
Mutant p53 Protein and the Hippo Transducers YAP and TAZ: A Critical Oncogenic Node in Human Cancers


Abstract
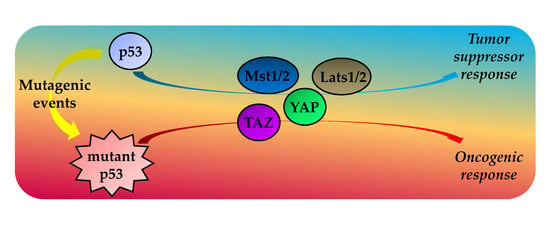
:
{kind=link}
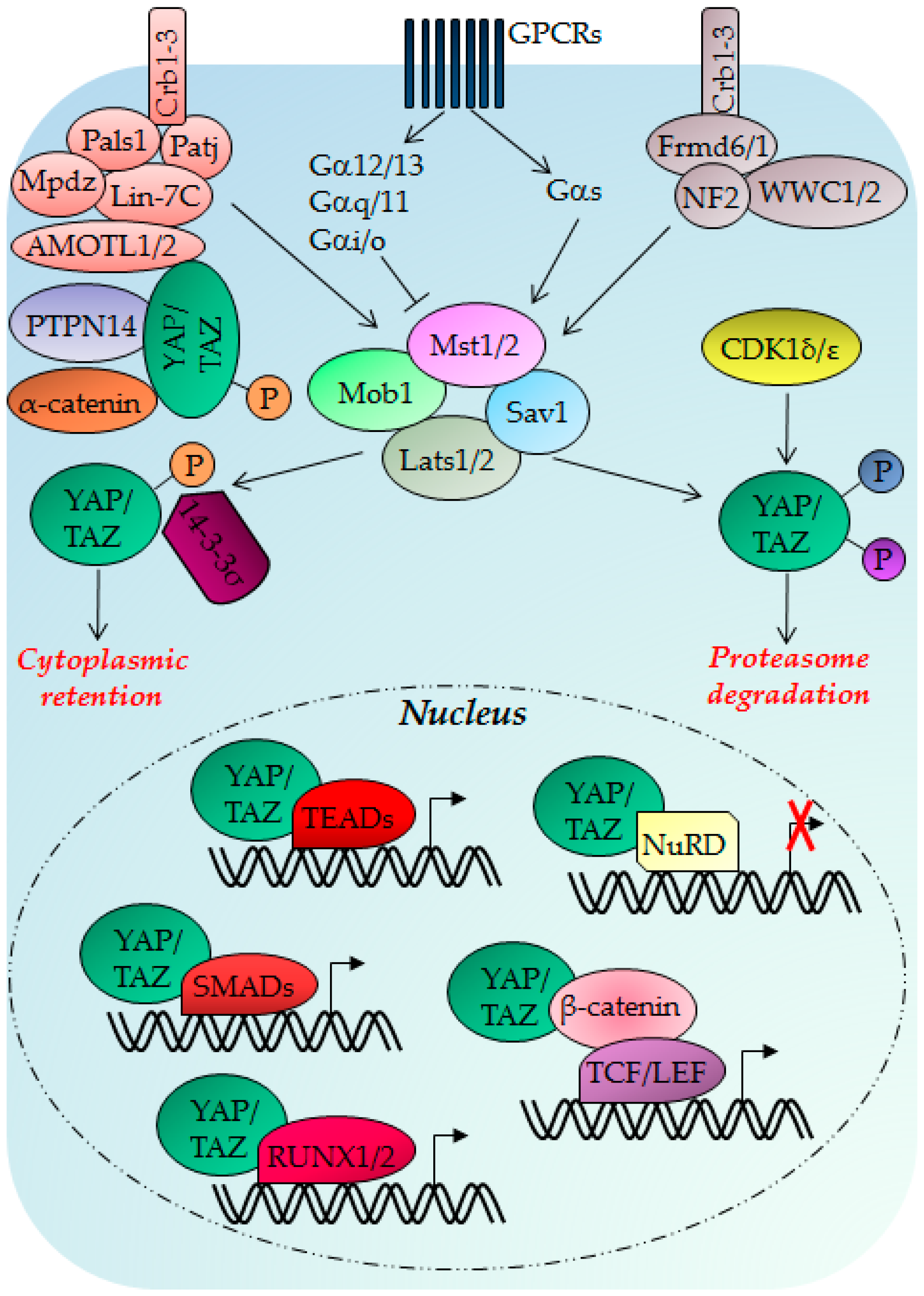{kind=link}
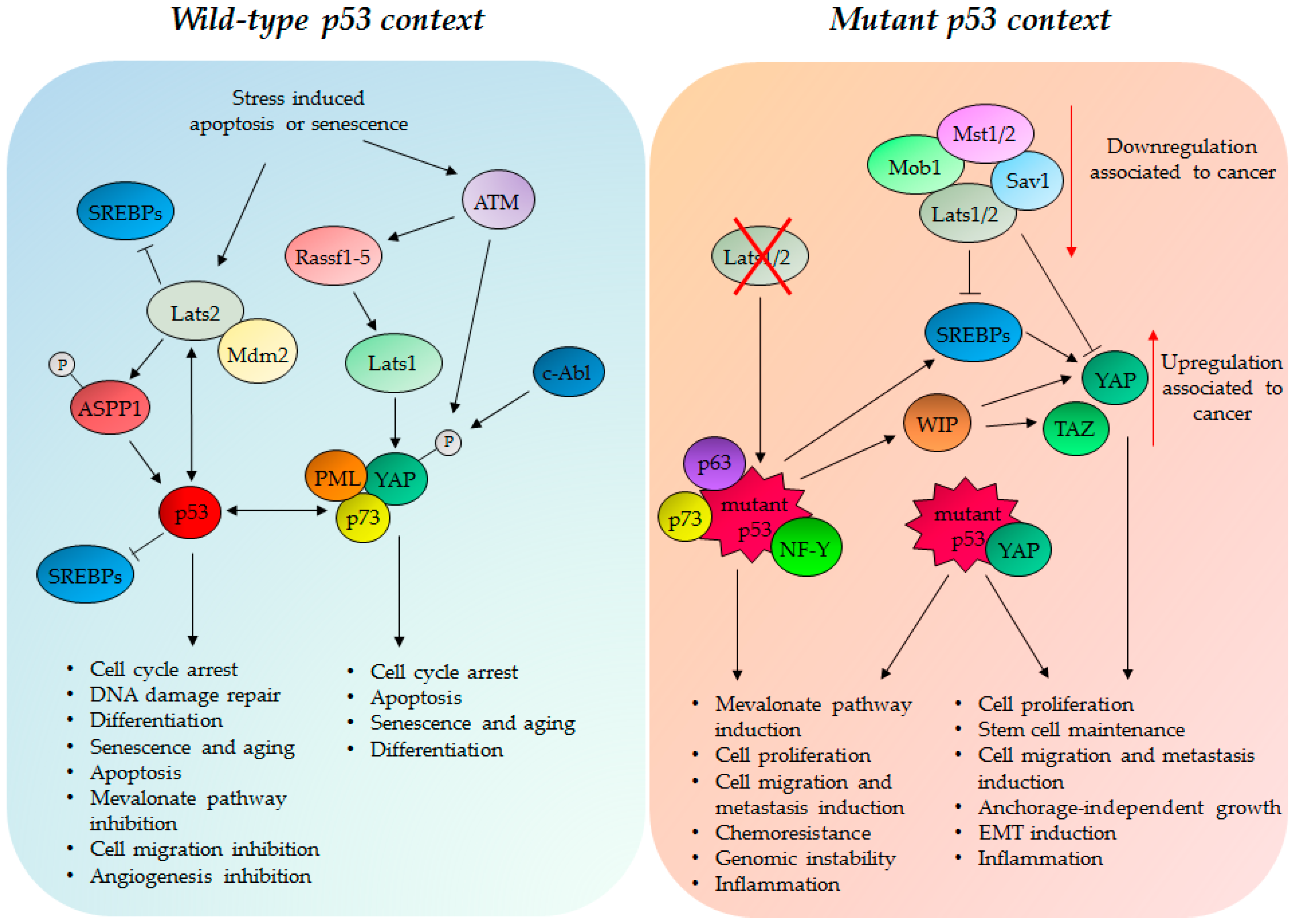{kind=link}
1. Introduction
2. p53 and Mutant p53 Protein Functions
2.1. Transcriptional Regulation
2.2. Cell Cycle Regulation
2.3. DNA Damage Response
2.4. Senescence and Aging
2.5. Apoptosis
2.6. Metabolic Regulation
2.7. Cell Migration and Angiogenesis Inhibition
2.8. Mutant p53 and Cancer
2.8.1. Mutant p53 Oncogenic Targets
2.8.2. Mutant p53 Partners
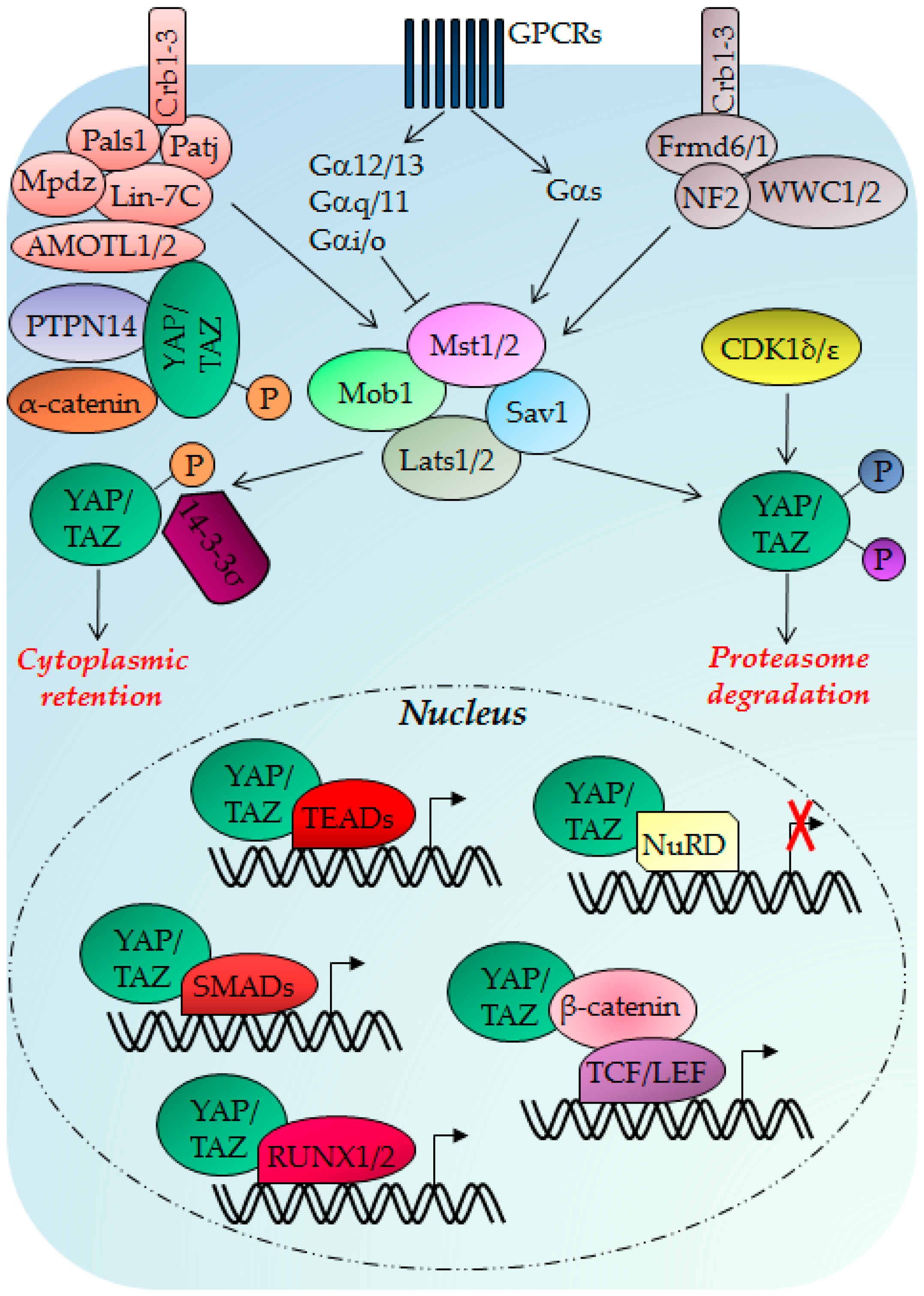
3. The Hippo Pathway
3.1. Hippo Pathway Regulation, Regulators and Functions
3.2. Hippo Pathway Deregulation in Cancer
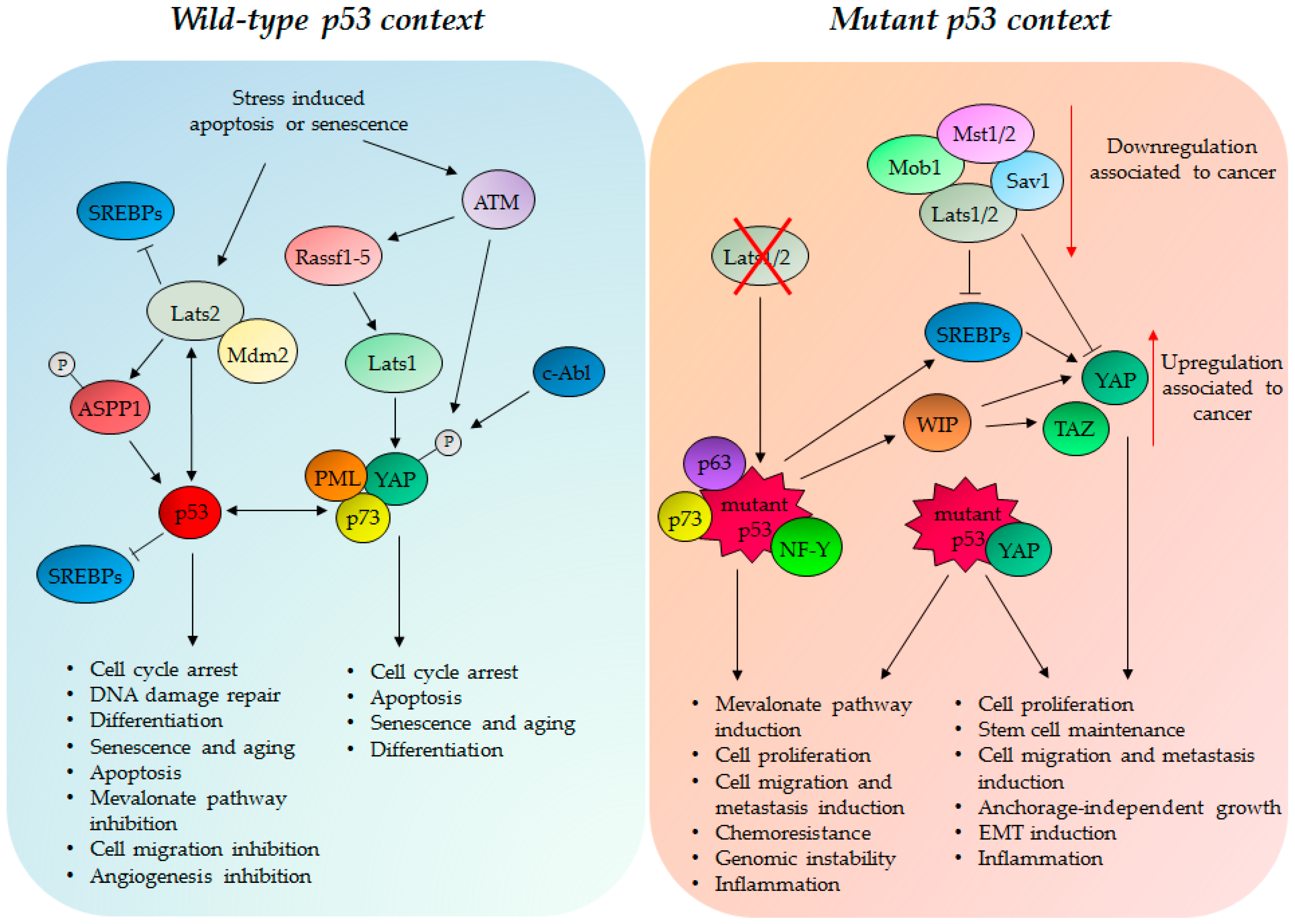
4. TP53 Status Impacts on the Hippo Pathway Activities
4.1. Wild Type p53 Protein and the Hippo Kinase Cassette in Tumor Suppression
4.2. GOF Mutant p53 Protein, YAP and TAZ in Tumorigenesis
5. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Acad11 | Acyl-CoA Dehydrogenase Family Member 11 |
| AFP | Alfa-fetoprotein |
| ALDH6A1 | Aldehyde Dehydrogenase 6 |
| α(II)PH | α(II) collagen Prolyl-4-Hydroxylase |
| AMOTL1/2 | Angiomotin-like proteins 1 and 2 |
| AMPK | AMP-activated Protein Kinase |
| ANGPT1 | Angiopoietin 1 |
| ANKRD1 | Ankyrin Repeat Domain 1 |
| API5 | Apoptosis Inhibitor 5 |
| AR | Aldose Reductase |
| AREG | Amphiregulin |
| ASNS | Asparagine Synthetase |
| ASPP1 | Apoptosis-Stimulating Protein of p53 1 |
| ATG12 | Autophagy-related protein 12 |
| ATM | Ataxia-Telangiectasia Mutated |
| ATP | Adenosine Triphosphate |
| ATR | ATM and Rad3-related protein kinase |
| Bad | Bcl-2-antagonist of cell death |
| BAG2 | BCL2 Associated Athanogene 2 |
| BAG5 | BCL2 Associated Athanogene 5 |
| Bax | Bcl-2-associated x protein |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma-extra Large |
| BECN1 | Beclin-1 |
| Birc2/cIAP1 | Baculoviral Inhibitor of the apoptosis repeat-containing protein 1 |
| Birc5 | Baculoviral Inhibitor of the apoptosis repeat-containing protein 5 |
| BRCA1 | BReast CAncer type 1 |
| CAMTA1 | Calmodulin-binding Transcription Activator 1 |
| CCNA | Cyclin A |
| CCNB | Cyclin B |
| Cdk1 | Cyclin dependent kinase 1 |
| CDK1 | Cyclin Dependent Kinase 1 |
| CHIP | Carboxyl terminus of Hsp70-Interacting Protein |
| Chk1 | Checkpoint kinase homologue 1 |
| Chk2 | Checkpoint kinase homologue 2 |
| CK1δ/ε | Casein Kinase 1 delta/epsilon |
| cMyc | V-Myc Avian Myelocytomatosis Viral Oncogene Homolog |
| CSCs | Cancer Stem Cells |
| CTGF | Connective Tissue Growth Factor |
| CYR61 | Cysteine-Rich angiogenic inducer 61 |
| DDIT4 | DNA-Damage-Inducible Transcript 4 |
| DRAM1 | DNA Damage Regulated Autophagy Modulator 1 |
| EEF1A2 | Elongation factor 1 A-2 |
| FoxA1 | Forkhead box A1 |
| Frmd6/1 | FERM domain-containing protein 6 and 1 |
| GADD45 | Growth Arrest and DNA Damage gene 45 |
| GLS2 | Phosphate-activated mitochondrial glutaminase |
| GPCRs | G protein-coupled receptors |
| H2AFX | H2A histone Family member X |
| hEGR1 | Early Growth Response protein 1 |
| HGF | Hepatocyte Growth Factor |
| HIPK2 | Homeodomain-Interacting Protein Kinase 2 |
| hsMAD1 | human Mitotic Arrest Deficiency protein 1 |
| ID2 | Inhibitor of DNA binding 2 |
| ID4 | Inhibitor of DNA binding 4 |
| IGF-1 | Insulin-Like Growth Factor-1 |
| IMPDH2 | Inosine-5′-Monophosphate Dehydrogenase 2 |
| ITGA6 | Integrin α-6 |
| KATA6 | Lysine Acetyltransferase 6A |
| KMT2A | Lysine Methyltransferase 2A |
| KMT2D | Lysine Methyltransferase 2D |
| Lats1/2 | Large tumor suppressor 1 and 2 |
| Lin-7C | Lin-7 homolog C |
| lncARSR | lncRNA Activated in RCC with Sunitinib Resistance, ENST00000424980 |
| LPA | Lysophosphatidic Acid |
| MALAT1 | Metastasis-Associated Lung Adenocarcinoma Transcript 1 |
| MARs | Matrix Attachment Regions DNA elements |
| MCD | Malonyl-CoA Decarboxylase |
| MCL1 | Human Myeloid Cell differentiation protein |
| Mdm2 | Mouse double minute 2 homolog |
| MDR1 | Multi Drug Reactivity 1 |
| MMP-1 | Matrix MetalloProteinase-1 |
| MMP5 | Membrane-associated Palmitoylated protein 5 |
| MOBKL1A/B | Mps one binder kinase activator-like A and B |
| Mpdz | Multiple PDZ domain protein |
| Msp | Macrophage stimulator protein |
| Mst1/2 | Mammalian Sterile-20 family Serine-Threonine kinases 1 and 2 |
| MT1DP | Metallothionein 1D Pseudogene |
| NER | Nucleotide Excision Repair |
| NF2 | Neurofibromin 2 |
| NF-Y | Nuclear transcription Factor Y |
| p53AIP1 | p53 Apoptosis Independent Protein 1 |
| PAI1 | Plasminogen stimulator Inhibitor 1 |
| P-AMPK | Phospho-AMP-activated Protein Kinase |
| Patj | Pals1-associated TJ protein |
| Pax3 | Paired box 3 |
| PCAF | P300/CBP-Associated Factor |
| PCNA | Proliferating Cell Nuclear Antigen |
| PDGFRb | Platelet Derived Growth Factor Receptor b |
| PHGDH | 3-Phosphoglycerate Dehydrogenase |
| Plk2 | Polo-Like Kinase-2 |
| PML | ProMyelocytic Leukemia |
| POLD2 | DNA Polymerase Delta subunit 2 |
| PPAR-γ | Peroxisome Proliferator-Activated Receptor γ |
| PTEN | Phosphatase and tensin homolog deleted on chromosome TEN |
| PTPN14 | Tyrosine-Protein Phosphatase Non-receptor type 14 |
| Puma | p53 upregulated modulator of apoptosis |
| Rassf | Ras-association domain family |
| RhoGAPs | Rho GTPase-Activating Proteins |
| RhoGEFs | Rho Guanine nucleotide Exchange Factors |
| RMRP | RNA component of Mitochondrial RNA Processing endoribonuclease |
| ROCK | Rho-associated protein Kinase |
| ROS | Reactive Oxygen Species |
| RUNX1 | Runt-related transcription factor 1 |
| RUNX2 | Runt-related transcription factor 2 |
| Sav1 | Salvador 1 |
| SESN1/2 | Sestrin 1/2 |
| SIRT1 | Sirtuin 1 |
| SNAI2 | Snail family transcriptional Inhibitor 2 |
| SOX2 | Sex determining region Y-box 2 |
| SREBPs | Sterol Regulatory Element Binding Proteins |
| TAFs | TBP Associated Factors |
| TAZ | Tafazzin |
| TBP | TATA-box Binding Proteins |
| Tbx5 | T-box transcription factor 5 |
| TCA | Tricarboxylic Acid |
| TEADs/TEFs | TEA Domain proteins/Transcription Enhancer Factors |
| TFIIH | Transcription Factor II Human |
| TIGAR | TP53-inducible glycolysis and apoptosis regulator |
| TRAIL | TNF-Related Apoptosis-inducing Ligand |
| TSC | Tuberous Sclerosis Complex 2 |
| TSP-1 | Thrombospondin-1 |
| TTF-1 | Thyroid Transcription Factor-1 |
| VEGF | Vascular Endothelial Growth Factor |
| WASP | Wiskott Aldrich Syndrome Protein |
| WWC1/2 | WW domain containing protein 1 and 2 |
| WWOX | WW domain-containing Oxidoreductase 1 |
| YAP | Yes-Associated Protein |
References
- Kress, M.; May, E.; Cassingena, R.; May, P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J. Virol. 1979, 31, 472–483. [Google Scholar] [PubMed]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Linzer, D.I.; Maltzman, W.; Levine, A.J. The SV40 a gene product is required for the production of a 54,000 MW cellular tumor antigen. Virology 1979, 98, 308–318. [Google Scholar] [CrossRef]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Iwakuma, T.; Lozano, G.; Flores, E.R. Li-Fraumeni syndrome: A p53 family affair. Cell Cycle 2005, 4, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; Hall, P.A. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Strano, S.; Rossi, M.; Fontemaggi, G.; Munarriz, E.; Soddu, S.; Sacchi, A.; Blandino, G. From p63 to p53 across p73. FEBS Lett. 2001, 490, 163–170. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Fausti, F.; Di Agostino, S.; Sudol, M.; Blandino, G. PML surfs into Hippo tumor suppressor pathway. Front. Oncol. 2013, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Baena-Lopez, L.A.; Rodriguez, I.; Baonza, A. The tumor suppressor genes dachsous and fat modulate different signalling pathways by regulating dally and dally-like. Proc. Natl. Acad. Sci. USA 2008, 105, 9645–9650. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Feng, Y.; Rauskolb, C.; Maitra, S.; Fehon, R.; Irvine, K.D. Delineation of a Fat tumor suppressor pathway. Nat. Genet. 2006, 38, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Karpowicz, P.; Perez, J.; Perrimon, N. The Hippo tumor suppressor pathway regulates intestinal stem cell regeneration. Development 2010, 137, 4135–4145. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The Hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Guan, K.L. Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol. Sci. 2013, 34, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Xu, Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ. 2003, 10, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Romer, L.; Klein, C.; Dehner, A.; Kessler, H.; Buchner, J. p53—A natural cancer killer: Structural insights and therapeutic concepts. Angew. Chem. 2006, 45, 6440–6460. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Nau, M.M.; Segal, S.; Minna, J.D. p53: A transdominant regulator of transcription whose function is ablated by mutations occurring in human cancer. EMBO J. 1992, 11, 1383–1390. [Google Scholar] [PubMed]
- Zhang, Y.; Xiong, Y. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science 2001, 292, 1910–1915. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Harris, C.C. Tp53 tumour suppressor gene: Clues to molecular carcinogenesis and cancer therapy. Cancer Surv. 1996, 28, 169–196. [Google Scholar] [PubMed]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Thut, C.J.; Chen, J.L.; Klemm, R.; Tjian, R. p53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science 1995, 267, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Deguin-Chambon, V.; Lelong, J.C.; Dessen, P.; May, P.; Debuire, B.; May, E. Further characterisation of the p53 responsive element—Identification of new candidate genes for trans-activation by p53. Oncogene 1997, 14, 85–94. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Mack, D.H.; Vartikar, J.; Pipas, J.M.; Laimins, L.A. Specific repression of TATA-mediated but not initiator-mediated transcription by wild-type p53. Nature 1993, 363, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Usheva, A.; Zambetti, G.P.; Momand, J.; Horikoshi, N.; Weinmann, R.; Levine, A.J.; Shenk, T. Wild-type p53 binds to the TATA-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 1992, 89, 12028–12032. [Google Scholar] [CrossRef] [PubMed]
- Adimoolam, S.; Ford, J.M. p53 and regulation of DNA damage recognition during nucleotide excision repair. Dna Repair 2003, 2, 947–954. [Google Scholar] [CrossRef]
- Zhou, J.X.; Niehans, G.A.; Shar, A.; Rubins, J.B.; Frizelle, S.P.; Kratzke, R.A. Mechanisms of G1 checkpoint loss in resected early stage non-small cell lung cancer. Lung Cancer 2001, 32, 27–38. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Jung-Hynes, B.; Ahmad, N. SIRT1 controls circadian clock circuitry and promotes cell survival: A connection with age-related neoplasms. FASEB J. 2009, 23, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Pines, J. Cell cycle. p21 inhibits cyclin shock. Nature 1994, 369, 520–521. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.Y.; Schwarz, J.K.; Piwnica-Worms, H.; Canman, C.E. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000, 60, 5934–5936. [Google Scholar] [PubMed]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H.; Lengauer, C.; Polyak, K.; He, T.C.; Zhang, L.; Thiagalingam, S.; Kinzler, K.W.; Vogelstein, B. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1997, 1, 3–11. [Google Scholar] [CrossRef]
- Cross, S.M.; Sanchez, C.A.; Morgan, C.A.; Schimke, M.K.; Ramel, S.; Idzerda, R.L.; Raskind, W.H.; Reid, B.J. A p53-dependent mouse spindle checkpoint. Science 1995, 267, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K.; Choi, T.; Kuriyama, R.; Rulong, S.; Vande Woude, G.F. Abnormal centrosome amplification in the absence of p53. Science 1996, 271, 1744–1747. [Google Scholar] [CrossRef] [PubMed]
- Del Sal, G.; Ruaro, E.M.; Utrera, R.; Cole, C.N.; Levine, A.J.; Schneider, C. Gas1-induced growth suppression requires a transactivation-independent p53 function. Mol. Cell. Biol. 1995, 15, 7152–7160. [Google Scholar] [CrossRef] [PubMed]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Oda, K.; Arakawa, H.; Tanaka, T.; Matsuda, K.; Tanikawa, C.; Mori, T.; Nishimori, H.; Tamai, K.; Tokino, T.; Nakamura, Y.; et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 2000, 102, 849–862. [Google Scholar] [CrossRef]
- Mummenbrauer, T.; Janus, F.; Muller, B.; Wiesmuller, L.; Deppert, W.; Grosse, F. p53 Protein exhibits 3′-to-5′ exonuclease activity. Cell 1996, 85, 1089–1099. [Google Scholar] [CrossRef]
- Morris, S.M. A role for p53 in the frequency and mechanism of mutation. Mutat. Res. 2002, 511, 45–62. [Google Scholar] [CrossRef]
- Hainaut, P.; Wiman, K.G. p53, Cell Cycle Arrest and Apoptosis. In 25 Years Of P53 Research; Springer: Berlin, Germany, 2005; pp. 141–163. [Google Scholar]
- Smith, M.L.; Chen, I.T.; Zhan, Q.; Bae, I.; Chen, C.Y.; Gilmer, T.M.; Kastan, M.B.; O’Connor, P.M.; Fornace, A.J., Jr. Interaction of the p53-regulated protein GADD45 with proliferating cell nuclear antigen. Science 1994, 266, 1376–1380. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Buratowski, S.; Svejstrup, J.Q.; Feaver, W.J.; Wu, X.; Kornberg, R.D.; Donahue, T.F.; Friedberg, E.C. The yeast TFB1 and SSL1 genes, which encode subunits of transcription factor IIH, are required for nucleotide excision repair and RNA polymerase II transcription. Mol. Cell. Biol. 1995, 15, 2288–2293. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Svejstrup, J.Q.; Feaver, W.J.; Wu, X.; Kornberg, R.D.; Friedberg, E.C. Transcription factor b (TFIIH) is required during nucleotide-excision repair in yeast. Nature 1994, 368, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Lozano, G. The p53 family grows old. Genes Dev. 2012, 26, 1997–2000. [Google Scholar] [CrossRef] [PubMed]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A. Does p53 affect organismal aging? J. Cell. Physiol. 2002, 192, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Cancer and ageing: Rival demons? Nat. Rev. Cancer 2003, 3, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Di Leonardo, A.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994, 8, 2540–2551. [Google Scholar] [CrossRef] [PubMed]
- Linke, S.P.; Harris, M.P.; Neugebauer, S.E.; Clarkin, K.C.; Shepard, H.M.; Maneval, D.C.; Wahl, G.M. p53-mediated accumulation of hypophosphorylated pRb after the G1 restriction point fails to halt cell cycle progression. Oncogene 1997, 15, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Mathon, N.F.; Lloyd, A.C. Cell senescence and cancer. Nat. Rev. Cancer 2001, 1, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Yonish-Rouach, E.; Resnitzky, D.; Lotem, J.; Sachs, L.; Kimchi, A.; Oren, M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991, 352, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.S.; Vousden, K.H. Complicating the complexity of p53. Carcinogenesis 2005, 26, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [PubMed]
- Chang, N.S.; Doherty, J.; Ensign, A.; Schultz, L.; Hsu, L.J.; Hong, Q. WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J. Biol. Chem. 2005, 280, 43100–43108. [Google Scholar] [CrossRef] [PubMed]
- Buckbinder, L.; Talbott, R.; Velasco-Miguel, S.; Takenaka, I.; Faha, B.; Seizinger, B.R.; Kley, N. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature 1995, 377, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Caelles, C.; Helmberg, A.; Karin, M. p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 1994, 370, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [PubMed]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Lozano, P.; Hixon, M.L.; Wagner, M.W.; Flores, A.I.; Ikawa, S.; Baldwin, A.S., Jr.; Chien, K.R.; Gualberto, A. p53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ. 1999, 10, 295–306. [Google Scholar] [PubMed]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chretien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Kulawiec, M.; Ayyasamy, V.; Singh, K.K. p53 regulates mtDNA copy number and mitocheckpoint pathway. J. Carcinog. 2009, 8, 8. [Google Scholar] [PubMed]
- Lebedeva, M.A.; Eaton, J.S.; Shadel, G.S. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim. Biophys. Acta 2009, 1787, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Okamura, S.; Ng, C.C.; Koyama, K.; Takei, Y.; Arakawa, H.; Monden, M.; Nakamura, Y. Identification of seven genes regulated by wild-type p53 in a colon cancer cell line carrying a well-controlled wild-type p53 expression system. Oncol. Res. 1999, 11, 281–285. [Google Scholar] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. p53 as a regulator of lipid metabolism in cancer. Int. J. Mol. Sci. 2016, 17, 2074. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. p53 activation in adipocytes of obese mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef] [PubMed]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Bist, A.; Fielding, C.J.; Fielding, P.E. p53 regulates caveolin gene transcription, cell cholesterol, and growth by a novel mechanism. Biochemistry 2000, 39, 1966–1972. [Google Scholar] [CrossRef] [PubMed]
- Deisenroth, C.; Itahana, Y.; Tollini, L.; Jin, A.; Zhang, Y. p53-Inducible DHRS3 is an endoplasmic reticulum protein associated with lipid droplet accumulation. J. Biol. Chem. 2011, 286, 28343–28356. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; LaGory, E.L.; Kenzelmann Broz, D.; Bieging, K.T.; Brady, C.A.; Link, N.; Abrams, J.M.; Giaccia, A.J.; Attardi, L.D. Analysis of p53 transactivation domain mutants reveals Acad11 as a metabolic target important for p53 pro-survival function. Cell Rep. 2015, 10, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, R.D.; Rother, K.; Muller, G.A.; Engeland, K. The retinal dehydrogenase/reductase retSDR1/DHRS3 gene is activated by p53 and p63 but not by mutants derived from tumors or EEC/ADULT malformation syndromes. Cell Cycle 2010, 9, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, Y.; Jin, A.; Tikunov, A.P.; Zhou, L.; Tollini, L.A.; Leslie, P.; Kim, T.H.; Li, L.O.; Coleman, R.A.; et al. Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, E2414–E2422. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 2004, 306, 2105–2108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, X.; Gao, X.; Mei, Y.; Wu, M. A new role of p53 in regulating lipid metabolism. J. Mol. Cell Biol. 2013, 5, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Napoli, M.; Flores, E.R. The p53 family orchestrates the regulation of metabolism: Physiological regulation and implications for cancer therapy. Br. J. Cancer 2017, 116, 149–155. [Google Scholar] [CrossRef] [PubMed]
- De Berardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Gadea, G.; Roux, P. Control of cell migration: A tumour suppressor function for p53? Biol. Cell 2006, 98, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Betson, M.; Braga, V.M. Tumor progression: Small GTPases and loss of cell-cell adhesion. Bioessays 2003, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.A.; Golub, T.R.; Lander, E.S.; Hynes, R.O. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000, 406, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Horiuchi, A.; Imai, T.; Wang, C.; Ohira, S.; Feng, Y.; Nikaido, T.; Konishi, I. Up-regulation of small GTPases, RhoA and RhoC, is associated with tumor progression in ovarian carcinoma. Lab. Investig. 2003, 83, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Yoshioka, K.; Akedo, H.; Uehata, M.; Ishizaki, T.; Narumiya, S. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat. Med. 1999, 5, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho GTPases in transformation and metastasis. Adv. Cancer Res. 2002, 84, 57–80. [Google Scholar] [PubMed]
- Comer, K.A.; Dennis, P.A.; Armstrong, L.; Catino, J.J.; Kastan, M.B.; Kumar, C.C. Human smooth muscle α-actin gene is a transcriptional target of the p53 tumor suppressor protein. Oncogene 1998, 16, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Iotsova, V.; Stehelin, D. Down-regulation of fibronectin gene expression by the p53 tumor suppressor protein. Cell Growth Differ. 1996, 7, 629–634. [Google Scholar] [PubMed]
- Mukhopadhyay, D.; Tsiokas, L.; Sukhatme, V.P. Wild-type p53 and v-Src exert opposing influences on human vascular endothelial growth factor gene expression. Cancer Res. 1995, 55, 6161–6165. [Google Scholar] [PubMed]
- Sun, Y.; Sun, Y.; Wenger, L.; Rutter, J.L.; Brinckerhoff, C.E.; Cheung, H.S. Human metalloproteinase-1 (collagenase-1) is a tumor suppressor protein p53 target gene. Ann. N. Y. Acad. Sci. 1999, 878, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Gish, K.; Murphy, M.; Yin, Y.; Notterman, D.; Hoffman, W.H.; Tom, E.; Mack, D.H.; Levine, A.J. The transcriptional program following p53 activation. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S. Cdc42—The centre of polarity. J. Cell Sci. 2004, 117, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Hall, A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell 2001, 106, 489–498. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature 2003, 421, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Lapasset, L.; Gauthier-Rouviere, C.; Roux, P. Regulation of Cdc42-mediated morphological effects: A novel function for p53. EMBO J. 2002, 21, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Roger, L.; Anguille, C.; de Toledo, M.; Gire, V.; Roux, P. TNFα induces sequential activation of Cdc42- and p38/p53-dependent pathways that antagonistically regulate filopodia formation. J. Cell Sci. 2004, 117, 6355–6364. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Zheng, Y. Rho family GTPases cooperate with p53 deletion to promote primary mouse embryonic fibroblast cell invasion. Oncogene 2004, 23, 5577–5585. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Izumi, H.; Onitsuka, T.; Miyamoto, N.; Kashiwagi, E.; Kidani, A.; Hirano, G.; Takahashi, M.; Naito, S.; Kohno, K. Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene 2008, 27, 5543–5553. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.A.; Peeper, D.S. Deregulating EMT and senescence: Double impact by a single twist. Cancer Cell 2008, 14, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Dameron, K.M.; Volpert, O.V.; Tainsky, M.A.; Bouck, N. The p53 tumor suppressor gene inhibits angiogenesis by stimulating the production of thrombospondin. Cold Spring Harb. Symp. Quant. Biol. 1994, 59, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.G.; Parker, A.E.; Zhu, X.; Green, M.R. p53-mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science 2006, 313, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Kemp, C.J.; Donehower, L.A.; Bradley, A.; Balmain, A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 1993, 74, 813–822. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002, 1, 289–298. [Google Scholar] [CrossRef]
- Schmitt, C.A.; McCurrach, M.E.; de Stanchina, E.; Wallace-Brodeur, R.R.; Lowe, S.W. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999, 13, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.C.; Park, S.H.; Timme, T.L.; Ren, C.; Eastham, J.A.; Donehower, L.A.; Bradley, A.; Kadmon, D.; Yang, G. Loss of p53 function leads to metastasis in ras+myc-initiated mouse prostate cancer. Oncogene 1995, 10, 869–879. [Google Scholar] [PubMed]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar] [PubMed]
- Hollstein, M.; Rice, K.; Greenblatt, M.S.; Soussi, T.; Fuchs, R.; Sorlie, T.; Hovig, E.; Smith-Sorensen, B.; Montesano, R.; Harris, C.C. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994, 22, 3551–3555. [Google Scholar] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Lutzker, S.G.; Levine, A.J. A functionally inactive p53 protein in teratocarcinoma cells is activated by either DNA damage or cellular differentiation. Nat. Med. 1996, 2, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Oliver, R.T.; Shamash, J.; Berney, D.M. p53 and MDM2 in germ cell cancer treatment response. J. Clin. Oncol. 2002, 20, 3928–3929. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Soussi, T.; Shomer, B.; Hollstein, M.; Greenblatt, M.; Hovig, E.; Harris, C.C.; Montesano, R. Database of p53 gene somatic mutations in human tumors and cell lines: Updated compilation and future prospects. Nucleic Acids Res. 1997, 25, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Soussi, T.; Thomas, G.; von Brevern, M.C. Bartsch, P53 gene alterations in human tumors: Perspectives for cancer control. Recent Results Cancer Res. 1997, 143, 369–389. [Google Scholar] [PubMed]
- Hussain, S.P.; Harris, C.C. Molecular epidemiology of human cancer: Contribution of mutation spectra studies of tumor suppressor genes. Cancer Res. 1998, 58, 4023–4037. [Google Scholar] [CrossRef]
- Walker, D.R.; Bond, J.P.; Tarone, R.E.; Harris, C.C.; Makalowski, W.; Boguski, M.S.; Greenblatt, M.S. Evolutionary conservation and somatic mutation hotspot maps of p53: Correlation with p53 protein structural and functional features. Oncogene 1999, 18, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Ang, H.C.; Veprintsev, D.B.; Blair, C.M.; Fersht, A.R. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J. Biol. Chem. 2005, 280, 16030–16037. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, A.; Fisher, D.E. p53 in life and death. Clin. Cancer Res. 1996, 2, 435–440. [Google Scholar] [PubMed]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar] [PubMed]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Lisek, K.; del Sal, G. Mutant p53: One, no one, and one hundred thousand. Front. Oncol. 2015, 5, 289. [Google Scholar] [CrossRef] [PubMed]
- Zambetti, G.P.; Levine, A.J. A comparison of the biological activities of wild-type and mutant p53. FASEB J. 1993, 7, 855–865. [Google Scholar] [PubMed]
- Gohler, T.; Jager, S.; Warnecke, G.; Yasuda, H.; Kim, E.; Deppert, W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res. 2005, 33, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef] [PubMed]
- Will, K.; Warnecke, G.; Wiesmuller, L.; Deppert, W. Specific interaction of mutant p53 with regions of matrix attachment region DNA elements (MARs) with a high potential for base-unpairing. Proc. Natl. Acad. Sci. USA 1998, 95, 13681–13686. [Google Scholar] [CrossRef] [PubMed]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.C.; Jost, C.A.; Brooks, L.A.; Irwin, M.S.; O’Nions, J.; Tidy, J.A.; James, N.; McGregor, J.M.; Harwood, C.A.; Yulug, I.G.; et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat. Genet. 2000, 25, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Munarriz, E.; Rossi, M.; Cristofanelli, B.; Shaul, Y.; Castagnoli, L.; Levine, A.J.; Sacchi, A.; Cesareni, G.; Oren, M.; et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J. Biol. Chem. 2000, 275, 29503–29512. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 2001, 276, 15164–15173. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Lozano, G. p53 mutation heterogeneity in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chang, D.; Ying, S.Y. Asymmetry of intronic pre-miRNA structures in functional RISC assembly. Gene 2005, 356, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Matas, D.; Sigal, A.; Stambolsky, P.; Milyavsky, M.; Weisz, L.; Schwartz, D.; Goldfinger, N.; Rotter, V. Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. EMBO J. 2001, 20, 4163–4172. [Google Scholar] [CrossRef] [PubMed]
- Frazier, M.W.; He, X.; Wang, J.; Gu, Z.; Cleveland, J.L.; Zambetti, G.P. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol. Cell. Biol. 1998, 18, 3735–3743. [Google Scholar] [CrossRef] [PubMed]
- Kollareddy, M.; Dimitrova, E.; Vallabhaneni, K.C.; Chan, A.; Le, T.; Chauhan, K.M.; Carrero, Z.I.; Ramakrishnan, G.; Watabe, K.; Haupt, Y.; et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat. Commun. 2015, 6, 7389. [Google Scholar] [CrossRef] [PubMed]
- Mizuarai, S.; Yamanaka, K.; Kotani, H. Mutant p53 induces the GEF-H1 oncogene, a guanine nucleotide exchange factor-H1 for RhoA, resulting in accelerated cell proliferation in tumor cells. Cancer Res. 2006, 66, 6319–6326. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, T.J.; Ghosh, P.; Dobashi, N.; Sasaki, C.Y.; Longo, D.L. Comparison of the effect of mutant and wild-type p53 on global gene expression. Cancer Res. 2004, 64, 8199–8207. [Google Scholar] [CrossRef] [PubMed]
- Scian, M.J.; Stagliano, K.E.; Ellis, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Modulation of gene expression by tumor-derived p53 mutants. Cancer Res. 2004, 64, 7447–7454. [Google Scholar] [CrossRef] [PubMed]
- Weisz, L.; Zalcenstein, A.; Stambolsky, P.; Cohen, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res. 2004, 64, 8318–8327. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Pfister, N.T.; Fomin, V.; Regunath, K.; Zhou, J.Y.; Zhou, W.; Silwal-Pandit, L.; Freed-Pastor, W.A.; Laptenko, O.; Neo, S.P.; Bargonetti, J.; et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 2015, 29, 1298–1315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Zalcenstein, A.; Weisz, L.; Stambolsky, P.; Bar, J.; Rotter, V.; Oren, M. Repression of the Msp/Mst-1 gene contributes to the antiapoptotic gain of function of mutant p53. Oncogene 2006, 25, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Margulies, L.; Sehgal, P.B. Modulation of the human interleukin-6 promoter (IL-6) and transcription factor C/EBP β (NF-IL6) activity by p53 species. J. Biol. Chem. 1993, 268, 15096–15100. [Google Scholar] [PubMed]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Jackson, C.T.; Subler, M.A.; Martin, D.W. Modulation of cellular and viral promoters by mutant human p53 proteins found in tumor cells. J. Virol. 1992, 66, 6164–6170. [Google Scholar] [PubMed]
- Iwanaga, Y.; Jeang, K.T. Expression of mitotic spindle checkpoint protein hsMAD1 correlates with cellular proliferation and is activated by a gain-of-function p53 mutant. Cancer Res. 2002, 62, 2618–2624. [Google Scholar] [PubMed]
- Ludes-Meyers, J.H.; Subler, M.A.; Shivakumar, C.V.; Munoz, R.M.; Jiang, P.; Bigger, J.E.; Brown, D.R.; Deb, S.P.; Deb, S. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol. Cell. Biol. 1996, 16, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Scian, M.J.; Stagliano, K.E.; Anderson, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Tumor-derived p53 mutants induce NF-κB2 gene expression. Mol. Cell. Biol. 2005, 25, 10097–10110. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi-Ishii, Y.; Tadokoro, K.; Hanaoka, F.; Tsuchida, N. Response of heat shock element within the human HSP70 promoter to mutated p53 genes. Cell Growth Differ. 1995, 6, 1–8. [Google Scholar] [PubMed]
- Weisz, L.; Damalas, A.; Liontos, M.; Karakaidos, P.; Fontemaggi, G.; Maor-Aloni, R.; Kalis, M.; Levrero, M.; Strano, S.; Gorgoulis, V.G.; et al. Mutant p53 enhances nuclear factor κB activation by tumor necrosis factor α in cancer cells. Cancer Res. 2007, 67, 2396–2401. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Fontemaggi, G.; Dell’Orso, S.; Muti, P.; Blandino, G.; Strano, S. Id2 gene is a transcriptional target of the protein complex mutant p53/E2F1. Cell Cycle 2010, 9, 2464–2466. [Google Scholar] [CrossRef] [PubMed]
- Fontemaggi, G.; Dell’Orso, S.; Trisciuoglio, D.; Shay, T.; Melucci, E.; Fazi, F.; Terrenato, I.; Mottolese, M.; Muti, P.; Domany, E.; et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol. 2009, 16, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Valenti, F.; Ganci, F.; Fontemaggi, G.; Sacconi, A.; Strano, S.; Blandino, G.; Di Agostino, S. Gain of function mutant p53 proteins cooperate with E2F4 to transcriptionally downregulate RAD17 and BRCA1 gene expression. Oncotarget 2015, 6, 5547–5566. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Wang, Z.; Fu, J.; Ji, L.; Liu, J.; Li, L.; Wang, H.; Chen, J.; Caulin, C.; Myers, J.N.; et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-β signalling and mutant p53 in cancer cells. Nat. Commun. 2013, 4, 2667. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Shah, A.S.; Ahmad, A. Gain-of-function of mutant p53: Mutant p53 enhances cancer progression by inhibiting KLF17 expression in invasive breast carcinoma cells. Cancer Lett. 2014, 354, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Fontemaggi, G.; Fazi, F.; di Agostino, S.; Padula, F.; Biagioni, F.; Muti, P.; Strano, S.; Blandino, G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012, 19, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Strano, S.; Blandino, G. microRNAs: Short non-coding bullets of gain of function mutant p53 proteins. Oncoscience 2014, 1, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Ganci, F.; Sacconi, A.; Bossel Ben-Moshe, N.; Manciocco, V.; Sperduti, I.; Strigari, L.; Covello, R.; Benevolo, M.; Pescarmona, E.; Domany, E.; et al. Expression of Tp53 mutation-associated microRNAs predicts clinical outcome in head and neck squamous cell carcinoma patients. Ann. Oncol. 2013, 24, 3082–3088. [Google Scholar] [CrossRef] [PubMed]
- Masciarelli, S.; Fontemaggi, G.; Di Agostino, S.; Donzelli, S.; Carcarino, E.; Strano, S.; Blandino, G. Gain-of-function mutant p53 downregulates miR-223 contributing to chemoresistance of cultured tumor cells. Oncogene 2014, 33, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Dell’Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant p53: An oncogenic transcription factor. Oncogene 2007, 26, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Gonfloni, S.; Caputo, V.; Iannizzotto, V. P63 in health and cancer. Int. J. Dev. Biol. 2015, 59, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Dell’Orso, S.; Mongiovi, A.M.; Monti, O.; Lapi, E.; Di Agostino, S.; Fontemaggi, G.; Blandino, G. Mutant p53 proteins: Between loss and gain of function. Head Neck 2007, 29, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.K.; Ha, J.H.; Lee, M.S.; Chi, S.W. Structure and apoptotic function of p73. BMB Rep. 2015, 48, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, M.; Di Agostino, S.; Blandino, G.; Strano, S. Oncogenic intra-p53 family member interactions in human cancers. Front. Oncol. 2016, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, A.; Aldape, K.; Kozakiewicz, K.; Tlsty, T.D. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc. Natl. Acad. Sci. USA 1998, 95, 5166–5171. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Kondo, K.; Marin, M.C.; Cheng, L.S.; Hahn, W.C.; Kaelin, W.G., Jr. Chemosensitivity linked to p73 function. Cancer Cell 2003, 3, 403–410. [Google Scholar] [CrossRef]
- Strano, S.; Blandino, G. p73-mediated chemosensitivity: A preferential target of oncogenic mutant p53. Cell Cycle 2003, 2, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; Del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 2002, 277, 18817–18826. [Google Scholar] [CrossRef] [PubMed]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.T.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Deppert, W. The versatile interactions of p53 with DNA: When flexibility serves specificity. Cell Death Differ. 2006, 13, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Deppert, W. Identification of genomic DNA sequences bound by mutant p53 protein (Gly245-->Ser) in vivo. Oncogene 2000, 19, 4178–4183. [Google Scholar] [CrossRef] [PubMed]
- Valenti, F.; Fausti, F.; Biagioni, F.; Shay, T.; Fontemaggi, G.; Domany, E.; Yaffe, M.B.; Strano, S.; Blandino, G.; Di Agostino, S. Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle 2011, 10, 4330–4340. [Google Scholar] [CrossRef] [PubMed]
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010, 17, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Huang, G.; Li, J.; Zhu, J.; Feng, Z.; Hu, W. A novel mutant p53 binding partner BAG5 stabilizes mutant p53 and promotes mutant p53 GOFs in tumorigenesis. Cell Discov. 2016, 2, 16039. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Liu, J.; Zhang, C.; Yu, H.; Wang, J.; Zheng, T.; Liu, L.; Li, J.; Feng, Z.; et al. BAG2 promotes tumorigenesis through enhancing mutant p53 protein levels and function. eLife 2015, 4, e08401. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.C.; Wei, X.; Shimizu, T.; Ramos, E.; Rohrbaugh, M.; Nikolaidis, N.; Ho, L.L.; Li, Y. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 2005, 120, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Staley, B.K.; Irvine, K.D. Hippo signaling in Drosophila: Recent advances and insights. Dev. Dyn. 2012, 241, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.; Schiripo, T.A.; Haber, D.; Hariharan, I.K. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef]
- Ferraiuolo, M.S.S.; Blandino, G. The Hippo Pathway. In Encyclopedia of Cell Biology; Stahl, P., Bradshaw, R., Eds.; Academic Press: Waltham, MA, USA, 2016; Volume 3, pp. 99–106. [Google Scholar]
- Yabuta, N.; Okada, N.; Ito, A.; Hosomi, T.; Nishihara, S.; Sasayama, Y.; Fujimori, A.; Okuzaki, D.; Zhao, H.; Ikawa, M.; et al. Lats2 is an essential mitotic regulator required for the coordination of cell division. J. Biol. Chem. 2007, 282, 19259–19271. [Google Scholar] [CrossRef] [PubMed]
- Yabuta, N.; Fujii, T.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Nishiguchi, H.; Endo, Y.; Toji, S.; Tanaka, H.; Nishimune, Y.; et al. Structure, expression, and chromosome mapping of LATS2, a mammalian homologue of the Drosophila tumor suppressor gene lats/warts. Genomics 2000, 63, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene 1994, 9, 2145–2152. [Google Scholar] [PubMed]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M.; Bork, P.; Einbond, A.; Kastury, K.; Druck, T.; Negrini, M.; Huebner, K.; Lehman, D. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J. Biol. Chem. 1995, 270, 14733–14741. [Google Scholar] [CrossRef] [PubMed]
- Yagi, R.; Chen, L.F.; Shigesada, K.; Murakami, Y.; Ito, Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999, 18, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Lei, Q.Y.; Guan, K.L. The Hippo-YAP pathway: New connections between regulation of organ size and cancer. Curr. Opin. Cell Biol. 2008, 20, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Hwang, E.S.; McManus, M.T.; Amsterdam, A.; Tian, Y.; Kalmukova, R.; Mueller, E.; Benjamin, T.; Spiegelman, B.M.; Sharp, P.A.; et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 2005, 309, 1074–108. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Morrison, C.D.; Liu, P.; Miecznikowski, J.; Bshara, W.; Han, S.; Zhu, Q.; Omilian, A.R.; Li, X.; Zhang, J. TAZ induces growth factor-independent proliferation through activation of EGFR ligand amphiregulin. Cell Cycle 2012, 11, 2922–2930. [Google Scholar] [CrossRef] [PubMed]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.; Zhao, B.; Yu, J.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the Hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hayashi, T.; Pan, W.W.; Fujita, Y.; Holt, M.V.; Qin, J.; Carson, D.A.; Guan, K.L. The Hippo pathway kinases LATS1/2 suppress cancer immunity. Cell 2016, 167, 1525–1539 e17. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Conrad, C.; Xia, F.; Park, J.S.; Payer, B.; Yin, Y.; Lauwers, G.Y.; Thasler, W.; Lee, J.T.; Avruch, J.; et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009, 16, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Hwang, K.L.; Brown, K.K.; Evason, K.J.; Beltz, S.; Tsomides, A.; O’Connor, K.; Galli, G.G.; Yimlamai, D.; Chhangawala, S.; et al. Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat. Cell Biol. 2016, 18, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Valis, K.; Talacko, P.; Grobarova, V.; Cerny, J.; Novak, P. Shikonin regulates C-MYC and GLUT1 expression through the MST1-YAP1-TEAD1 axis. Exp. Cell Res. 2016, 349, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; de Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, A.M.; Ludwig, R.L.; Vousden, K.H. Cytoplasmic ASPP1 inhibits apoptosis through the control of YAP. Genes Dev. 2010, 24, 2430–2439. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Chen, J.; Feng, H.; Peng, S.; Adams, U.; Bai, Y.; Huang, L.; Li, J.; Huang, J.; Meng, S.; et al. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res. 2013, 73, 3615–3624. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Fallahi, E.; O’Driscoll, N.A.; Matallanas, D. The MST/Hippo pathway and cell death: A non-canonical affair. Genes 2016, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Lo Sardo, F.; Forcato, M.; Sacconi, A.; Capaci, V.; Zanconato, F.; Di Agostino, S.; Del Sal, G.; Pandolfi, P.P.; Strano, S.; Bicciato, S.; et al. MCM7 and its hosted miR-25, 93 and 106b cluster elicit YAP/TAZ oncogenic activity in lung cancer. Carcinogenesis 2017, 38, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Chaulk, S.G.; Lattanzi, V.J.; Hiemer, S.E.; Fahlman, R.P.; Varelas, X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J. Biol. Chem. 2014, 289, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Qiao, Y.; Tang, X.; Ma, L.; Wang, Y.; Zhang, X.; Weng, W.; Pan, Q.; Yu, Y.; Sun, F.; et al. Tumor suppressor long non-coding RNA, MT1DP is negatively regulated by YAP and Runx2 to inhibit FoxA1 in liver cancer cells. Cell. Signal. 2014, 26, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jeong, S. Wnt activated β-catenin and YAP proteins enhance the expression of non-coding RNA component of RNase MRP in colon cancer cells. Oncotarget 2015, 6, 34658–34668. [Google Scholar] [PubMed]
- Aylon, Y.; Gershoni, A.; Rotkopf, R.; Biton, I.E.; Porat, Z.; Koh, A.P.; Sun, X.; Lee, Y.; Fiel, M.I.; Hoshida, Y.; et al. The LATS2 tumor suppressor inhibits SREBP and suppresses hepatic cholesterol accumulation. Genes Dev. 2016, 30, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Zhang, C.; Liang, N.; Zhang, Z.; Chang, A.; Yin, J.; Li, Z.; Li, N.; Tan, X.; Luo, N.; et al. Yes-associated protein (YAP) increases chemosensitivity of hepatocellular carcinoma cells by modulation of p53. Cancer Biol. Ther. 2013, 14, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Sarver, A.; Tovy, A.; Ainbinder, E.; Oren, M. Lats2 is critical for the pluripotency and proper differentiation of stem cells. Cell Death Differ. 2014, 21, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Ofir-Rosenfeld, Y.; Yabuta, N.; Lapi, E.; Nojima, H.; Lu, X.; Oren, M. The Lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of ASPP1. Genes Dev. 2010, 24, 2420–2429. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 2003, 11, 11–23. [Google Scholar] [CrossRef]
- Downward, J.; Basu, S. YAP and p73: A complex affair. Mol. Cell. Biol. 2008, 32, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Lapi, E.; Di Agostino, S.; Donzelli, S.; Gal, H.; Domany, E.; Rechavi, G.; Pandolfi, P.P.; Givol, D.; Strano, S.; Lu, X.; et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol. Cell 2008, 32, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Fausti, F.; Di Agostino, S.; Cioce, M.; Bielli, P.; Sette, C.; Pandolfi, P.; Oren, M.; Sudol, M.; Strano, S.; Blandino, G. ATM kinase enables the functional axis of YAP, PML and p53 to ameliorate loss of Werner protein-mediated oncogenic senescence. Cell Death Differ. 2013, 20, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Escoll, M.; Gargini, R.; Cuadrado, A.; Anton, I.M.; Wandosell, F. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. The Hippo pathway, p53 and cholesterol. Cell Cycle 2016, 15, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Ishikawa, T.; Sirianni, R.; Tang, H.; McDonald, J.G.; Yuhanna, I.S.; Thompson, B.; Girard, L.; Mineo, C.; Brekken, R.A.; et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep. 2013, 5, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Salomoni, P.; Luo, J.; Shih, A.; Zhong, S.; Gu, W.; Pandolfi, P.P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000, 2, 730–736. [Google Scholar] [PubMed]
- Pearson, M.; Pelicci, P.G. PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene 2001, 20, 7250–7256. [Google Scholar] [CrossRef] [PubMed]
- Girardini, J.E.; del Sal, G. Improving pharmacological rescue of p53 function: RITA targets mutant p53. Cell Cycle 2010, 9, 2062. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Patlolla, J.M.; Qian, L.; Zhang, Y.; Brewer, M.; Mohammed, A.; Desai, D.; Amin, S.; Lightfoot, S.; Kopelovich, L. Chemopreventive effects of the p53-modulating agents CP-31398 and Prima-1 in tobacco carcinogen-induced lung tumorigenesis in A/J mice. Neoplasia 2013, 15, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Cortese, G.; Monti, O.; Dell’Orso, S.; Sacchi, A.; Eisenstein, M.; Citro, G.; Strano, S.; Blandino, G. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle 2008, 7, 3440–3447. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Chen, Y.; Chen, M.H.; Chen, G.; Chang, H. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Br. J. Cancer 2014, 110, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, F.T.; Chade, D.C.; Reis, S.T.; Piantino, C.; Dall’ Oglio, M.F.; Srougi, M.; Leite, K.R. Curcumin, but not Prima-1, decreased tumor cell proliferation in the syngeneic murine orthotopic bladder tumor model. Clinics 2011, 66, 2121–2124. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J. PRIMA-1 as a cancer therapy restoring mutant p53: A review. Biosciencehorizons 2015, 8, 1–5. [Google Scholar] [CrossRef]
- Bou-Hanna, C.; Jarry, A.; Lode, L.; Schmitz, I.; Schulze-Osthoff, K.; Kury, S.; Bezieau, S.; Mosnier, J.F.; Laboisse, C.L. Acute cytotoxicity of MIRA-1/NSC19630, a mutant p53-reactivating small molecule, against human normal and cancer cells via a caspase-9-dependent apoptosis. Cancer Lett. 2015, 359, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Piantino, C.B.; Reis, S.T.; Viana, N.I.; Silva, I.A.; Morais, D.R.; Antunes, A.A.; Dip, N.; Srougi, M.; Leite, K.R. Prima-1 induces apoptosis in bladder cancer cell lines by activating p53. Clinics 2013, 68, 297–303. [Google Scholar] [CrossRef]
- Saha, M.N.; Jiang, H.; Yang, Y.; Reece, D.; Chang, H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol. Cancer Ther. 2013, 12, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Feuerstein, R.; Serror, L.; Aberdam, E.; Muller, F.J.; van Bokhoven, H.; Wiman, K.G.; Zhou, H.; Aberdam, D.; Petit, I. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc. Natl. Acad. Sci. USA 2013, 110, 2152–2156. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, M.; Abdi, J.; Manujendra, S.N.; Chen, C.; Chang, H. PRIMA-1Met induces apoptosis in Waldenstrom’s Macroglobulinemia cells independent of p53. Cancer Biol. Ther. 2015, 16, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Zache, N.; Lambert, J.M.; Wiman, K.G.; Bykov, V.J. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell. Oncol. 2008, 30, 411–418. [Google Scholar] [PubMed]
- Zhang, S.; Zhou, L.; Hong, B.; van den Heuvel, A.P.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Small-molecule NSC59984 restores p53 pathway signaling and antitumor effects against colorectal cancer via p73 activation and degradation of mutant p53. Cancer Res. 2015, 75, 3842–3852. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.; Prives, C. Targeting mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio. 2015, 5, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Ying, M.Y. Small molecule drug Verteporfin inhibits TAZ/YAP-driven signaling and tumorigenicity of breast cancer cells. Cancer Res. 2015, 75. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Cordenonsi, M.; Piccolo, S. YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 2016, 29, 26–33. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferraiuolo, M.; Verduci, L.; Blandino, G.; Strano, S. Mutant p53 Protein and the Hippo Transducers YAP and TAZ: A Critical Oncogenic Node in Human Cancers. Int. J. Mol. Sci. 2017, 18, 961. https://doi.org/10.3390/ijms18050961
Ferraiuolo M, Verduci L, Blandino G, Strano S. Mutant p53 Protein and the Hippo Transducers YAP and TAZ: A Critical Oncogenic Node in Human Cancers. International Journal of Molecular Sciences. 2017; 18(5):961. https://doi.org/10.3390/ijms18050961
Chicago/Turabian StyleFerraiuolo, Maria, Lorena Verduci, Giovanni Blandino, and Sabrina Strano. 2017. "Mutant p53 Protein and the Hippo Transducers YAP and TAZ: A Critical Oncogenic Node in Human Cancers" International Journal of Molecular Sciences 18, no. 5: 961. https://doi.org/10.3390/ijms18050961
APA StyleFerraiuolo, M., Verduci, L., Blandino, G., & Strano, S. (2017). Mutant p53 Protein and the Hippo Transducers YAP and TAZ: A Critical Oncogenic Node in Human Cancers. International Journal of Molecular Sciences, 18(5), 961. https://doi.org/10.3390/ijms18050961