.; Lange, K.W.; Robbins, T.W. Executive and mnemonic functions in early Huntington’s disease. Brain 1996, 119 Pt 5, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Lemiere, J.; Decruyenaere, M.; Evers-Kiebooms, G.; Vandenbussche, E.; Dom, R. Cognitive changes in patients with Huntington’s disease (HD) and asymptomatic carriers of the HD mutation—A longitudinal follow-up study. J. Neurol. 2004, 251, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Lakics, V.; Karran, E.H.; Boess, F.G. Corrigendum to “quantitative comparison of phosphodiesterase mrna distribution in human brain and peripheral tissues”. Neuropharmacology 2013, 67, 532. [Google Scholar] [CrossRef]
- Xie, Z.; Adamowicz, W.O.; Eldred, W.D.; Jakowski, A.B.; Kleiman, R.J.; Morton, D.G.; Stephenson, D.T.; Strick, C.A.; Williams, R.D.; Menniti, F.S. Cellular and subcellular localization of PDE10A, a striatum-enriched phosphodiesterase. Neuroscience 2006, 139, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Hebb, A.L.; Robertson, H.A.; Denovan-Wright, E.M. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington’s disease transgenic mice prior to the onset of motor symptoms. Neuroscience 2004, 123, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Saavedra, A.; Carreton, O.; Xifro, X.; Alberch, J.; Perez-Navarro, E. Increased PKA signaling disrupts recognition memory and spatial memory: Role in Huntington’s disease. Hum. Mol. Genet. 2011, 20, 4232–4247. [Google Scholar] [CrossRef] [PubMed]
- Polli, J.W.; Kincaid, R.L. Expression of a calmodulin-dependent phosphodiesterase isoform (PDE1B1) correlates with brain regions having extensive dopaminergic innervation. J. Neurosci. 1994, 14, 1251–1261. [Google Scholar] [PubMed]
- Nishi, A.; Snyder, G.L. Advanced research on dopamine signaling to develop drugs for the treatment of mental disorders: Biochemical and behavioral profiles of phosphodiesterase inhibition in dopaminergic neurotransmission. J. Pharmacol. Sci. 2010, 114, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Boldrini, M.; Underwood, M.D.; Hen, R.; Rosoklija, G.B.; Dwork, A.J.; John Mann, J.; Arango, V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009, 34, 2376–2389. [Google Scholar] [CrossRef] [PubMed]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.H.; Miller, H.L.; Charney, D.S. Hippocampal volume reduction in major depression. Am. J. Psychiatry 2000, 157, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Sheline, Y.I.; Gado, M.H.; Kraemer, H.C. Untreated depression and hippocampal volume loss. Am. J. Psychiatry 2003, 160, 1516–1518. [Google Scholar] [CrossRef] [PubMed]
- Sheline, Y.I.; Wang, P.W.; Gado, M.H.; Csernansky, J.G.; Vannier, M.W. Hippocampal atrophy in recurrent major depression. Proc. Natl. Acad. Sci. USA 1996, 93, 3908–3913. [Google Scholar] [CrossRef] [PubMed]
- Tsopelas, C.; Stewart, R.; Savva, G.M.; Brayne, C.; Ince, P.; Thomas, A.; Matthews, F.E.; Medical Research Council Cognitive Function; Ageing Study. Neuropathological correlates of late-life depression in older people. Br. J. Psychiatry 2011, 198, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, R.F.; Marcusson, J.O.; Eriksson, A.; Wiehager, B.; O’Neill, C. Adenylyl cyclase activity and G-protein subunit levels in postmortem frontal cortex of suicide victims. Brain Res. 1994, 633, 297–304. [Google Scholar] [CrossRef]
- Dowlatshahi, D.; MacQueen, G.M.; Wang, J.F.; Young, L.T. Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet 1998, 352, 1754–1755. [Google Scholar] [PubMed]
- Dwivedi, Y.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. [(3)h]cAMP binding sites and protein kinase a activity in the prefrontal cortex of suicide victims. Am. J. Psychiatry 2002, 159, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Reiach, J.S.; Li, P.P.; Warsh, J.J.; Kish, S.J.; Young, L.T. Reduced adenylyl cyclase immunolabeling and activity in postmortem temporal cortex of depressed suicide victims. J. Affect. Disord. 1999, 56, 141–151. [Google Scholar] [CrossRef]
- Fujita, M.; Hines, C.S.; Zoghbi, S.S.; Mallinger, A.G.; Dickstein, L.P.; Liow, J.S.; Zhang, Y.; Pike, V.W.; Drevets, W.C.; Innis, R.B.; et al. Downregulation of brain phosphodiesterase type IV measured with 11C-(R)-rolipram positron emission tomography in major depressive disorder. Biol. Psychiatry 2012, 72, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.R.; Wu, G.S.; Dong, C.; Arcos-Burgos, M.; Ribeiro, L.; Licinio, J.; Wong, M.L. Association of PDE11A global haplotype with major depression and antidepressant drug response. Neuropsychiatr. Dis. Treat. 2009, 5, 163–170. [Google Scholar] [PubMed]
- Barad, M.; Bourtchouladze, R.; Winder, D.G.; Golan, H.; Kandel, E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc. Natl. Acad. Sci. USA 1998, 95, 15020–15025. [Google Scholar] [CrossRef] [PubMed]
- Rutten, K.; Prickaerts, J.; Blokland, A. Rolipram reverses scopolamine-induced and time-dependent memory deficits in object recognition by different mechanisms of action. Neurobiol. Learn. Mem. 2006, 85, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Ramos, B.P.; Birnbaum, S.G.; Lindenmayer, I.; Newton, S.S.; Duman, R.S.; Arnsten, A.F. Dysregulation of protein kinase A signaling in the aged prefrontal cortex: New strategy for treating age-related cognitive decline. Neuron 2003, 40, 835–845. [Google Scholar] [CrossRef]
- Gomez, C.D.; Buijs, R.M.; Sitges, M. The anti-seizure drugs vinpocetine and carbamazepine, but not valproic acid, reduce inflammatory IL-1β and TNF-α expression in rat hippocampus. J. Neurochem. 2014, 130, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Molnar, P.; Gaal, L. Effect of different subtypes of cognition enhancers on long-term potentiation in the rat dentate gyrus in vivo. Eur. J. Pharmacol. 1992, 215, 17–22. [Google Scholar] [CrossRef]
- DeNoble, V.J. Vinpocetine enhances retrieval of a step-through passive avoidance response in rats. Pharmacol. Biochem. Behav. 1987, 26, 183–186. [Google Scholar] [CrossRef]
- Deshmukh, R.; Sharma, V.; Mehan, S.; Sharma, N.; Bedi, K.L. Amelioration of intracerebroventricular streptozotocin induced cognitive dysfunction and oxidative stress by vinpocetine—A PDE1 inhibitor. Eur. J. Pharmacol. 2009, 620, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hindmarch, I.; Fuchs, H.H.; Erzigkeit, H. Efficacy and tolerance of vinpocetine in ambulant patients suffering from mild to moderate organic psychosyndromes. Int. Clin. Psychopharmacol. 1991, 6, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Zaitone, S.A.; Abo-Elmatty, D.M.; Elshazly, S.M. Piracetam and vinpocetine ameliorate rotenone-induced parkinsonism in rats. Indian J. Pharmacol. 2012, 44, 774–779. [Google Scholar] [PubMed]
- Ratra, M.; Sharma, P.; Gupta, R. Neuroprotective effect of vinpocetine against 3-NP induced reduction of body weight and oxidative stress in rats. Int. J. Phytomed. 2012, 3, 8. [Google Scholar]
- Nunes, F.; Ferreira-Rosa, K.; Pereira Mdos, S.; Kubrusly, R.C.; Manhaes, A.C.; Abreu-Villaca, Y.; Filgueiras, C.C. Acute administration of vinpocetine, a phosphodiesterase type 1 inhibitor, ameliorates hyperactivity in a mice model of fetal alcohol spectrum disorder. Drug Alcohol. Depend. 2011, 119, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Laddha, S.S.; Bhatnagar, S.P. A new therapeutic approach in Parkinson’s disease: Some novel quinazoline derivatives as dual selective phosphodiesterase 1 inhibitors and anti-inflammatory agents. Bioorg. Med. Chem. 2009, 17, 6796–6802. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, R.; Raju, R.V.; Rajput, A.H.; Sharma, R.K. Inhibition of bovine brain calmodulin-dependent cyclic nucleotide phosphodiesterase isozymes by deprenyl. Life Sci. 1996, 59, PL337–PL341. [Google Scholar] [CrossRef]
- Domek-Lopacinska, K.; Strosznajder, J.B. The effect of selective inhibition of cyclic GMP hydrolyzing phosphodiesterases 2 and 5 on learning and memory processes and nitric oxide synthase activity in brain during aging. Brain Res. 2008, 1216, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Terpolilli, N.A.; Kim, S.W.; Thal, S.C.; Kuebler, W.M.; Plesnila, N. Inhaled nitric oxide reduces secondary brain damage after traumatic brain injury in mice. J. Cereb. Blood Flow Metab. 2013, 33, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Terpolilli, N.A.; Feiler, S.; Dienel, A.; Muller, F.; Heumos, N.; Friedrich, B.; Stover, J.; Thal, S.; Scholler, K.; Plesnila, N. Nitric oxide inhalation reduces brain damage, prevents mortality, and improves neurological outcome after subarachnoid hemorrhage by resolving early pial microvasospasms. J. Cereb. Blood Flow Metab. 2016, 36, 2096–2107. [Google Scholar] [CrossRef] [PubMed]
- Terpolilli, N.A.; Kim, S.W.; Thal, S.C.; Kataoka, H.; Zeisig, V.; Nitzsche, B.; Klaesner, B.; Zhu, C.; Schwarzmaier, S.; Meissner, L.; et al. Inhalation of nitric oxide prevents ischemic brain damage in experimental stroke by selective dilatation of collateral arterioles. Circ. Res. 2012, 110, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Terpolilli, N.A.; Moskowitz, M.A.; Plesnila, N. Nitric oxide: Considerations for the treatment of ischemic stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Reneerkens, O.A.; Rutten, K.; Bollen, E.; Hage, T.; Blokland, A.; Steinbusch, H.W.; Prickaerts, J. Inhibition of phoshodiesterase type 2 or type 10 reverses object memory deficits induced by scopolamine or MK-801. Behav. Brain Res. 2013, 236, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.S.; Rutten, K.; Sydlik, S.; Rostamian, S.; Steinbusch, H.W.; van den Hove, D.L.; Prickaerts, J. Chronic phosphodiesterase type 2 inhibition improves memory in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neuropharmacology 2013, 64, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Tanaka, M.; Hasebe, S.; Yamaguchi, T.; Shiba, T.; Ogita, K. Beneficial effect of cilostazol-mediated neuronal repair following trimethyltin-induced neuronal loss in the dentate gyrus. J. Neurosci. Res. 2015, 93, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Yanai, S.; Semba, Y.; Ito, H.; Endo, S. Cilostazol improves hippocampus-dependent long-term memory in mice. Psychopharmacology 2014, 231, 2681–2693. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, M.; Takiguchi, O.; Nishiyama, A.; Mori, H. Cilostazol prevents amyloid β peptide(25–35)-induced memory impairment and oxidative stress in mice. Br. J. Pharmacol. 2010, 161, 1899–1912. [Google Scholar] [CrossRef] [PubMed]
- Schaal, S.M.; Garg, M.S.; Ghosh, M.; Lovera, L.; Lopez, M.; Patel, M.; Louro, J.; Patel, S.; Tuesta, L.; Chan, W.M.; et al. The therapeutic profile of rolipram, PDE target and mechanism of action as a neuroprotectant following spinal cord injury. PLoS ONE 2012, 7, e43634. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.M.; Pereira, J.E.; Filipe, V.M.; Magalhaes, L.G.; Couto, P.A.; Gonzalo-Orden, J.M.; Raimondo, S.; Geuna, S.; Mauricio, A.C.; Nikulina, E.; et al. Rolipram promotes functional recovery after contusive thoracic spinal cord injury in rats. Behav. Brain Res. 2013, 243, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, C.M.; Beaumont, E.; Wells, M.J.; Magnuson, D.S.; Hetman, M.; Onifer, S.M. Rolipram attenuates acute oligodendrocyte death in the adult rat ventrolateral funiculus following contusive cervical spinal cord injury. Neurosci. Lett. 2008, 438, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Bao, F.; Fleming, J.C.; Golshani, R.; Pearse, D.D.; Kasabov, L.; Brown, A.; Weaver, L.C. A selective phosphodiesterase-4 inhibitor reduces leukocyte infiltration, oxidative processes, and tissue damage after spinal cord injury. J. Neurotrauma 2011, 28, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Flora, G.; Joseph, G.; Patel, S.; Singh, A.; Bleicher, D.; Barakat, D.J.; Louro, J.; Fenton, S.; Garg, M.; Bunge, M.B.; et al. Combining neurotrophin-transduced schwann cells and rolipram to promote functional recovery from subacute spinal cord injury. Cell Transpl. 2013, 22, 2203–2217. [Google Scholar] [CrossRef] [PubMed]
- Jindal, A.; Mahesh, R.; Bhatt, S. Etazolate, a phosphodiesterase-4 enzyme inhibitor produces antidepressant-like effects by blocking the behavioral, biochemical, neurobiological deficits and histological abnormalities in hippocampus region caused by olfactory bulbectomy. Psychopharmacology 2015, 232, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Bruno, O.; Fedele, E.; Prickaerts, J.; Parker, L.A.; Canepa, E.; Brullo, C.; Cavallero, A.; Gardella, E.; Balbi, A.; Domenicotti, C.; et al. GEBR-7B, a novel PDE4D selective inhibitor that improves memory in rodents at non-emetic doses. Br. J. Pharmacol. 2011, 164, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.S.; van den Hove, D.L.; Pfau, F.; Philippens, M.; Bruno, O.; Fedele, E.; Ricciarelli, R.; Steinbusch, H.W.; Vanmierlo, T.; Prickaerts, J. Improvement of spatial memory function in APPswe/PS1dE9 mice after chronic inhibition of phosphodiesterase type 4D. Neuropharmacology 2014, 77, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Rutter, A.R.; Poffe, A.; Cavallini, P.; Davis, T.G.; Schneck, J.; Negri, M.; Vicentini, E.; Montanari, D.; Arban, R.; Gray, F.A.; et al. GSK356278, a potent, selective, brain-penetrant phosphodiesterase 4 inhibitor that demonstrates anxiolytic and cognition-enhancing effects without inducing side effects in preclinical species. J. Pharmacol. Exp. Ther. 2014, 350, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Gallant, M.; Aspiotis, R.; Day, S.; Dias, R.; Dube, D.; Dube, L.; Friesen, R.W.; Girard, M.; Guay, D.; Hamel, P.; et al. Discovery of MK-0952, a selective PDE4 inhibitor for the treatment of long-term memory loss and mild cognitive impairment. Bioorg. Med. Chem. Lett. 2010, 20, 6387–6393. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Dias, R.; Jones, T.; Liu, S.; Styhler, A.; Claveau, D.; Otu, F.; Ng, K.; Laliberte, F.; Zhang, L.; et al. L-454,560, a potent and selective PDE4 inhibitor with in vivo efficacy in animal models of asthma and cognition. Biochem. Pharmacol. 2007, 73, 1971–1981. [Google Scholar] [CrossRef] [PubMed]
- Rutten, K.; Misner, D.L.; Works, M.; Blokland, A.; Novak, T.J.; Santarelli, L.; Wallace, T.L. Enhanced long-term potentiation and impaired learning in phosphodiesterase 4D-knockout (PDE4D) mice. Eur. J. Neurosci. 2008, 28, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Sawa, A.; Ichimaru, Y.; Miyashiro, M.; Kato, S.; Yamamoto, T.; Ueki, S. Ameliorating effects of rolipram on experimentally induced impairments of learning and memory in rodents. Eur. J. Pharmacol. 1997, 321, 273–278. [Google Scholar] [CrossRef]
- Hulley, P.; Hartikka, J.; Abdel’Al, S.; Engels, P.; Buerki, H.-R.; Wiederhold, K.-H.; Müller, T.; Kelly, P.; Lowe, D.; Lübbert, H. Inhibitors of type IV phosphodiesterases reduce the toxicity of MPTP in substantia nigra neuronsin vivo. Eur. J. Neurosci. 1995, 7, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Calingasan, N.Y.; Lorenzo, B.J.; Beal, M.F. Attenuation of mptp neurotoxicity by rolipram, a specific inhibitor of phosphodiesterase IV. Exp. Neurol. 2008, 211, 311–314. [Google Scholar] [CrossRef] [PubMed]
- DeMarch, Z.; Giampa, C.; Patassini, S.; Bernardi, G.; Fusco, F.R. Beneficial effects of rolipram in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2008, 30, 375–387. [Google Scholar] [CrossRef] [PubMed]
- DeMarch, Z.; Giampa, C.; Patassini, S.; Martorana, A.; Bernardi, G.; Fusco, F.R. Beneficial effects of rolipram in a quinolinic acid model of striatal excitotoxicity. Neurobiol. Dis. 2007, 25, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Giampa, C.; Middei, S.; Patassini, S.; Borreca, A.; Marullo, F.; Laurenti, D.; Bernardi, G.; Ammassari-Teule, M.; Fusco, F.R. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington’s disease. Eur. J. Neurosci. 2009, 29, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Hashimoto, K.; Inada, T.; Fukui, S.; Iyo, M. Suppression of oro-facial movements by rolipram, a cAMP phosphodiesterase inhibitor, in rats chronically treated with haloperidol. Eur. J. Pharmacol. 1995, 282, 71–76. [Google Scholar] [CrossRef]
- MacDonald, E.; van der Lee, H.; Pocock, D.; Cole, C.; Thomas, N.; VandenBerg, P.M.; Bourtchouladze, R.; Kleim, J.A. A novel phosphodiesterase type 4 inhibitor, HT-0712, enhances rehabilitation-dependent motor recovery and cortical reorganization after focal cortical ischemia. Neurorehabil. Neural Repair 2007, 21, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Dart-Neuroscience. Clinical Trials. Available online: http://www.dartneuroscience.com/ClinicalTrials.php (accessed on 22 March 2017).
- Atkins, C.M.; Cepero, M.L.; Kang, Y.; Liebl, D.J.; Dietrich, W.D. Effects of early rolipram treatment on histopathological outcome after controlled cortical impact injury in mice. Neurosci. Lett. 2013, 532, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Staniszewski, A.; Deng, S.X.; Privitera, L.; Leznik, E.; Liu, S.; Zhang, H.; Feng, Y.; Palmeri, A.; Landry, D.W.; et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-β load in an Alzheimer’s disease mouse model. J. Neurosci. 2009, 29, 8075–8086. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Hervias, I.; Ricobaraza, A.; Puerta, E.; Perez-Roldan, J.M.; Garcia-Barroso, C.; Franco, R.; Aguirre, N.; Garcia-Osta, A. Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 2029–2041. [Google Scholar] [CrossRef] [PubMed]
- Devan, B.D.; Sierra-Mercado, D., Jr.; Jimenez, M.; Bowker, J.L.; Duffy, K.B.; Spangler, E.L.; Ingram, D.K. Phosphodiesterase inhibition by sildenafil citrate attenuates the learning impairment induced by blockade of cholinergic muscarinic receptors in rats. Pharmacol. Biochem. Behav. 2004, 79, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Devan, B.D.; Pistell, P.J.; Duffy, K.B.; Kelley-Bell, B.; Spangler, E.L.; Ingram, D.K. Phosphodiesterase inhibition facilitates cognitive restoration in rodent models of age-related memory decline. NeuroRehabilitation 2014, 34, 101–111. [Google Scholar] [PubMed]
- Charriaut-Marlangue, C.; Nguyen, T.; Bonnin, P.; Duy, A.P.; Leger, P.L.; Csaba, Z.; Pansiot, J.; Bourgeois, T.; Renolleau, S.; Baud, O. Sildenafil mediates blood-flow redistribution and neuroprotection after neonatal hypoxia-ischemia. Stroke 2014, 45, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Charriaut-Marlangue, C.; Bonnin, P.; Gharib, A.; Leger, P.L.; Villapol, S.; Pocard, M.; Gressens, P.; Renolleau, S.; Baud, O. Inhaled nitric oxide reduces brain damage by collateral recruitment in a neonatal stroke model. Stroke 2012, 43, 3078–3084. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.; Vottier, G.; Pansiot, J.; Duong-Quy, S.; Bollen, B.; Dalous, J.; Gallego, J.; Mercier, J.C.; Dinh-Xuan, A.T.; Bonnin, P.; et al. Inhaled no prevents hyperoxia-induced white matter damage in neonatal rats. Exp. Neurol. 2014, 252, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.; Duy, A.P.; Pansiot, J.; Bollen, B.; Gallego, J.; Charriaut-Marlangue, C.; Baud, O. Impact of inhaled nitric oxide on white matter damage in growth-restricted neonatal rats. Pediatr. Res. 2015, 77, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Phan Duy, A.; Pham, H.; Pansiot, J.; Gressens, P.; Charriaut-Marlangue, C.; Baud, O. Nitric oxide pathway and proliferation of neural progenitors in the neonatal rat. Dev. Neurosci. 2015, 37, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, A.; Giralt, A.; Arumi, H.; Alberch, J.; Perez-Navarro, E. Regulation of hippocampal cGMP levels as a candidate to treat cognitive deficits in Huntington’s disease. PLoS ONE 2013, 8, e73664. [Google Scholar] [CrossRef] [PubMed]
- Puerta, E.; Hervias, I.; Barros-Minones, L.; Jordan, J.; Ricobaraza, A.; Cuadrado-Tejedor, M.; Garcia-Osta, A.; Aguirre, N. Sildenafil protects against 3-nitropropionic acid neurotoxicity through the modulation of calpain, CREB, and BDNF. Neurobiol. Dis. 2010, 38, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Barroso, C.; Ricobaraza, A.; Pascual-Lucas, M.; Unceta, N.; Rico, A.J.; Goicolea, M.A.; Salles, J.; Lanciego, J.L.; Oyarzabal, J.; Franco, R.; et al. Tadalafil crosses the blood–brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology 2013, 64, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Picconi, B.; Bagetta, V.; Ghiglieri, V.; Paille, V.; di Filippo, M.; Pendolino, V.; Tozzi, A.; Giampa, C.; Fusco, F.R.; Sgobio, C.; et al. Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain 2011, 134, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Mazzon, E.; Gil, C.; Impellizzeri, D.; Palomo, V.; Redondo, M.; Perez, D.I.; Esposito, E.; Martinez, A.; Cuzzocrea, S. Pde 7 inhibitors: New potential drugs for the therapy of spinal cord injury. PLoS ONE 2011, 6, e15937. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rodriguez, E.M.; Arenzana, F.J.; Pastor, J.; Redondo, M.; Palomo, V.; Garcia de Sola, R.; Gil, C.; Martinez, A.; Bribian, A.; de Castro, F. Inhibition of endogenous phosphodiesterase 7 promotes oligodendrocyte precursor differentiation and survival. Cell. Mol. Life Sci. 2013, 70, 3449–3462. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garcia, C.; Bravo, B.; Ballester, A.; Gomez-Perez, R.; Eguiluz, C.; Redondo, M.; Martinez, A.; Gil, C.; Ballester, S. Comparative assessment of PDE4 and 7 inhibitors as therapeutic agents in experimental autoimmune encephalomyelitis. Br. J. Pharmacol. 2013, 170, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Redondo, M.; Zarruk, J.G.; Ceballos, P.; Perez, D.I.; Perez, C.; Perez-Castillo, A.; Moro, M.A.; Brea, J.; Val, C.; Cadavid, M.I.; et al. Neuroprotective efficacy of quinazoline type phosphodiesterase 7 inhibitors in cellular cultures and experimental stroke model. Eur. J. Med. Chem. 2012, 47, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Morales-Garcia, J.A.; Redondo, M.; Alonso-Gil, S.; Gil, C.; Perez, C.; Martinez, A.; Santos, A.; Perez-Castillo, A. Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of parkinson disease. PLoS ONE 2011, 6, e17240. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gonzalez, R.; Pascual, C.; Antequera, D.; Bolos, M.; Redondo, M.; Perez, D.I.; Perez-Grijalba, V.; Krzyzanowska, A.; Sarasa, M.; Gil, C.; et al. Phosphodiesterase 7 inhibitor reduced cognitive impairment and pathological hallmarks in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2133–2145. [Google Scholar] [CrossRef] [PubMed]
- Morales-Garcia, J.A.; Palomo, V.; Redondo, M.; Alonso-Gil, S.; Gil, C.; Martinez, A.; Perez-Castillo, A. Crosstalk between phosphodiesterase 7 and glycogen synthase kinase-3: Two relevant therapeutic targets for neurological disorders. ACS Chem. Neurosci. 2014, 5, 194–204. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Shulman, J.M.; Chibnik, L.B.; Keenan, B.T.; Raj, T.; Wilson, R.S.; Yu, L.; Leurgans, S.E.; Tran, D.; Aubin, C.; et al. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol. Aging 2012, 33, 1017e1–1017e15. [Google Scholar] [CrossRef] [PubMed]
- Hutson, P.H.; Finger, E.N.; Magliaro, B.C.; Smith, S.M.; Converso, A.; Sanderson, P.E.; Mullins, D.; Hyde, L.A.; Eschle, B.K.; Turnbull, Z.; et al. The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943 (6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one) enhances synaptic plasticity and cognitive function in rodents. Neuropharmacology 2011, 61, 665–676. [Google Scholar] [PubMed]
- Verhoest, P.R.; Fonseca, K.R.; Hou, X.; Proulx-Lafrance, C.; Corman, M.; Helal, C.J.; Claffey, M.M.; Tuttle, J.B.; Coffman, K.J.; Liu, S.; et al. Design and discovery of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (PF-04447943), a selective brain penetrant PDE9A inhibitor for the treatment of cognitive disorders. J. Med. Chem. 2012, 55, 9045–9054. [Google Scholar] [PubMed]
- Kroker, K.S.; Mathis, C.; Marti, A.; Cassel, J.C.; Rosenbrock, H.; Dorner-Ciossek, C. PDE9A inhibition rescues amyloid β-induced deficits in synaptic plasticity and cognition. Neurobiol. Aging 2014, 35, 2072–2078. [Google Scholar] [CrossRef] [PubMed]
- Threlfell, S.; Sammut, S.; Menniti, F.S.; Schmidt, C.J.; West, A.R. Inhibition of phosphodiesterase 10A increases the responsiveness of striatal projection neurons to cortical stimulation. J. Pharmacol. Exp. Ther. 2009, 328, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Threlfell, S.; West, A.R. Review: Modulation of striatal neuron activity by cyclic nucleotide signaling and phosphodiesterase inhibition. Basal Ganglia 2013, 3, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Chappie, T.; Humphrey, J.; Menniti, F.; Schmidt, C. PDE10A inhibitors: An assessment of the current cns drug discovery landscape. Curr. Opin. Drug Discov. Dev. 2009, 12, 458–467. [Google Scholar]
- Banerjee, A.; Narayana, L.; Raje, F.A.; Pisal, D.V.; Kadam, P.A.; Gullapalli, S.; Kumar, H.; More, S.V.; Bajpai, M.; Sangana, R.R.; et al. Discovery of benzo[d]imidazo[5,1-b]thiazole as a new class of phosphodiesterase 10a inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6747–6754. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Saavedra, A.; Carreton, O.; Arumi, H.; Tyebji, S.; Alberch, J.; Perez-Navarro, E. PDE10 inhibition increases glua1 and creb phosphorylation and improves spatial and recognition memories in a Huntington’s disease mouse model. Hippocampus 2013, 23, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Giampa, C.; Laurenti, D.; Anzilotti, S.; Bernardi, G.; Menniti, F.S.; Fusco, F.R. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS ONE 2010, 5, e13417. [Google Scholar] [CrossRef] [PubMed]
- Nishi, A.; Kuroiwa, M.; Miller, D.B.; O’Callaghan, J.P.; Bateup, H.S.; Shuto, T.; Sotogaku, N.; Fukuda, T.; Heintz, N.; Greengard, P.; et al. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J. Neurosci. 2008, 28, 10460–10471. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.X.; Hassell, A.M.; Vanderwall, D.; Lambert, M.H.; Holmes, W.D.; Luther, M.A.; Rocque, W.J.; Milburn, M.V.; Zhao, Y.D.; Ke, H.M.; et al. Atomic structure of PDE4: Insights into phosphodiesterase mechanism and specificity. Science 2000, 288, 1822–1825. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ye, M.; Robinson, H.; Francis, S.H.; Ke, H. Conformational variations of both phosphodiesterase-5 and inhibitors provide the structural basis for the physiological effects of vardenafil and sildenafil. Mol. Pharmacol. 2008, 73, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Feng, Q.; Cote, R.H. Efficacy and selectivity of phosphodiesterase-targeted drugs in inhibiting photoreceptor phosphodiesterase (PDE6) in retinal photoreceptors. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3060–3066. [Google Scholar] [CrossRef] [PubMed]
- Cote, R.H. Cyclic guanosine 5′-monophosphate binding to regulatory gaf domains of photoreceptor phosphodiesterase. Methods Mol Biol 2005, 307, 141–154. [Google Scholar] [PubMed]
- Lukowski, R.; Rybalkin, S.D.; Loga, F.; Leiss, V.; Beavo, J.A.; Hofmann, F. Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase i in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 5646–5651. [Google Scholar] [CrossRef] [PubMed]
- Bazhin, A.V.; Tambor, V.; Dikov, B.; Philippov, P.P.; Schadendorf, D.; Eichmuller, S.B. cGMP-phosphodiesterase 6, transducin and Wnt5a/Frizzled-2-signaling control cGMP and Ca(2+) homeostasis in melanoma cells. Cell. Mol. Life Sci. 2010, 67, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Morris, G.Z.; Corbin, J.D. Molecular mechanisms that could contribute to prolonged effectiveness of pDE5 inhibitors to improve erectile function. Int. J. Impot. Res. 2008, 20, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Heaslip, R.J.; Evans, D.Y. Emetic, central nervous system, and pulmonary activities of rolipram in the dog. Eur. J. Pharmacol. 1995, 286, 281–290. [Google Scholar] [CrossRef]
- Pearse, D.D.; Hughes, Z.A. PDE4B as a microglia target to reduce neuroinflammation. Glia 2016, 64, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br. J. Pharmacol. 2011, 163, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Robichaud, A.; Tattersall, F.D.; Choudhury, I.; Rodger, I.W. Emesis induced by inhibitors of type IV cyclic nucleotide phosphodiesterase (PDE IV) in the ferret. Neuropharmacology 1999, 38, 289–297. [Google Scholar] [CrossRef]
- Degerman, E.; In’t Zandt, R.; Palbrink, A.; Eliasson, L.; Caye-Thomasen, P.; Magnusson, M. Inhibition of phosphodiesterase 3, 4, and 5 induces endolymphatic hydrops in mouse inner ear, as evaluated with repeated 9.4T MRI. Acta Oto-Laryngol. 2017, 137, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Baye, J. Roflumilast (Daliresp): A novel phosphodiesterase-4 inhibitor for the treatment of severe chronic obstructive pulmonary disease. P T. 2012, 37, 149–161. [Google Scholar] [PubMed]
- Zhang, M.Z.; Zhou, Z.Z.; Yuan, X.; Cheng, Y.F.; Bi, B.T.; Gong, M.F.; Chen, Y.P.; Xu, J.P. Chlorbipram: A novel PDE4 inhibitor with improved safety as a potential antidepressant and cognitive enhancer. Eur. J. Pharmacol. 2013, 721, 56–63. [Google Scholar] [CrossRef] [PubMed]
- GlaxoSmithKline. An Open Label Positron Emission Tomography Study in Healthy Male Subjects to Investigate Brain PDE4 Engagement, Pharmacokinetics and Safety of Single Oral Doses of GSK356278, Using 11C-(R)-Rolipram as a PET Ligand(s). Available online: https://www.gsk-clinicalstudyregister.com/study/116038 (accessed on 22 March 2017).
- GlaxoSmithKline. A Randomised, Placebo Controlled, Ascending, Repeat Dose Study in Healthy Volunteers Investigating Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of GSK356278. Available online: https://www.gsk-clinicalstudyregister.com/study/115719 (accessed on 22 March 2017).
- Robichaud, A.; Stamatiou, P.B.; Jin, S.L.; Lachance, N.; MacDonald, D.; Laliberte, F.; Liu, S.; Huang, Z.; Conti, M.; Chan, C.C. Deletion of phosphodiesterase 4D in mice shortens α(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J. Clin. Investig. 2002, 110, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; D’Amato, E.C.; Burgin, A.B. Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer’s disease. Neurotherapeutics 2015, 12, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Partners, T.D. A Multiple Ascending Dose Study of BPN14770 in Healthy Young and Elderly Male or Female Subjects. Available online: https://clinicaltrials.gov/ct2/show/NCT02840279 (accessed on 22 March 2017).
- Guariento, S.; Bruno, O.; Fossa, P.; Cichero, E. New insights into PDE4B inhibitor selectivity: COMFA analyses and molecular docking studies. Mol. Divers. 2016, 20, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Sluka, K.A. Activation of the cAMP transduction cascade contributes to the mechanical hyperalgesia and allodynia induced by intradermal injection of capsaicin. Br. J. Pharmacol. 1997, 122, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Song, X.S.; Cao, J.L.; Xu, Y.B.; He, J.H.; Zhang, L.C.; Zeng, Y.M. Activation of ERK/CREB pathway in spinal cord contributes to chronic constrictive injury-induced neuropathic pain in rats. Acta Pharmacol. Sin. 2005, 26, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Hatzis, C.; Eisenach, J.C. Intrathecal injection of cAMP response element binding protein (CREB) antisense oligonucleotide attenuates tactile allodynia caused by partial sciatic nerve ligation. Brain Res. 2003, 988, 97–104. [Google Scholar] [CrossRef]
- Moalem-Taylor, G.; Li, M.; Allbutt, H.N.; Wu, A.; Tracey, D.J. A preconditioning nerve lesion inhibits mechanical pain hypersensitivity following subsequent neuropathic injury. Mol. Pain 2011, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Mancini, J.A. Phosphodiesterase 4 inhibitors for the treatment of asthma and COPD. Curr. Med. Chem. 2006, 13, 3253–3262. [Google Scholar] [CrossRef] [PubMed]
- Giembycz, M.A. An update and appraisal of the cilomilast phase III clinical development programme for chronic obstructive pulmonary disease. Br. J. Clin. Pharmacol. 2006, 62, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Leung, B.; Poole, P. Phosphodiesterase 4 inhibitors for chronic obstructive pulmonary disease. In Cochrane Database of Systematic Reviews; Wiley-Blackwell: Hoboken, NJ, USA, 2013. [Google Scholar]
- Rabe, K.F.; Bateman, E.D.; O’Donnell, D.; Witte, S.; Bredenbroker, D.; Bethke, T.D. Roflumilast—An oral anti-inflammatory treatment for chronic obstructive pulmonary disease: A randomised controlled trial. Lancet 2005, 366, 563–571. [Google Scholar] [CrossRef]
- Calverley, P.M.; Sanchez-Toril, F.; McIvor, A.; Teichmann, P.; Bredenbroeker, D.; Fabbri, L.M. Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007, 176, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Rennard, S.; Knobil, K.; Rabe, K.F.; Morris, A.; Schachter, N.; Locantore, N.; Canonica, W.G.; Zhu, Y.; Barnhart, F. The efficacy and safety of cilomilast in COPD. Drugs 2008, 68 (Suppl. S2), 3–57. [Google Scholar] [CrossRef] [PubMed]
- Puhan, M.A.; Hansel, N.N.; Sobradillo, P.; Enright, P.; Lange, P.; Hickson, D.; Menezes, A.M.; ter Riet, G.; Held, U.; Domingo-Salvany, A.; et al. Large-scale international validation of the ado index in subjects with COPD: An individual subject data analysis of 10 cohorts. BMJ Open 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Hatzelmann, A.; Morcillo, E.J.; Lungarella, G.; Adnot, S.; Sanjar, S.; Beume, R.; Schudt, C.; Tenor, H. The preclinical pharmacology of roflumilast—A selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2010, 23, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Burgin, A.B.; Magnusson, O.T.; Singh, J.; Witte, P.; Staker, B.L.; Bjornsson, J.M.; Thorsteinsdottir, M.; Hrafnsdottir, S.; Hagen, T.; Kiselyov, A.S.; et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 2010, 28, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wunder, F.; Quednau, R.; Geerts, A.; Barg, M.; Tersteegen, A. Characterization of the cellular activity of PDE4 inhibitors using two novel PDE4 reporter cell lines. Mol. Pharm. 2013, 10, 3697–3705. [Google Scholar] [CrossRef] [PubMed]
- Fox, D., 3rd; Burgin, A.B.; Gurney, M.E. Structural basis for the design of selective phosphodiesterase 4B inhibitors. Cell. Signal. 2014, 26, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Ariga, M.; Neitzert, B.; Nakae, S.; Mottin, G.; Bertrand, C.; Pruniaux, M.P.; Jin, S.L.; Conti, M. Nonredundant function of phosphodiesterases 4D and 4B in neutrophil recruitment to the site of inflammation. J. Immunol. 2004, 173, 7531–7538. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.L.C.; Swinnen, J.V.; Conti, M. Characterization of the structure of a low km, rolipram-sensitive cAMP phosphodiesterase—Mapping of the catalytic domain. J. Biol. Chem. 1992, 267, 18929–18939. [Google Scholar] [PubMed]
- Mehats, C.; Jin, S.L.; Wahlstrom, J.; Law, E.; Umetsu, D.T.; Conti, M. PDE4D plays a critical role in the control of airway smooth muscle contraction. FASEB J. 2003, 17, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T. Cyclic AMP-specific phosphodiesterase-4 as a target for the development of antidepressant drugs. Curr. Pharm. Des. 2009, 15, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D.; Schafer, P.; Zhang, K.Y.J. Keynote review: Phosphodiesterase-4 as a therapeutic target. Drug Discov. Today 2005, 10, 1503–1519. [Google Scholar] [CrossRef]
- McCahill, A.; McSorley, T.; Huston, E.; Hill, E.V.; Lynch, M.J.; Gall, I.; Keryer, G.; Lygren, B.; Tasken, K.; van Heeke, G.; et al. In resting COS1 cells a dominant negative approach shows that specific, anchored PDE4 cAMP phosphodiesterase isoforms gate the activation, by basal cyclic AMP production, of AKAP-tethered protein kinase a type II located in the centrosomal region. Cell. Signal. 2005, 17, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, Y.H.; Cheng, Y.K.; Lu, X.; Shao, Y.X.; Li, X.; Liu, M.; Liu, P.; Luo, H.B. Identification of novel phosphodiesterase-4D inhibitors prescreened by molecular dynamics-augmented modeling and validated by bioassay. J. Chem. Inf. Model. 2013, 53, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Peter, D.; Goggel, R.; Colbatzky, F.; Nickolaus, P. Inhibition of cyclooxygenase-2 prevents adverse effects induced by phosphodiesterase type 4 inhibitors in rats. Br. J. Pharmacol. 2011, 162, 415–427. [Google Scholar] [CrossRef] [PubMed]
- NIH-USA. Available online: https://clinicaltrials.gov (accessed on 22 March 2017).


{kind=link}
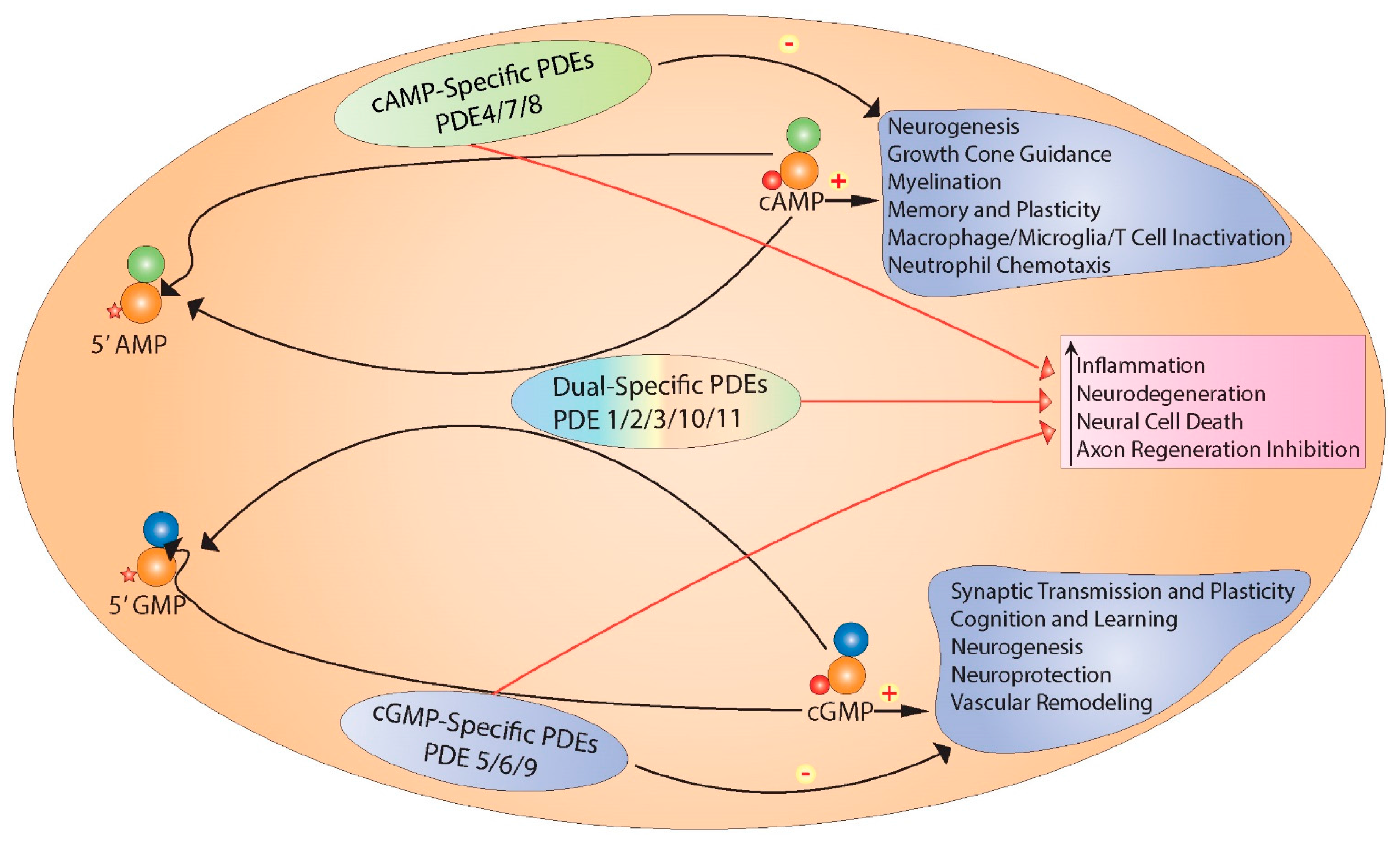{kind=link}
| Disease Name | Intervention | Target | Trial Phase | Trial Number |
|---|---|---|---|---|
| Traumatic brain injury | Sildenafil | PDE5 | - | NCT02990078 |
| Alzheimer’s disease | Cilostazol | PDE3 | 2 | NCT02491268 |
| Roflumilast | PDE4 | - | NCT02835716 | |
| BPN14770 | PDE4D | 1 | NCT02840279 | |
| BPN14770 | PDE4D | 1 | NCT02648672 | |
| Stroke | Cilostazol | PDE3 | 3 | NCT02481323 |
| Cilostazol | PDE3 | 3 | NCT01995370 | |
| Tadalafil | PDE5 | 2 | NCT02801032 | |
| Sildenafil | PDE5 | 1 | NCT02628847 | |
| Amyotrophic lateral sclerosis | Ibudilast (MN-166) | PDE4 | 1/2 | NCT02714036 |
| Multiple sclerosis | Ibudilast (MN-166) | PDE4 | 2 | NCT01982942 |
| Autonomic nervous system failure and supine hypotension | Sildenafil | PDE5 | 1/2 | NCT00223717 |
| Neonatal encephalopathy | Sildenafil | PDE5 | 1 | NCT02812433 |
| Cerebral vasospasm following sub arachnoid hemorrhage | Milrinone | PDE3 | 2 | NCT02712788 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knott, E.P.; Assi, M.; Rao, S.N.R.; Ghosh, M.; Pearse, D.D. Phosphodiesterase Inhibitors as a Therapeutic Approach to Neuroprotection and Repair. Int. J. Mol. Sci. 2017, 18, 696. https://doi.org/10.3390/ijms18040696
Knott EP, Assi M, Rao SNR, Ghosh M, Pearse DD. Phosphodiesterase Inhibitors as a Therapeutic Approach to Neuroprotection and Repair. International Journal of Molecular Sciences. 2017; 18(4):696. https://doi.org/10.3390/ijms18040696
Chicago/Turabian StyleKnott, Eric P., Mazen Assi, Sudheendra N. R. Rao, Mousumi Ghosh, and Damien D. Pearse. 2017. "Phosphodiesterase Inhibitors as a Therapeutic Approach to Neuroprotection and Repair" International Journal of Molecular Sciences 18, no. 4: 696. https://doi.org/10.3390/ijms18040696
APA StyleKnott, E. P., Assi, M., Rao, S. N. R., Ghosh, M., & Pearse, D. D. (2017). Phosphodiesterase Inhibitors as a Therapeutic Approach to Neuroprotection and Repair. International Journal of Molecular Sciences, 18(4), 696. https://doi.org/10.3390/ijms18040696