New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis

Abstract
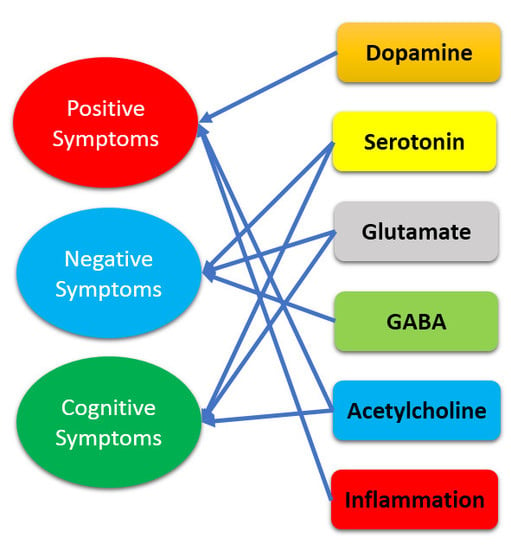
1. Introduction
2. Dopamine Hypothesis and Beyond
2.1. Glutamate
2.2. Serotonin
2.3. Acetylcholine
2.4. Gamma-Aminobutyric Acid
2.5. Inflammation
2.6. Summary of the Hypothesis beyond Dopamine Mechanism
3. Novel Treatment Strategies
3.1. Specific Treatments for Negative Symptoms
3.2. Specific Treatments for Cognitive Deficits
3.3. Specific Treatment for Different Phases of the Illness
4. Novel Treatment Targets
4.1. Dopaminergic Antagonists and Stabilizers
4.2. Glutamatergic Agents
4.3. Serotonin Agents
4.4. Gamma-Aminobutyric Acid (GABA) Allosteric Modulators
4.5. Cholinergic Agonists
4.6. Neuropeptides
4.7. Anti-Inflammatory Approaches
4.8. Genetic-Based Approaches
4.9. Other Approaches
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Howes, O.D.; Kambeitz, J.; Kim, E.; Stahl, D.; Slifstein, M.; Abi-Dargham, A.; Kapur, S. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch. Gen. Psychiatry 2012, 69, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S. Psychosis as a state of aberrant salience: A framework linking biology, phenomenology, and pharmacology in schizophrenia. Am. J. Psychiatry 2003, 160, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Berridge, K.C. From prediction error to incentive salience: Mesolimbic computation of reward motivation. Eur. J. Neurosci. 2012, 35, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Kapur, S. The dopamine hypothesis of schizophrenia: Version III—The final common pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Laruelle, M.; Abi-Dargham, A.; van Dyck, C.H.; Gil, R.; D’Souza, C.D.; Erdos, J.; McCance, E.; Rosenblatt, W.; Fingado, C.; Zoghbi, S.S.; et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc. Natl. Acad. Sci. USA 1996, 93, 9235–9240. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Williams, M.; Ibrahim, K.; Leung, G.; Egerton, A.; McGuire, P.K.; Turkheimer, F. Midbrain dopamine function in schizophrenia and depression: A post-mortem and positron emission tomographic imaging study. Brain 2013, 136 Pt 11, 3242–3251. [Google Scholar] [CrossRef] [PubMed]
- Egerton, A.; Chaddock, C.A.; Winton-Brown, T.T.; Bloomfield, M.A.; Bhattacharyya, S.; Allen, P.; McGuire, P.K.; Howes, O.D. Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: Findings in a second cohort. Biol. Psychiatry 2013, 74, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.C.; Bacanu, S.A.; Bigdeli, T.B.; Moscati, A.; Kendler, K.S. Evaluating the dopamine hypothesis of schizophrenia in a large-scale genome-wide association study. Schizophr. Res. 2016, 176, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, P.C.; Frith, C.D. Perceiving is believing: A Bayesian approach to explaining the positive symptoms of schizophrenia. Nat. Rev. 2009, 10, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Urs, N.M.; Peterson, S.M.; Caron, M.G. New Concepts in Dopamine D2 Receptor Biased Signaling and Implications for Schizophrenia Therapy. Biol. Psychiatry 2017, 81, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Moran, R.J.; Jones, M.W.; Blockeel, A.J.; Adams, R.A.; Stephan, K.E.; Friston, K.J. Losing control under ketamine: Suppressed cortico-hippocampal drive following acute ketamine in rats. Neuropsychopharmacology 2015, 40, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Glantz, L.A.; Lewis, D.A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 2000, 57, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Grunder, G.; Cumming, P. The dopamine hypothesis of schizophrenia: Current status. In The Neurobiology of Schizophrenia; Abel, T., Nickl-Jockschat, T., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 109–124. [Google Scholar]
- Moghaddam, B. Bringing order to the glutamate chaos in schizophrenia. Neuron 2003, 40, 881–884. [Google Scholar] [CrossRef]
- Olney, J.W.; Newcomer, J.W.; Farber, N.B. NMDA receptor hypofunction model of schizophrenia. J. Psychiatry Res. 1999, 33, 523–533. [Google Scholar] [CrossRef]
- Goff, D.C.; Coyle, J.T. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am. J. Psychiatry 2001, 158, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kornhuber, H.H.; Schmid-Burgk, W.; Holzmuller, B. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci. Lett. 1980, 20, 379–382. [Google Scholar] [CrossRef]
- Farber, N.B. The NMDA receptor hypofunction model of psychosis. Ann. N. Y. Acad. Sci. 2003, 1003, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, G.K.; Marek, G.J. Serotonin model of schizophrenia: Emerging role of glutamate mechanisms. Brain Res. 2000, 31, 302–312. [Google Scholar] [CrossRef]
- Kapur, S.; Remington, G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am. J. Psychiatry 1996, 153, 466–476. [Google Scholar] [PubMed]
- Abi-Dargham, A. Alterations of serotonin transmission in schizophrenia. Int. Rev. Neurobiol. 2007, 78, 133–164. [Google Scholar] [PubMed]
- Dalack, G.W.; Becks, L.; Hill, E.; Pomerleau, O.F.; Meador-Woodruff, J.H. Nicotine withdrawal and psychiatric symptoms in cigarette smokers with schizophrenia. Neuropsychopharmacology 1999, 21, 195–202. [Google Scholar] [CrossRef]
- Tandon, R. Cholinergic aspects of schizophrenia. Br. J. Psychiatry 1999, 174 (Suppl. S37), 7–11. [Google Scholar]
- Raedler, T.J.; Bymaster, F.P.; Tandon, R.; Copolov, D.; Dean, B. Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry 2007, 12, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Brunzell, D.H.; McIntosh, J.M. α7 nicotinic acetylcholine receptors modulate motivation to self-administer nicotine: Implications for smoking and schizophrenia. Neuropsychopharmacology 2012, 37, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Forchuk, C.; Norman, R.; Malla, A.; Martin, M.L.; McLean, T.; Cheng, S.; Diaz, K.; McIntosh, E.; Rickwood, A.; Vos, S.; et al. Schizophrenia and the motivation for smoking. Perspect. Psychiatr. Care 2002, 38, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.; Olincy, A.; Ross, R.G.; Waldo, M.C.; Stevens, K.E.; Adler, L.E.; Leonard, S. The genetics of sensory gating deficits in schizophrenia. Curr. Psychiatry Rep. 2003, 5, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Olincy, A.; Stevens, K.E. Treating schizophrenia symptoms with an α7 nicotinic agonist, from mice to men. Biochem. Pharmacol. 2007, 74, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M. The GABA system in schizophrenia: Cells, molecules and microcircuitry. Schizophr. Res. 2015, 167, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Garbutt, J.C.; van Kammen, D.P. The interaction between GABA and dopamine: Implications for schizophrenia. Schizophr. Bull. 1983, 9, 336–353. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Lim, B.; Matzilevich, D.; Walsh, J.P.; Subburaju, S.; Minns, M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc. Natl. Acad. Sci. USA 2007, 104, 10164–10169. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Singer, W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. 2010, 11, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Cardin, J.A.; Carlen, M.; Meletis, K.; Knoblich, U.; Zhang, F.; Deisseroth, K.; Tsai, L.H.; Moore, C.I. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 2009, 459, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Gonzalez-Burgos, G. Pathophysiologically based treatment interventions in schizophrenia. Nat. Med. 2006, 12, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Wassef, A.; Baker, J.; Kochan, L.D. GABA and schizophrenia: A review of basic science and clinical studies. J. Clin. Psychopharmacol. 2003, 23, 601–640. [Google Scholar] [CrossRef] [PubMed]
- Feigenson, K.A.; Kusnecov, A.W.; Silverstein, S.M. Inflammation and the two-hit hypothesis of schizophrenia. Neurosci. Biobehav. Rev. 2014, 38, 72–93. [Google Scholar] [CrossRef] [PubMed]
- Na, K.S.; Jung, H.Y.; Kim, Y.K. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2014, 48, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Mayilyan, K.R.; Weinberger, D.R.; Sim, R.B. The complement system in schizophrenia. Drug News Perspect. 2008, 21, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Oga, T.; Nakagaki, K.; Sakai, K.; Sumida, K.; Hoshino, K.; Miyawaki, I.; Saito, K.; Suto, F.; Ichinohe, N. Developmental expression profiles of axon guidance signaling and the immune system in the marmoset cortex: Potential molecular mechanisms of pruning of dendritic spines during primate synapse formation in late infancy and prepuberty (I). Biochem. Biophys. Res. Commun. 2014, 444, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Ann. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Kayser, M.S.; Dalmau, J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr. Res. 2016, 176, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef]
- Jackson, J.; Eaton, W.; Cascella, N.; Fasano, A.; Warfel, D.; Feldman, S.; Richardson, C.; Vyas, G.; Linthicum, J.; Santora, D.; et al. A gluten-free diet in people with schizophrenia and anti-tissue transglutaminase or anti-gliadin antibodies. Schizophr. Res. 2012, 140, 262–263. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Tomasetti, C.; Buonaguro, E.F.; de Bartolomeis, A. The glutamatergic aspects of schizophrenia molecular pathophysiology: Role of the postsynaptic density, and implications for treatment. Curr. Neuropharmacol. 2014, 12, 219–238. [Google Scholar] [CrossRef] [PubMed]
- De Bartolomeis, A.; Buonaguro, E.F.; Iasevoli, F. Serotonin-glutamate and serotonin-dopamine reciprocal interactions as putative molecular targets for novel antipsychotic treatments: From receptor heterodimers to postsynaptic scaffolding and effector proteins. Psychopharmacology 2013, 225, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Steullet, P.; Cabungcal, J.H.; Monin, A.; Dwir, D.; O’Donnell, P.; Cuenod, M.; Do, K.Q. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: A “central hub” in schizophrenia pathophysiology? Schizophr. Res. 2016, 176, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.; Morris, B.J.; Pratt, J.A. Functional brain connectivity phenotypes for schizophrenia drug discovery. J. Psychopharmacol. 2015, 29, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Mirnics, K. Schizophrenia as a disorder of molecular pathways. Biol. Psychiatry 2015, 77, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, J.L.; Tandon, N.; Haller, C.S.; Mathew, I.T.; Eack, S.M.; Clementz, B.A.; Pearlson, G.D.; Sweeney, J.A.; Tamminga, C.A.; Keshavan, M.S. Correlations between brain structure and symptom dimensions of psychosis in schizophrenia, schizoaffective, and psychotic bipolar I disorders. Schizophr. Bull. 2015, 41, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.G.; Steiner, J.; Guest, P.C.; Dobrowolny, H.; Bogerts, B. Glial cells as key players in schizophrenia pathology: Recent insights and concepts of therapy. Schizophr. Res. 2015, 161, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Trent, S.; Thomas, K.L.; O’Donovan, M.C.; Owen, M.J. Genetic risk for schizophrenia: Convergence on synaptic pathways involved in plasticity. Biol. Psychiatry 2015, 77, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Pickard, B.S. Schizophrenia biomarkers: Translating the descriptive into the diagnostic. J. Psychopharmacol. 2015, 29, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr. Res. 2010, 122, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Gaebel, W.; Barch, D.M.; Bustillo, J.; Gur, R.E.; Heckers, S.; Malaspina, D.; Owen, M.J.; Schultz, S.; Tsuang, M.; et al. Definition and description of schizophrenia in the DSM-5. Schizophr. Res. 2013, 150, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, B.; Fenton, W.S.; Carpenter, W.T., Jr.; Marder, S.R. The NIMH-MATRICS consensus statement on negative symptoms. Schizophr. Bull. 2006, 32, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.C.; Horan, W.P.; Marder, S.R. Psychopharmacology of the negative symptoms: Current status and prospects for progress. Eur. Neuropsychopharmacol. 2014, 24, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Umbricht, D.; Alberati, D.; Martin-Facklam, M.; Borroni, E.; Youssef, E.A.; Ostland, M.; Wallace, T.L.; Knoflach, F.; Dorflinger, E.; Wettstein, J.G.; et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: A randomized, double-blind, proof-of-concept study. JAMA Psychiatry 2014, 71, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, V.L.; Millen, B.A.; Andersen, S.; Kinon, B.J.; Lagrandeur, L.; Lindenmayer, J.P.; Gomez, J.C. Pomaglumetad methionil: No significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr. Res. 2013, 150, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.K.; Byun, N.; Bubser, M. Muscarinic and nicotinic acetylcholine receptor agonists and allosteric modulators for the treatment of schizophrenia. Neuropsychopharmacology 2012, 37, 16–42. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, I.B.; Hallak, J.; Husain, N.; Minhas, F.; Stirling, J.; Richardson, P.; Dursun, S.; Dunn, G.; Deakin, B. Minocycline benefits negative symptoms in early schizophrenia: A randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J. Psychopharmacol. 2012, 26, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Modabbernia, A.; Rezaei, F.; Salehi, B.; Jafarinia, M.; Ashrafi, M.; Tabrizi, M.; Hosseini, S.M.; Tajdini, M.; Ghaleiha, A.; Akhondzadeh, S. Intranasal oxytocin as an adjunct to risperidone in patients with schizophrenia: An 8-week, randomized, double-blind, placebo-controlled study. CNS Drugs 2013, 27, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Usall, J.; Huerta-Ramos, E.; Iniesta, R.; Cobo, J.; Araya, S.; Roca, M.; Serrano-Blanco, A.; Teba, F.; Ochoa, S. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: A double-blind, randomized, placebo-controlled trial. J. Clin. Psychiatry 2011, 72, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.E.; Bradford, D.W.; Hamer, R.M.; Naylor, J.C.; Allen, T.B.; Lieberman, J.A.; Strauss, J.L.; Kilts, J.D. Pregnenolone as a novel therapeutic candidate in schizophrenia: Emerging preclinical and clinical evidence. Neuroscience 2011, 191, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Green, M.F. Cognitive impairment and functional outcome in schizophrenia and bipolar disorder. J. Clin. Psychiatry 2006, 67 (Suppl. S9), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Paulman, R.G.; Devous, M.D., Sr.; Gregory, R.R.; Herman, J.H.; Jennings, L.; Bonte, F.J.; Nasrallah, H.A.; Raese, J.D. Hypofrontality and cognitive impairment in schizophrenia: Dynamic single-photon tomography and neuropsychological assessment of schizophrenic brain function. Biol. Psychiatry 1990, 27, 377–399. [Google Scholar] [CrossRef]
- Ibrahim, H.M.; Tamminga, C.A. Treating impaired cognition in schizophrenia. Curr. Pharm. Biotechnol. 2012, 13, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.R.; Mercer, L.; Casetti, V.; Sendt, K.V.; Giaroli, G.; Shergill, S.S.; Tracy, D.K. Dementia praecox redux: A systematic review of the nicotinic receptor as a target for cognitive symptoms of schizophrenia. J. Psychopharmacol. 2015, 29, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Keshavan, M.S.; Tandon, R.; Boutros, N.N.; Nasrallah, H.A. Schizophrenia, “just the facts”: What we know in 2008 Part 3: Neurobiology. Schizophr. Res. 2008, 106, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Amminger, G.P.; Schafer, M.R.; Papageorgiou, K.; Klier, C.M.; Cotton, S.M.; Harrigan, S.M.; Mackinnon, A.; McGorry, P.D.; Berger, G.E. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: A randomized, placebo-controlled trial. Arch. Gen. Psychiatry 2010, 67, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Tsuang, M.T.; Van Os, J.; Tandon, R.; Barch, D.M.; Bustillo, J.; Gaebel, W.; Gur, R.E.; Heckers, S.; Malaspina, D.; Owen, M.J.; et al. Attenuated psychosis syndrome in DSM-5. Schizophr. Res. 2013, 150, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.; Maes, M.; Anderson, G.; Dean, O.M.; Moylan, S.; Berk, M. Putative neuroprotective agents in neuropsychiatric disorders. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2013, 42, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Isaacson, S.; Mills, R.; Williams, H.; Chi-Burris, K.; Corbett, A.; Dhall, R.; Ballard, C. Pimavanserin for patients with Parkinson’s disease psychosis: A randomised, placebo-controlled phase 3 trial. Lancet 2014, 383, 533–540. [Google Scholar] [CrossRef]
- Alberati, D.; Moreau, J.L.; Lengyel, J.; Hauser, N.; Mory, R.; Borroni, E.; Pinard, E.; Knoflach, F.; Schlotterbeck, G.; Hainzl, D.; et al. Glycine reuptake inhibitor RG1678: A pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology 2012, 62, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Chaki, S.; Hikichi, H. Targeting of metabotropic glutamate receptors for the treatment of schizophrenia. Curr. Pharm. Des. 2011, 17, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Satow, A.; Maehara, S.; Ise, S.; Hikichi, H.; Fukushima, M.; Suzuki, G.; Kimura, T.; Tanak, T.; Ito, S.; Kawamoto, H.; et al. Pharmacological effects of the metabotropic glutamate receptor 1 antagonist compared with those of the metabotropic glutamate receptor 5 antagonist and metabotropic glutamate receptor 2/3 agonist in rodents: Detailed investigations with a selective allosteric metabotropic glutamate receptor 1 antagonist, FTIDC [4-[1-(2-fluoropyridine-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methy l-3,6-dihydropyridine-1(2H)-carboxamide]. J. Pharmacol. Exp. Ther. 2008, 326, 577–586. [Google Scholar] [PubMed]
- Moghaddam, B. Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacology 2004, 174, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.T.; Zhang, L.; Martenyi, F.; Lowe, S.L.; Jackson, K.A.; Andreev, B.V.; Avedisova, A.S.; Bardenstein, L.M.; Gurovich, I.Y.; Morozova, M.A.; et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: A randomized Phase 2 clinical trial. Nat. Med. 2007, 13, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Kinon, B.J.; Zhang, L.; Millen, B.A.; Osuntokun, O.O.; Williams, J.E.; Kollack-Walker, S.; Jackson, K.; Kryzhanovskaya, L.; Jarkova, N.; Group, H.S. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J. Clin. Psychopharmacol. 2011, 31, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C. Drug development in schizophrenia: Are glutamatergic targets still worth aiming at? Curr. Opin. Psychiatry 2015, 28, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Downing, A.C.; Munsie, L.M.; Chen, P.; Reed, M.R.; Ruble, C.L.; Landschulz, K.T.; Kinon, B.J.; Nisenbaum, L.K. Pharmacogenetic analysis of the mGlu2/3 agonist LY2140023 monohydrate in the treatment of schizophrenia. Pharm. J. 2012, 12, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Garay, R.P.; Bourin, M.; de Paillette, E.; Samalin, L.; Hameg, A.; Llorca, P.M. Potential serotonergic agents for the treatment of schizophrenia. Expert Opin. Investig. Drugs 2016, 25, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, B.A.; Prinssen, E.P. Can 5-HT3 antagonists contribute toward the treatment of schizophrenia? Behav. Pharmacol. 2015, 26, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, I.B.; Husain, N.; Drake, R.; Dunn, G.; Husain, M.O.; Kazmi, A.; Hamirani, M.M.; Rahman, R.; Stirling, J.; Deakin, W. Add-on clinical effects of simvastatin and ondansetron in patients with schizophrenia stabilized on antipsychotic treatment: Pilot study. Ther. Adv. Psychopharmacol. 2014, 4, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Mukai, T.; Matsuda, Y.; Iwata, N. Selective serotonin 3 receptor antagonist treatment for schizophrenia: Meta-analysis and systematic review. Neuromol. Med. 2014, 16, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Samadi, R.; Soluti, S.; Daneshmand, R.; Assari, S.; Manteghi, A.A. Efficacy of Risperidone Augmentation with Ondansetron in the Treatment of Negative and Depressive Symptoms in Schizophrenia: A Randomized Clinical Trial. Iran. J. Med. Sci. 2017, 42, 14–23. [Google Scholar] [PubMed]
- Gill, K.M.; Grace, A.A. The role of α5 GABAA receptor agonists in the treatment of cognitive deficits in schizophrenia. Curr. Pharm. Des. 2014, 20, 5069–5076. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; DeQuardo, J.R.; Goodson, J.; Mann, N.A.; Greden, J.F. Effect of anticholinergics on positive and negative symptoms in schizophrenia. Psychopharmacol. Bull. 1992, 28, 297–302. [Google Scholar] [PubMed]
- Freedman, R. α7-nicotinic acetylcholine receptor agonists for cognitive enhancement in schizophrenia. Ann. Rev. Med. 2014, 65, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Dencker, D.; Thomsen, M.; Wortwein, G.; Weikop, P.; Cui, Y.; Jeon, J.; Wess, J.; Fink-Jensen, A. Muscarinic Acetylcholine Receptor Subtypes as Potential Drug Targets for the Treatment of Schizophrenia, Drug Abuse and Parkinson’s Disease. ACS Chem. Neurosci. 2012, 3, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, A.; Dean, B. The Cholinergic System: An Emerging Drug Target for Schizophrenia. Curr. Pharm. Des. 2016, 22, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Money, T.T.; Scarr, E.; Udawela, M.; Gibbons, A.S.; Jeon, W.J.; Seo, M.S.; Dean, B. Treating schizophrenia: Novel targets for the cholinergic system. CNS Neurol. Disord. Drug Targets 2010, 9, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E.; Dean, B. Role of the cholinergic system in the pathology and treatment of schizophrenia. Expert Rev. Neurother. 2009, 9, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.A.; Green, M.C. The use of cholecystokinin in schizophrenia: A review. Psychol. Med. 1988, 18, 593–603. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, B.C.; Pushpa-Rajah, J.A.; Gillies, D.; Rathbone, J.; Variend, H.; Kalakouti, E.; Kyprianou, K. Cannabis and schizophrenia. Cochrane Database Syst. Rev. 2014, CD004837. [Google Scholar] [CrossRef]
- Pushpa-Rajah, J.A.; McLoughlin, B.C.; Gillies, D.; Rathbone, J.; Variend, H.; Kalakouti, E.; Kyprianou, K. Cannabis and schizophrenia. Schizophr. Bull. 2015, 41, 336–337. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.C.; Sewell, R.A.; Ranganathan, M. Cannabis and psychosis/schizophrenia: Human studies. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Zuardi, A.W.; Crippa, J.A.; Hallak, J.E.; Bhattacharyya, S.; Atakan, Z.; Martin-Santos, R.; McGuire, P.K.; Guimaraes, F.S. A critical review of the antipsychotic effects of cannabidiol: 30 years of a translational investigation. Curr. Pharm. Des. 2012, 18, 5131–5140. [Google Scholar] [CrossRef] [PubMed]
- Caceda, R.; Kinkead, B.; Nemeroff, C.B. Neurotensin: Role in psychiatric and neurological diseases. Peptides 2006, 27, 2385–2404. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Na, K.S.; Myint, A.M.; Leonard, B.E. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2016, 64, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Kenk, M.; Selvanathan, T.; Rao, N.; Suridjan, I.; Rusjan, P.; Remington, G.; Meyer, J.H.; Wilson, A.A.; Houle, S.; Mizrahi, R. Imaging neuroinflammation in gray and white matter in schizophrenia: An in vivo PET study with [18F]-FEPPA. Schizophr. Bull. 2015, 41, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Krause, D.; Dehning, S.; Musil, R.; Schennach-Wolff, R.; Obermeier, M.; Moller, H.J.; Klauss, V.; Schwarz, M.J.; Riedel, M. Celecoxib treatment in an early stage of schizophrenia: Results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr. Res. 2010, 121, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Javitt, D. From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Li, S.; Chen, S.; Lipina, T.V.; Wang, M.; Lai, T.K.; Lee, F.H.; Zhang, H.; Zhai, D.; Ferguson, S.S.; et al. A dopamine D2 receptor-DISC1 protein complex may contribute to antipsychotic-like effects. Neuron 2014, 84, 1302–1316. [Google Scholar] [CrossRef] [PubMed]
- Feldcamp, L.A.; Boutros, P.C.; Raymond, R.; Fletcher, P.J.; Nobrega, J.N.; Wong, A.H.C. Pdxdc1 modulates prepulse inhibition of acoustic startle in the mouse. Transl. Psychiatry 2017, 7, e1125. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.J.; Leweke, F.M.; Kapur, S.; Meyer-Lindenberg, A. The neurobiology of schizophrenia: New leads and avenues for treatment. Curr. Opin. Neurobiol. 2010, 20, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Wockner, L.F.; Morris, C.P.; Noble, E.P.; Lawford, B.R.; Whitehall, V.L.; Young, R.M.; Voisey, J. Brain-specific epigenetic markers of schizophrenia. Transl. Psychiatry 2015, 5, e680. [Google Scholar] [CrossRef] [PubMed]
- Gavin, D.P.; Floreani, C. Epigenetics of schizophrenia: An open and shut case. Int. Rev. Neurobiol. 2014, 115, 155–201. [Google Scholar] [PubMed]
- Guidotti, A.; Auta, J.; Chen, Y.; Davis, J.M.; Dong, E.; Gavin, D.P.; Grayson, D.R.; Matrisciano, F.; Pinna, G.; Satta, R.; et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 2011, 60, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.; Barragan, I.; Ingelman-Sundberg, M. Epigenetic mechanisms of importance for drug treatment. Trends Pharmacol. Sci. 2014, 35, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [PubMed]
- Millan, M.J. An epigenetic framework for neurodevelopmental disorders: From pathogenesis to potential therapy. Neuropharmacology 2013, 68, 2–82. [Google Scholar] [CrossRef] [PubMed]
- Arranz, M.J.; Kapur, S. Pharmacogenetics in psychiatry: Are we ready for widespread clinical use? Schizophr. Bull. 2008, 34, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Lotrich, F.E. The emerging potential of pharmacogenetics in psychiatry. Am. J. Psychiatry 2012, 169, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.K.; Zhang, J.P.; Lencz, T. Pharmacogenetics in psychiatry: Translating research into clinical practice. Mol. Psychiatry 2012, 17, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, J.; Brandon, N.J. Schizophrenia drug discovery and development in an evolving era: Are new drug targets fulfilling expectations? J. Psychopharmacol. 2015, 29, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Poels, E.M.; Kegeles, L.S.; Kantrowitz, J.T.; Slifstein, M.; Javitt, D.C.; Lieberman, J.A.; Abi-Dargham, A.; Girgis, R.R. Imaging glutamate in schizophrenia: Review of findings and implications for drug discovery. Mol. Psychiatry 2014, 19, 20–29. [Google Scholar] [CrossRef] [PubMed]

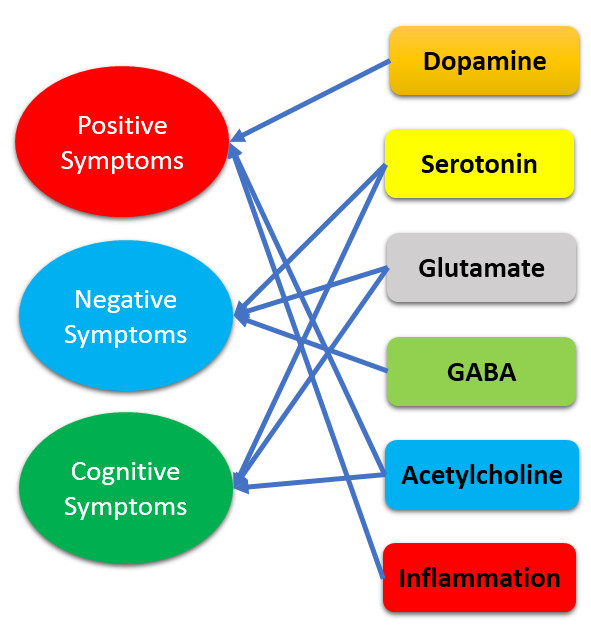{kind=link}
| Hypothesis | Target | Strategy |
|---|---|---|
| Dopamine | Dopaminergic stabilizers | Improve medication adherence |
| Glutamate | NMDAR, AMPA receptor, or metabotropic receptors | Improve negative symptoms and cognitive impairments |
| Serotonin | 5-HT1A agonists, 5-HT reuptake inhibitors, 5-HT2C antagonists and agonists, 5-HT3 antagonists, 5-HT6 antagonists, and 5HT7 antagonists | Reduce the extrapyramidal effects Improve negative symptoms and cognitive impairments Potential treatment for different phases of the illness |
| Acetylcholine | α-7 nicotinic and M1 muscarinic agonists and positive allosteric modulators | Nicotinic agonists for cognitive symptoms Muscarinic agonists for positive symptoms |
| Gamma-aminobutyric acid | Selective GABA-A agonists, GABA-B antagonists, and allosteric modulators at GABA-A receptor subtypes | Augmentation of psychosis treatment |
| Inflammation | Cytokines | Possibly the early period of the psychosis |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, A.C.; Tsai, S.-J. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. Int. J. Mol. Sci. 2017, 18, 1689. https://doi.org/10.3390/ijms18081689
Yang AC, Tsai S-J. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. International Journal of Molecular Sciences. 2017; 18(8):1689. https://doi.org/10.3390/ijms18081689
Chicago/Turabian StyleYang, Albert C., and Shih-Jen Tsai. 2017. "New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis" International Journal of Molecular Sciences 18, no. 8: 1689. https://doi.org/10.3390/ijms18081689
APA StyleYang, A. C., & Tsai, S.-J. (2017). New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. International Journal of Molecular Sciences, 18(8), 1689. https://doi.org/10.3390/ijms18081689