
Inferring Genes and Biological Functions That Are Sensitive to the Severity of Toxicity Symptoms


Abstract
:1. Introduction
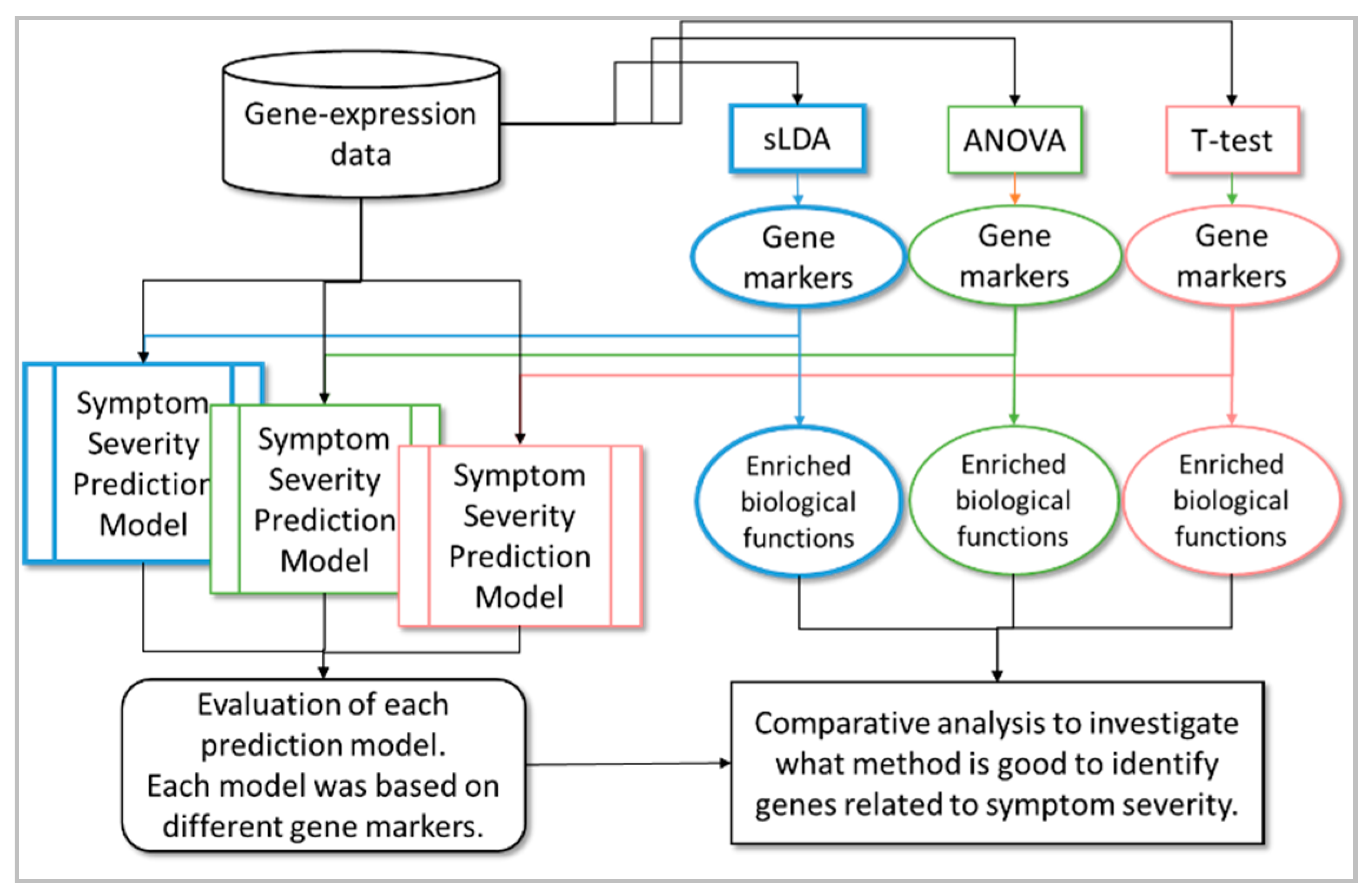
2. Results
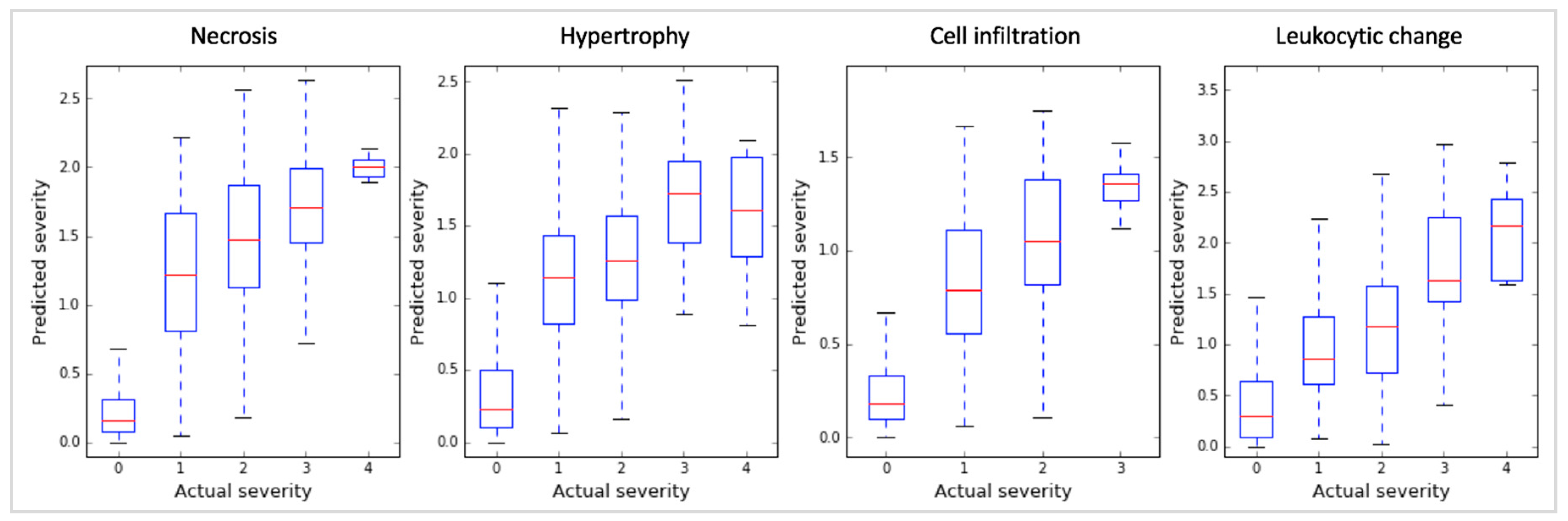
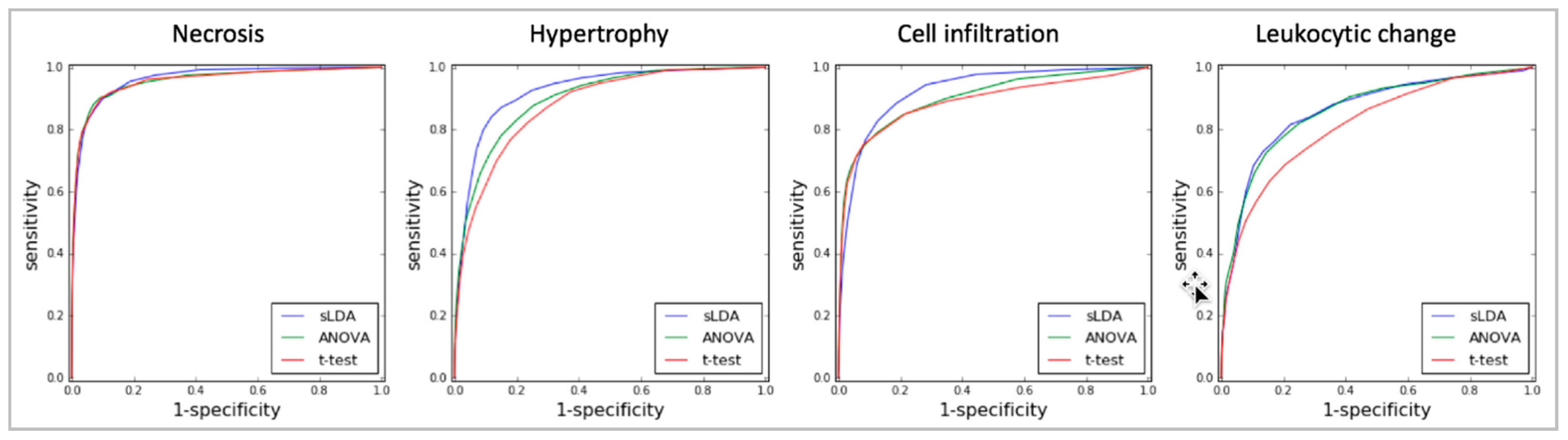
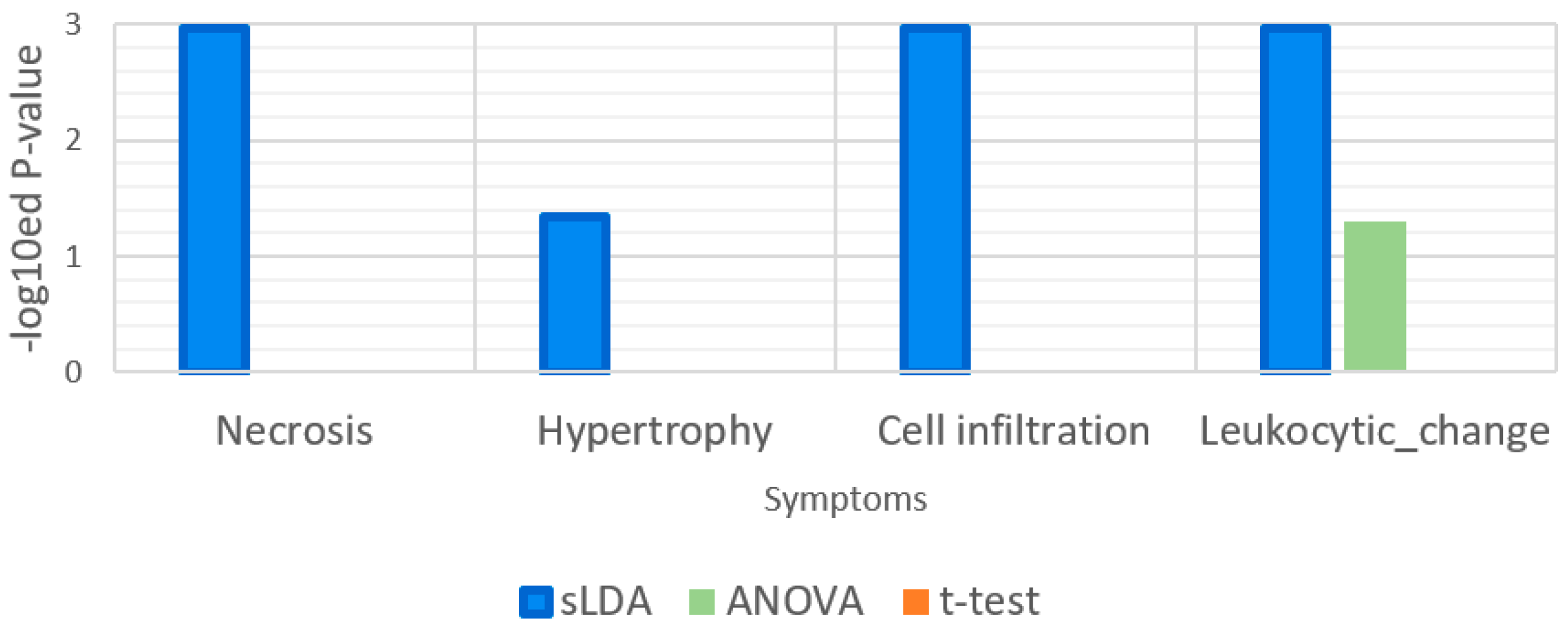
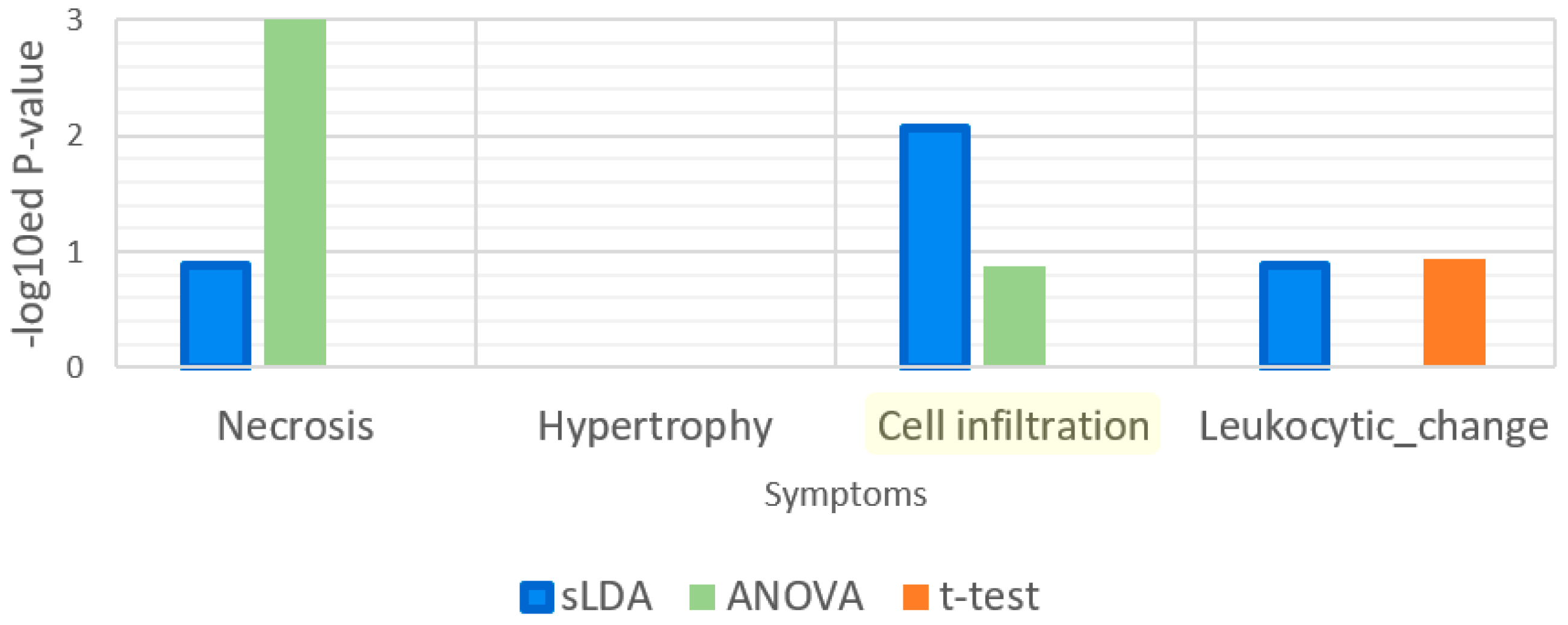
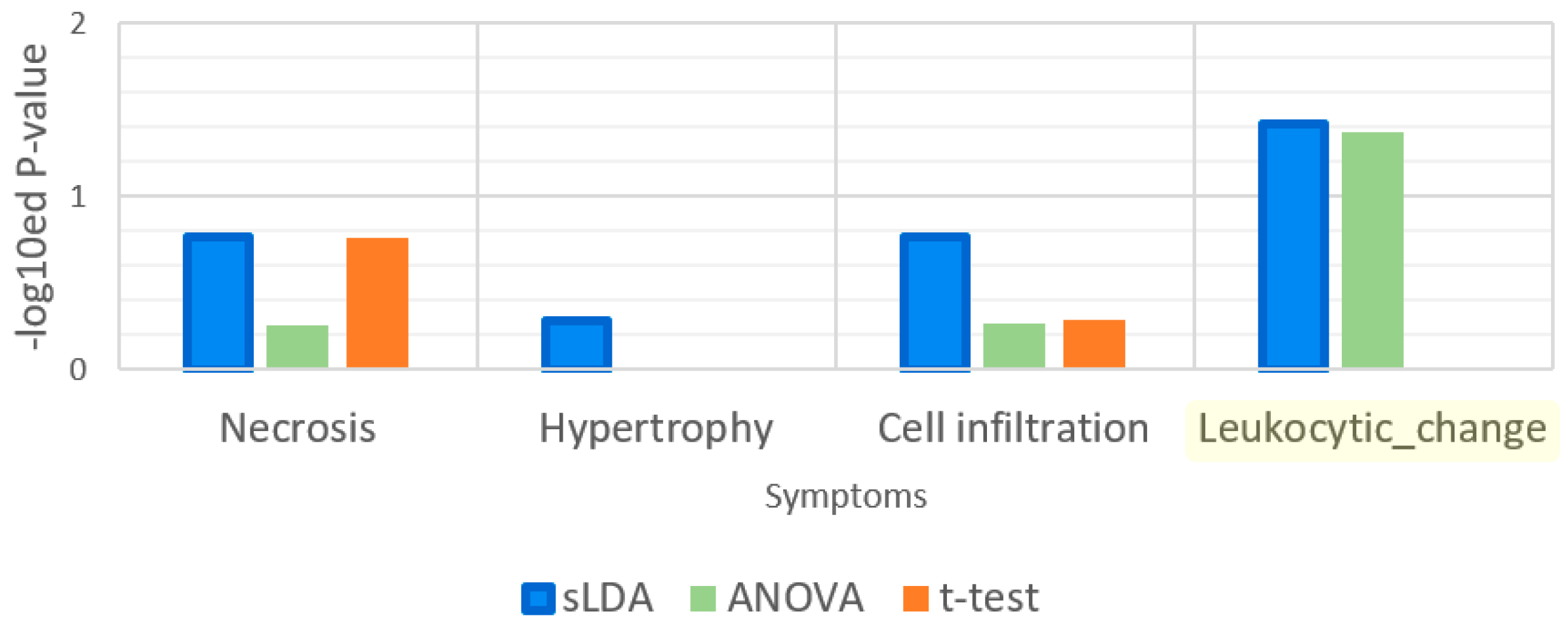
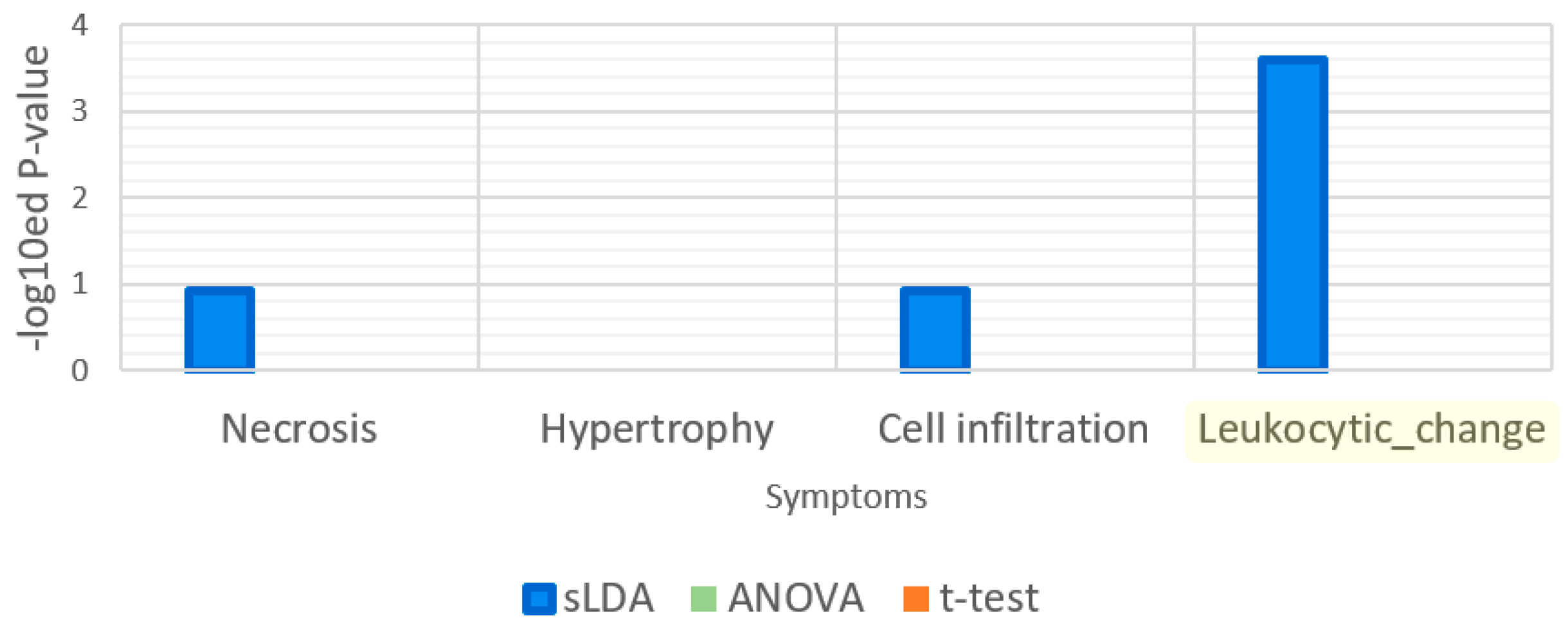
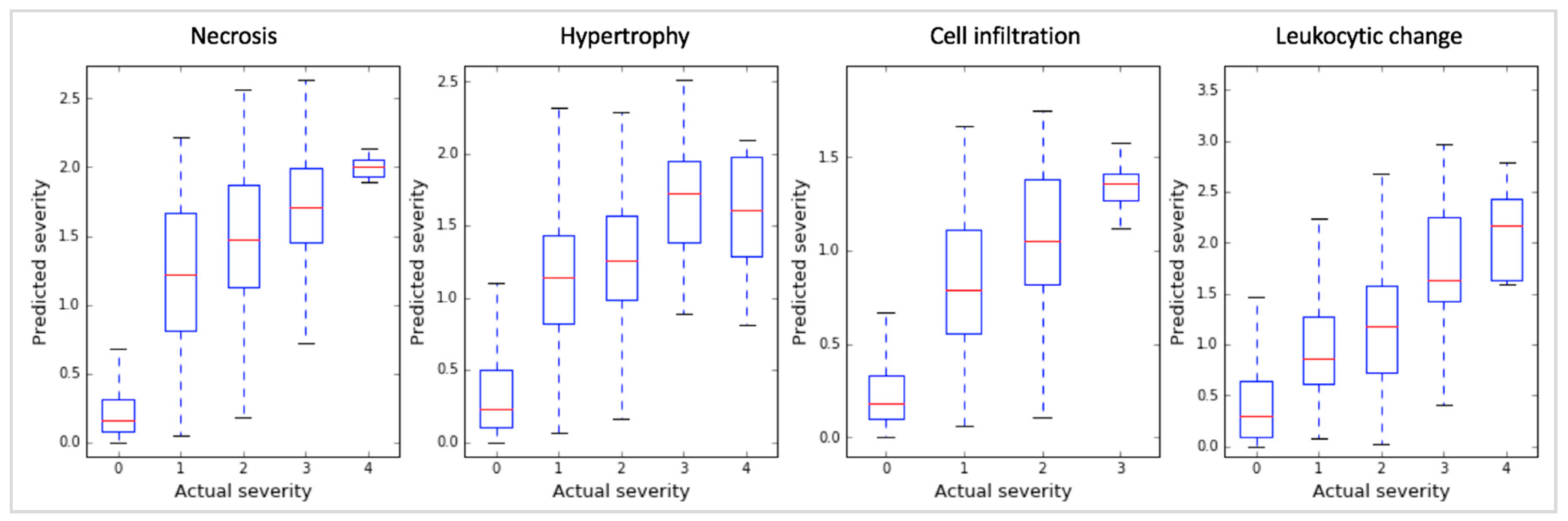
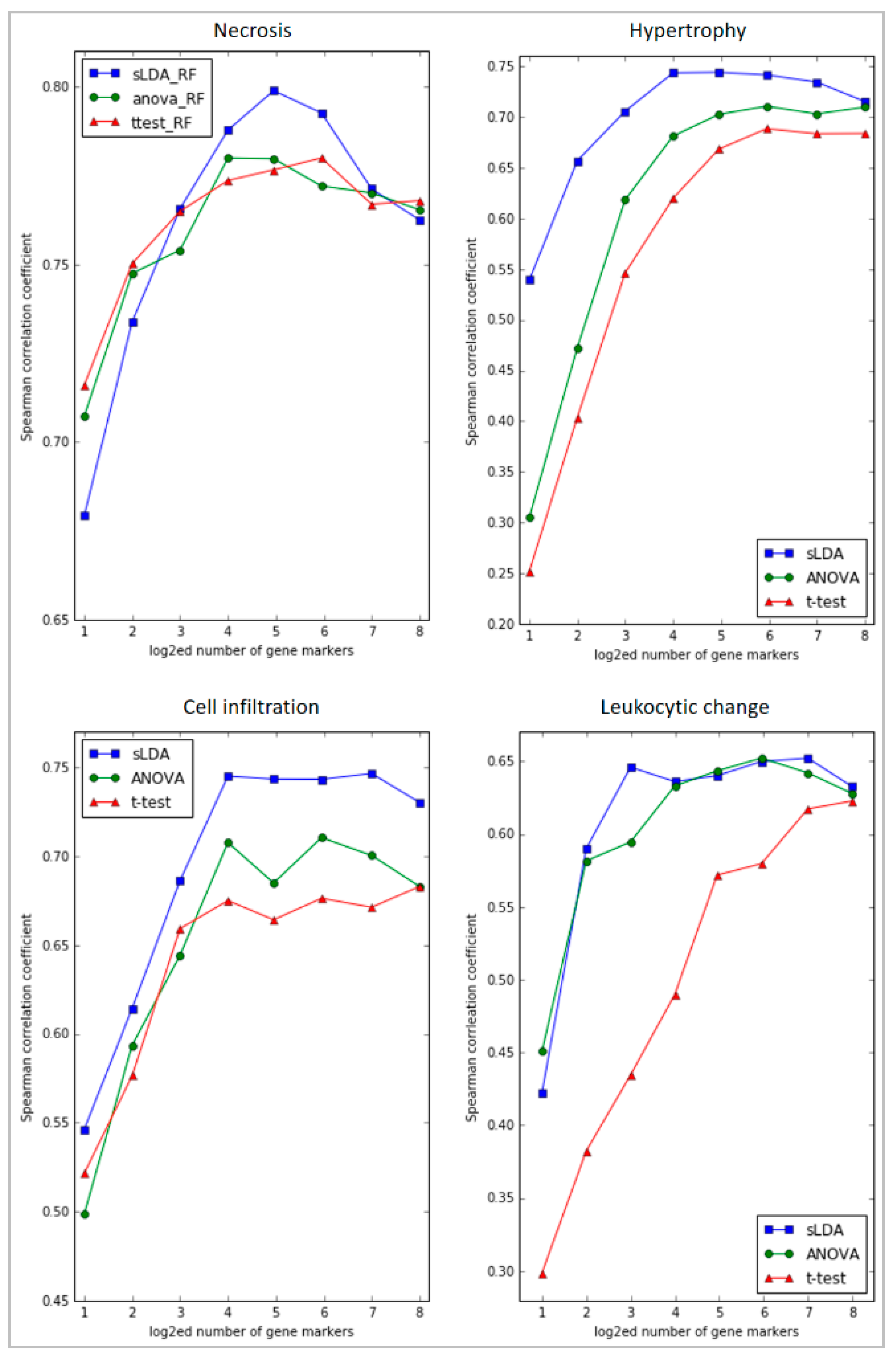
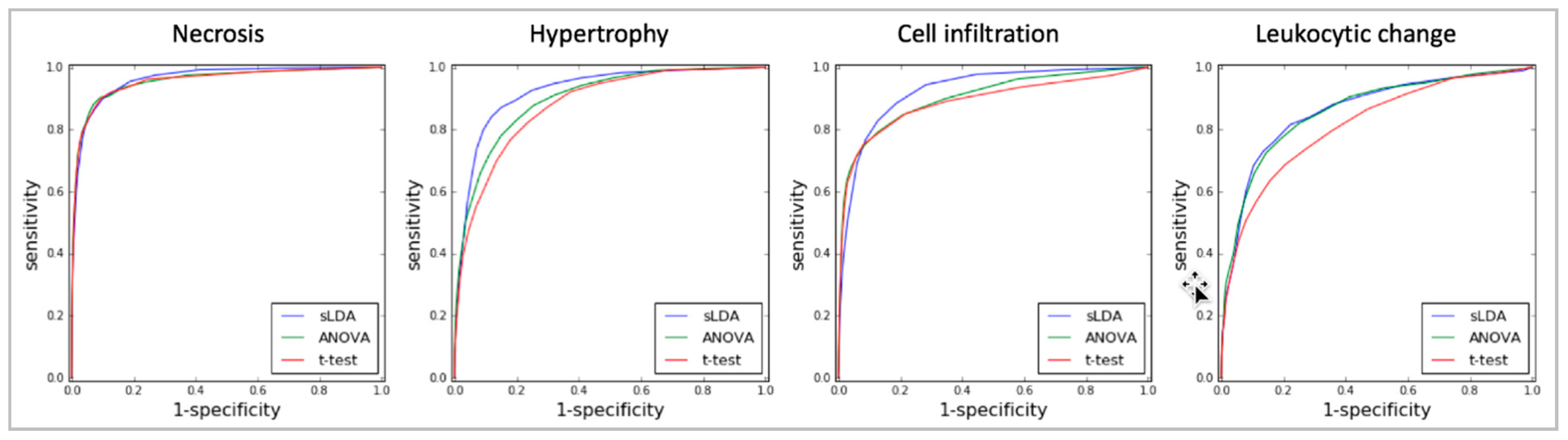
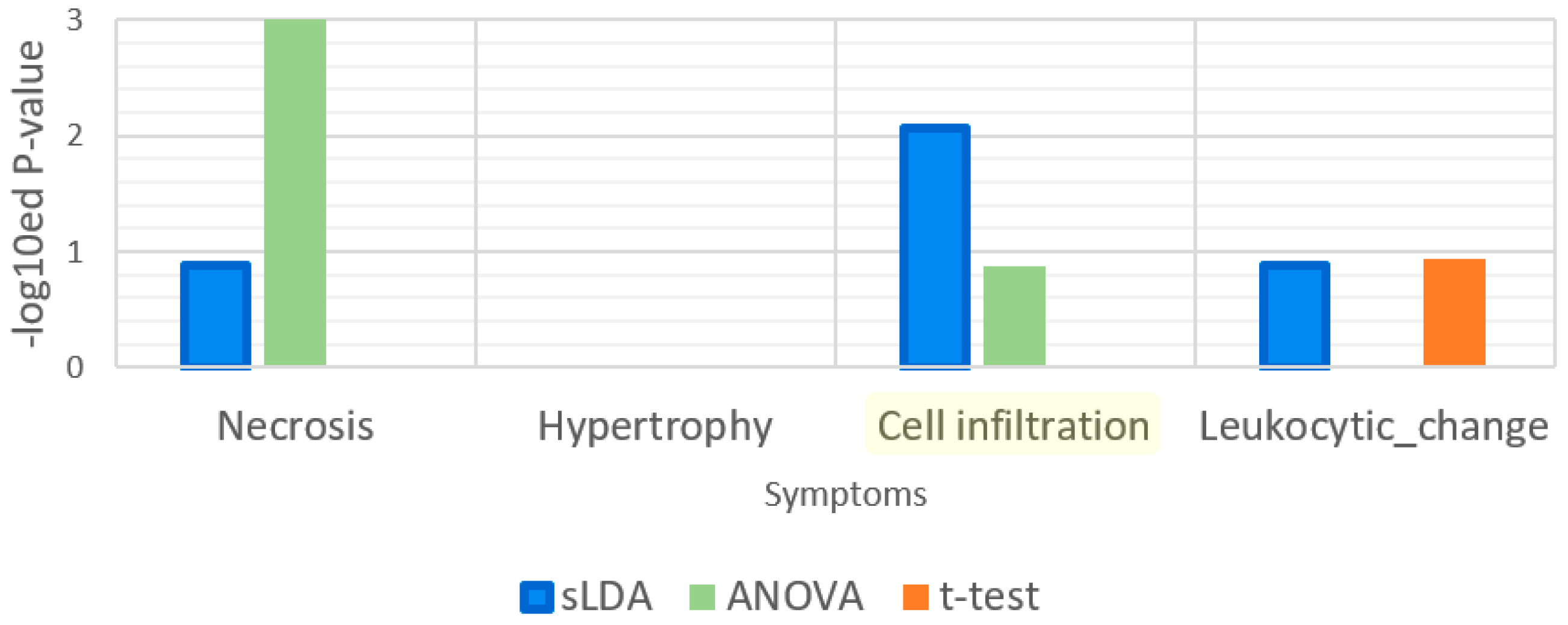
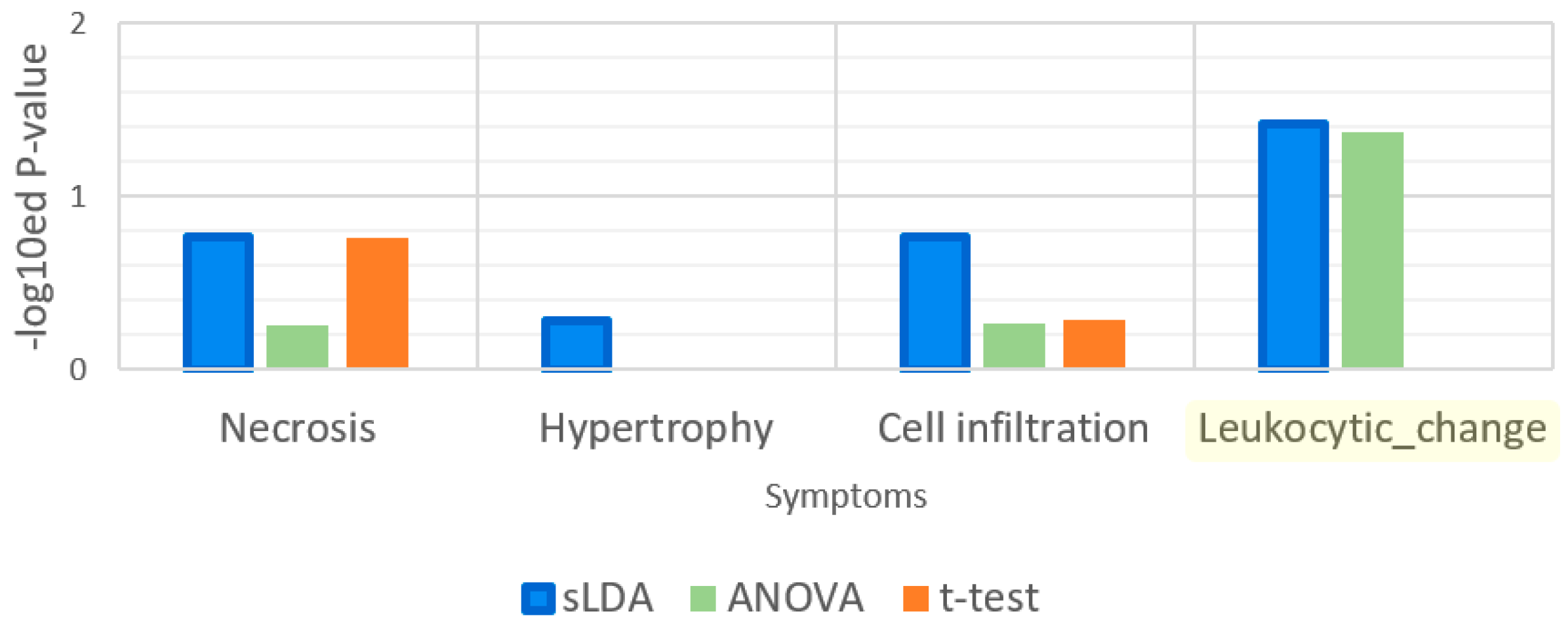
2.1. Toxicity Severity Prediction Ability of Inferred Gene Markers
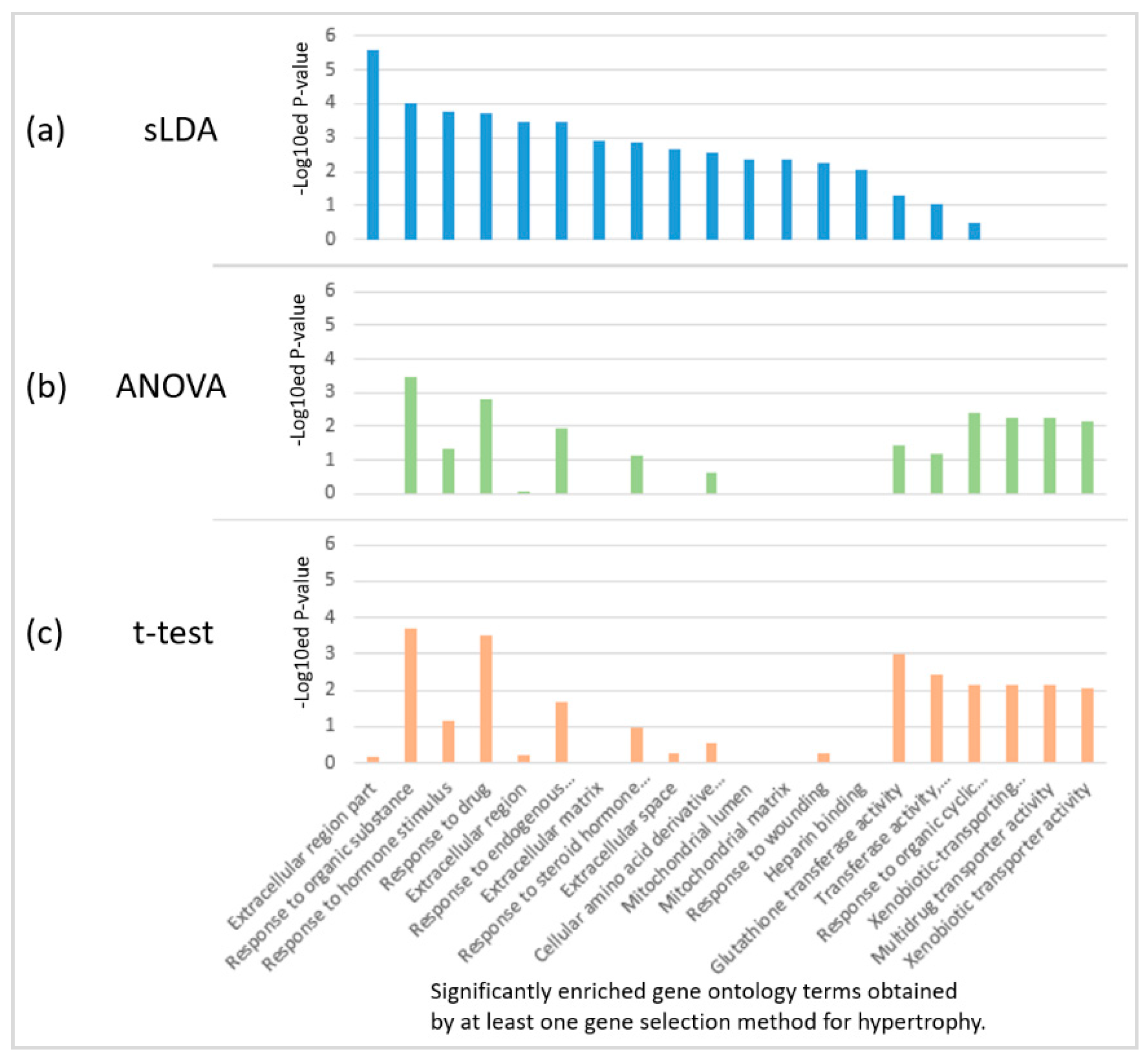
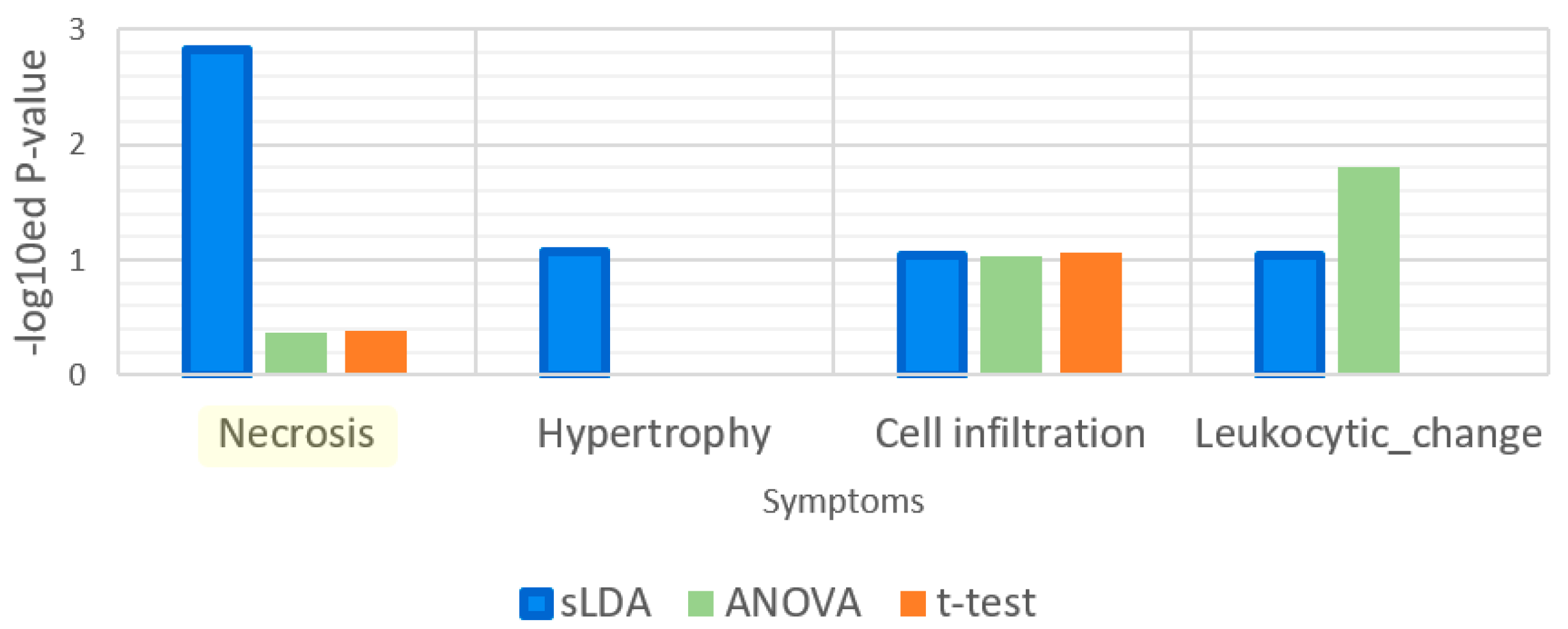
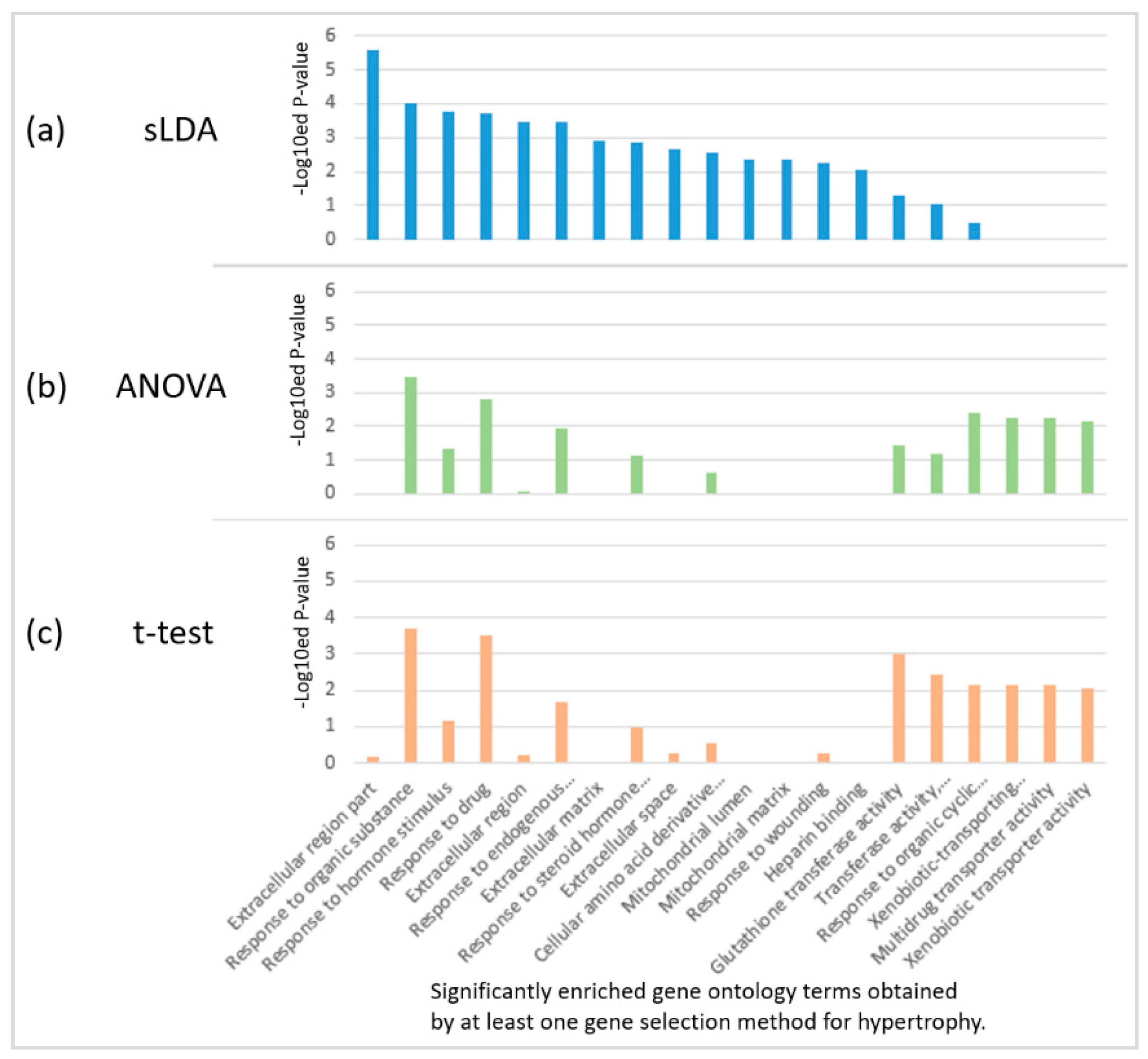
2.2. Biological Investigation of Inferred Gene Markers with Respect to Toxicity Symptom Severity
3. Discussion
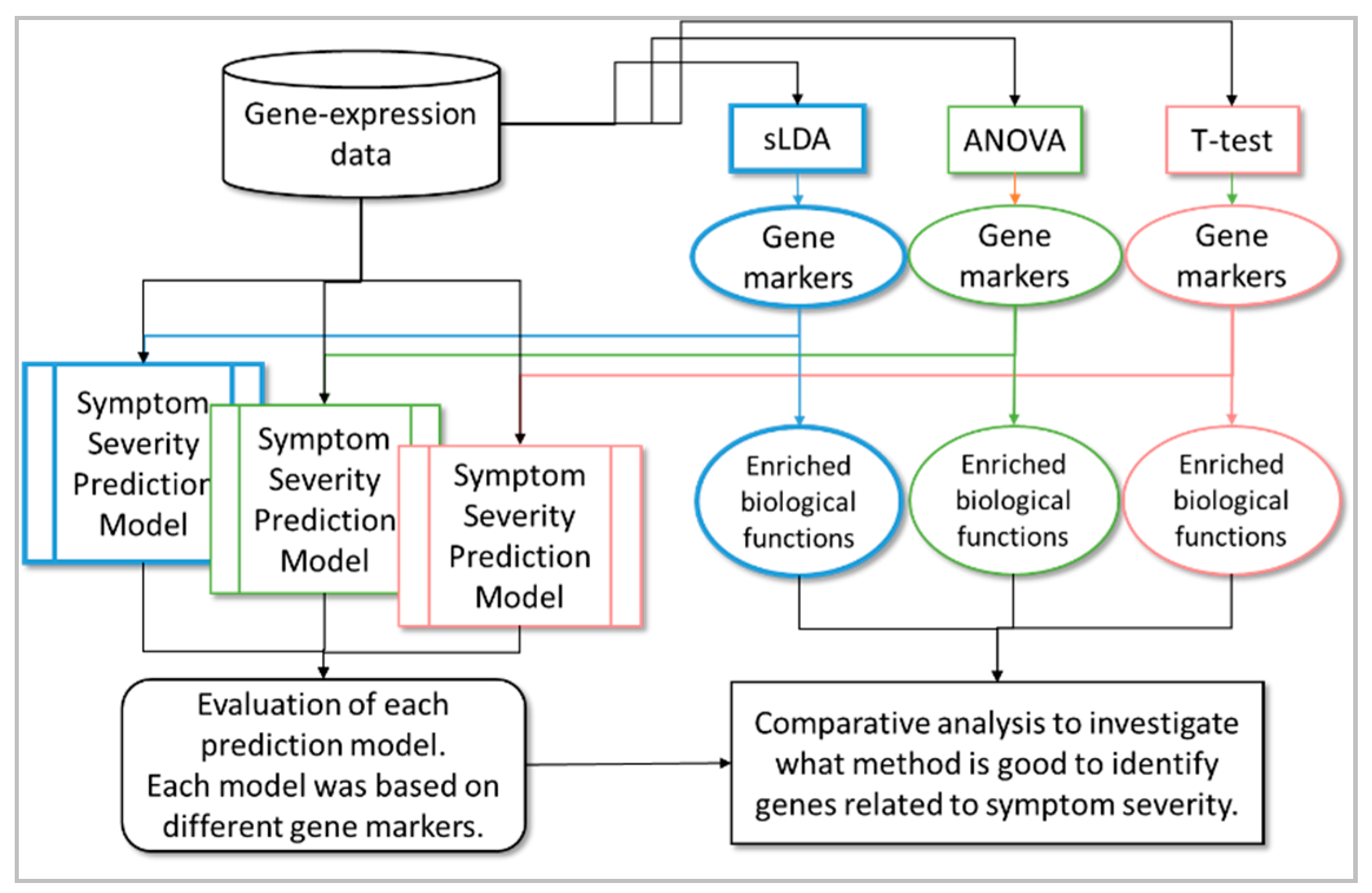
4. Materials and Methods
4.1. Data Description
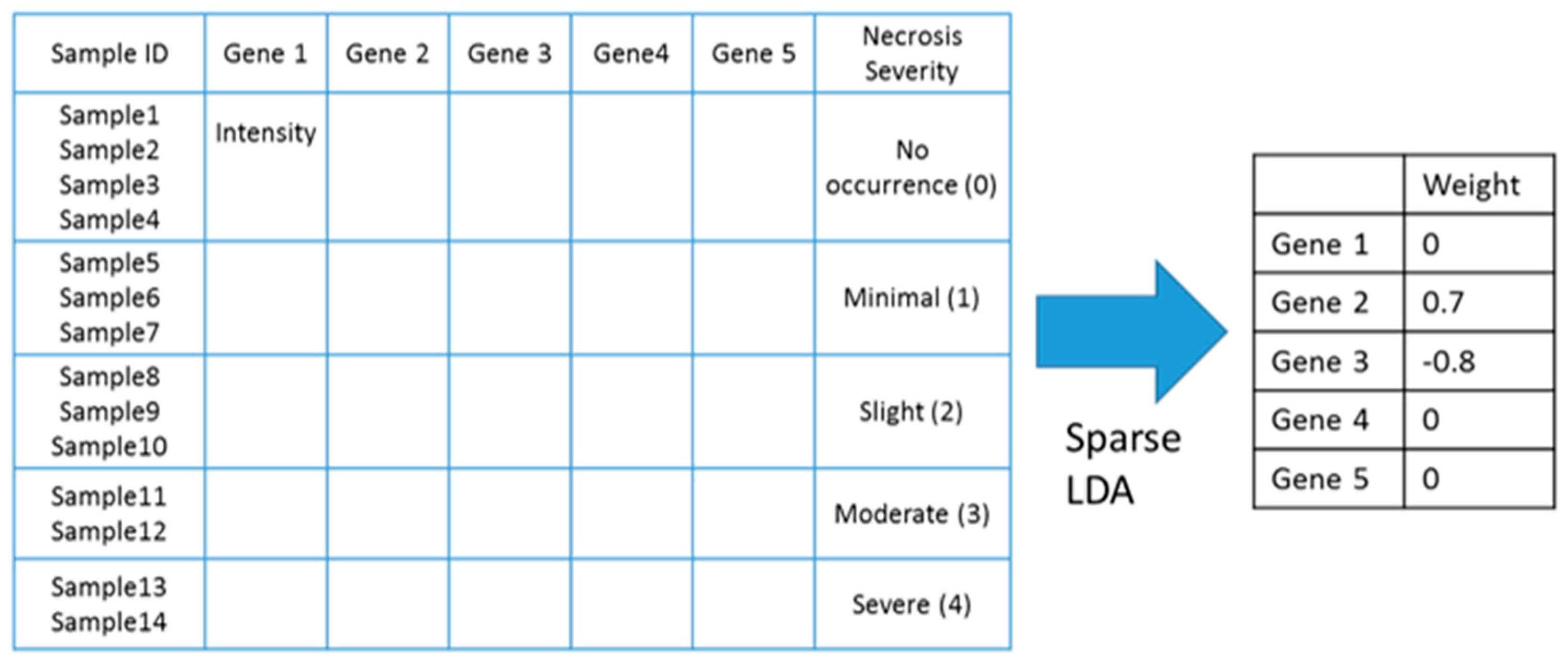
4.2. Identifying Gene Markers Sensitive to Symptom Severity
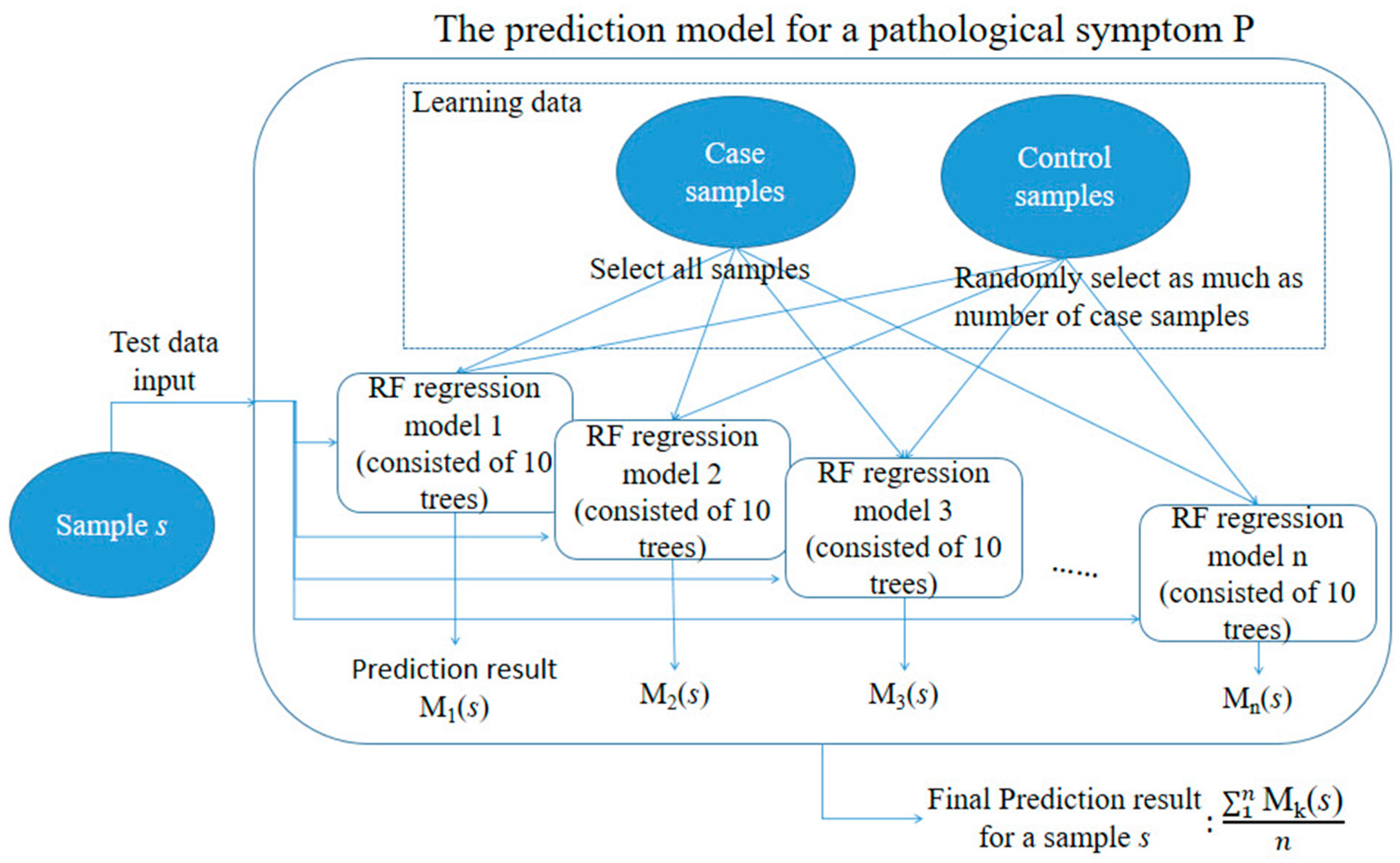
4.3. Construction of the Toxicity Severity Prediction Model
4.4. Performance Evaluation of the RF Models in Toxicity Severity Prediction
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| sLDA | Sparse linear discriminant analysis |
| RF | Random forest |
| SCC | Spearman’s correlation coefficient |
| ROC | Receiver operating characteristic |
| AUC | Area under curve |
References
- Zhang, J.D.; Berntenis, N.; Roth, A.; Ebeling, M. Data mining reveals a network of early-response genes as a consensus signature of drug-induced in vitro and in vivo toxicity. Pharmacol. J. 2014, 14, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Suvitaival, T.; Parkkinen, J.A.; Virtanen, S.; Kaski, S. Cross-organism toxicogenomics with group factor analysis. Syst. Biomed. 2014, 2, 71–80. [Google Scholar] [CrossRef]
- Huang, L.; Heinloth, A.N.; Zeng, Z.B.; Paules, R.S.; Bushel, P.R. Genes related to apoptosis predict necrosis of the liver as a phenotype observed in rats exposed to a compendium of hepatotoxicants. BMC Genom. 2008, 9, 288. [Google Scholar] [CrossRef] [PubMed]
- Bowles, M.; Shigeta, R. Statistical models for predicting liver toxicity from genomic data. Syst. Biomed. 2013, 1, 144–149. [Google Scholar] [CrossRef]
- Wu, M.C.; Zhang, L.; Wang, Z.; Christiani, D.C.; Lin, X. Sparse linear discriminant analysis for simultaneous testing for the significance of a gene set/pathway and gene selection. Bioinformatics 2009, 25, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Clemmensen, L.; Hastie, T.; Witten, D.; Ersbøll, B. Sparse discriminant analysis. Technometrics 2011, 53, 406–413. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and regression by randomforest. R News 2002, 2, 18–22. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology, C. Gene ontology consortium: Going forward. Nucleic. Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Omiecinski, C.J.; Heuvel, J.P.V.; Perdew, G.H.; Peters, J.M. Xenobiotic metabolism, disposition, and regulation by receptors: From biochemical phenomenon to predictors of major toxicities. Toxicol. Sci. 2011, 120, S49–S75. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, L.; Lister, S. The Royal Marsden Manual of Clinical Nursing Procedures; Wiley-Blackwell: Hoboken, NJ, USA, 2015. [Google Scholar]
- Stone, K.D.; Prussin, C.; Metcalfe, D.D. IgE, Mast Cells, Basophils, and Eosinophils. J. Allergy Clin. Immunol. 2010, 125, S73–S80. [Google Scholar] [CrossRef] [PubMed]
- Amador-Noguez, D.; Dean, A.; Huang, W.D.; Setchell, K.; Moore, D.; Darlington, G. Alterations in xenobiotic metabolism in the long-lived little mice. Aging Cell 2007, 6, 453–470. [Google Scholar] [CrossRef] [PubMed]
- 1Miao, W.M.; Hu, L.G.; Kandouz, M.; Batist, G. Oltipraz is a bifunctional inducer activating both phase I and phase II drug-metabolizing enzymes via the xenobiotic responsive element. Mol. Pharmacol. 2003, 64, 346–354. [Google Scholar]
- Noble, D.D.; McCafferty, J.B.; Greening, A.P.; Innes, J.A. Respiratory heat and moisture loss is associated with eosinophilic inflammation in asthma. Eur. Respir. J. 2007, 29, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Yu, M.C.; Lee, C.H.; Lee, T.C.; Yang, S.Y. High eosinophil cationic protein level in asthmatic patients with “heat” zheng. Am. J. Chin. Med. 2003, 31, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.I.; Choi, I.S.; Lee, S.H.; Lee, J.B.; Park, C.H.; Hong, S.N. Localized heat urticaria associated with mast cell and eosinophil degranulation. J. Allergy Clin. Immunol. 2002, 109, 714–715. [Google Scholar] [CrossRef] [PubMed]
- Bachelet, M.; Marchand, F.; Souil, E.; Francois, D.; Mariethoz, E.; Weyer, A.; Polla, B.S. Expression and localization of heat shock proteins in rat basophilic leukemia cells: Differential modulation by degranulation, thermal or oxidative stress. Allergy 2002, 57, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Elsas, M.I.; Elsas, P.X.; Joseph, D.; Havet, N.; da Silva, L.A.; Salgado, D.R.; Saliou, B.; Vargaftig, B.B. Stimulation of early eosinophil progenitors by a heat stable alveolar macrophage product from ovalbumin-sensitized and non-sensitized guinea pigs. J. Br. Soc. Allergy Clin. Immunol. 1997, 27, 208–217. [Google Scholar] [CrossRef]
- Iwaki, T.; Hamada, Y.; Tateishi, J. Advanced glycosylation end-products and heat shock proteins accumulate in the basophilic degeneration of the myocardium and the corpora amylacea of the glia. Pathol. Int. 1996, 46, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Resnik, S.; Helley, W.B. Heat stress degranulation of the basophil leucocyte in man. Nature 1966, 209, 812. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Xie, X.Q.; Liu, Y.; Han, Z.P.; Zhao, X.; Cai, N.; Zhang, S.S.; Song, J.R.; Wei, L.X. Autophagy lessens ischemic liver injury by reducing oxidative damage. Cell Biosci. 2013, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, M.; Jiang, H.; Yu, Y.; Yu, P.; Tong, R.; Wu, J.; Zhang, S.; Yao, K.; Zou, Y.; et al. Extracellular high-mobility group box 1 mediates pressure overload-induced cardiac hypertrophy and heart failure. J. Cell. Mol. Med. 2016, 20, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Sheikh, F. Scaffold proteins regulating extracellular regulated kinase function in cardiac hypertrophy and disease. Front. Pharmacol. 2016, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Mutlak, M.; Kehat, I. Extracellular signal-regulated kinases 1/2 as regulators of cardiac hypertrophy. Front. Pharmacol. 2015, 6, 149. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Gonzalez, S.; Shah, S.; Kyupelyan, L.; Petrigliano, F.A.; McAllister, D.R.; Adams, J.S.; Karperien, M.; Tuan, T.L.; Benya, P.D.; et al. Extracellular matrix domain formation as an indicator of chondrocyte dedifferentiation and hypertrophy. Tissue Eng. Part C Methods 2014, 20, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Orfanidou, T.; Malizos, K.N.; Varitimidis, S.; Tsezou, A. 1,25-Dihydroxyvitamin D3 and extracellular inorganic phosphate activate mitogen-activated protein kinase pathway through fibroblast growth factor 23 contributing to hypertrophy and mineralization in osteoarthritic chondrocytes. Exp. Biol. Med. 2012, 237, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Cheng, C.F.; Djoko, B.; Lian, W.S.; Tu, C.F.; Tsai, M.T.; Chen, Y.H.; Chen, C.C.; Cheng, C.J.; Yang, R.B. Transgenic overexpression of the secreted, extracellular EGF-CUB domain-containing protein SCUBE3 induces cardiac hypertrophy in mice. Cardiovasc. Res. 2007, 75, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, E.; Vitellozzi, R.; Fernandez, P.; Falcieri, E.; Battistelli, M.; Burattini, S.; Facchini, A.; Flamigni, F.; Santi, S.; Facchini, A.; et al. Chondrocyte hypertrophy and apoptosis induced by groalpha require three-dimensional interaction with the extracellular matrix and a co-receptor role of chondroitin sulfate and are associated with the mitochondrial splicing variant of cathepsin b. J. Cell. Phys. 2007, 210, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Baudry, M.; Davis, J.L. Long-Term Potentiation, Vol. 1: A Debate of Current Issues; A Bradford Book: Cambridge, MA, USA, 1991. [Google Scholar]
- Igarashi, Y.; Nakatsu, N.; Yamashita, T.; Ono, A.; Ohno, Y.; Urushidani, T.; Yamada, H. Open TG-GATEs: A large-scale toxicogenomics database. Nucleic Acids Res. 2015, 43, D921–D927. [Google Scholar] [CrossRef] [PubMed]
- Harbron, C.; Chang, K.M.; South, M.C. Refplus: An R package extending the RMA algorithm. Bioinformatics 2007, 23, 2493–2494. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Selection Method | Does It Consider Each Symptom Severity as Each Group? | Can It Be Used to Investigate Mutual Relationships between the Expression of Genes? | The Use of This Paper |
|---|---|---|---|
| Sparse LDA | Yes | Yes | Used for gene markers related to increases or decreases in toxicity symptom severity. Groups of samples were divided by the severity values. |
| ANOVA | Yes | No | |
| t-Test | No | No | Used to find gene markers related to toxicity symptom occurrence. Groups of samples were divided by the occurrences of samples. |
| Gene Selection Method | Inferred Markers Involved in “Response to Xenobiotic Stimulus” | Symptoms | Detoxification Genes? | ||||
|---|---|---|---|---|---|---|---|
| ProbesetIDs | Gene Symbols | Necrosis | Hypertrophy | Cell Infiltration | Leukocytic Changes | ||
| sLDA | 1370269_at | Cyp1a1 | ● | Yes [13] | |||
| 1387759_s_at | Ugt1a1, Ugt1a2, …, Ugt1a9 | ● | Yes [13] | ||||
| 1370613_s_at | ● | ● | |||||
| 1369921_at | Gstm3 | ● | ● | ● | ● | Yes [13] | |
| 1371089_at | Gsta5 | ● | ● | ● | Yes [14] | ||
| ANOVA | 1388153_at, 1370939_at | ACSL1 | ● | Unknown | |||
| 1398282_at | Kynu | ● | Unknown | ||||
| t-Test | No markers | ||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Shin, M. Inferring Genes and Biological Functions That Are Sensitive to the Severity of Toxicity Symptoms. Int. J. Mol. Sci. 2017, 18, 755. https://doi.org/10.3390/ijms18040755
Kim J, Shin M. Inferring Genes and Biological Functions That Are Sensitive to the Severity of Toxicity Symptoms. International Journal of Molecular Sciences. 2017; 18(4):755. https://doi.org/10.3390/ijms18040755
Chicago/Turabian StyleKim, Jinwoo, and Miyoung Shin. 2017. "Inferring Genes and Biological Functions That Are Sensitive to the Severity of Toxicity Symptoms" International Journal of Molecular Sciences 18, no. 4: 755. https://doi.org/10.3390/ijms18040755
APA StyleKim, J., & Shin, M. (2017). Inferring Genes and Biological Functions That Are Sensitive to the Severity of Toxicity Symptoms. International Journal of Molecular Sciences, 18(4), 755. https://doi.org/10.3390/ijms18040755