Sample Size Estimation for Detection of Splicing Events in Transcriptome Sequencing Data


Abstract
:1. Introduction
Observation of Splicing Events
2. Results
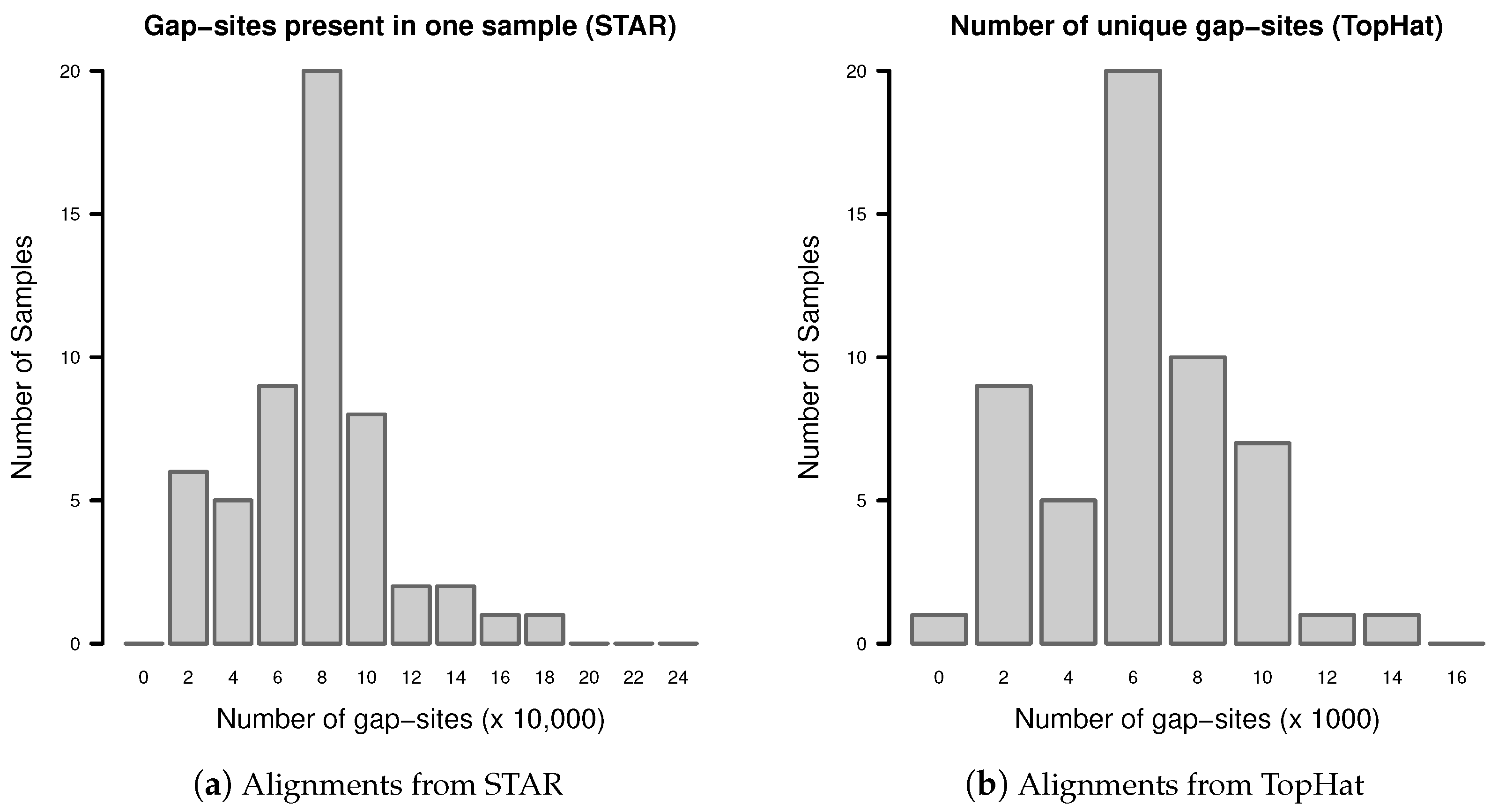
2.1. Sample Size Estimation
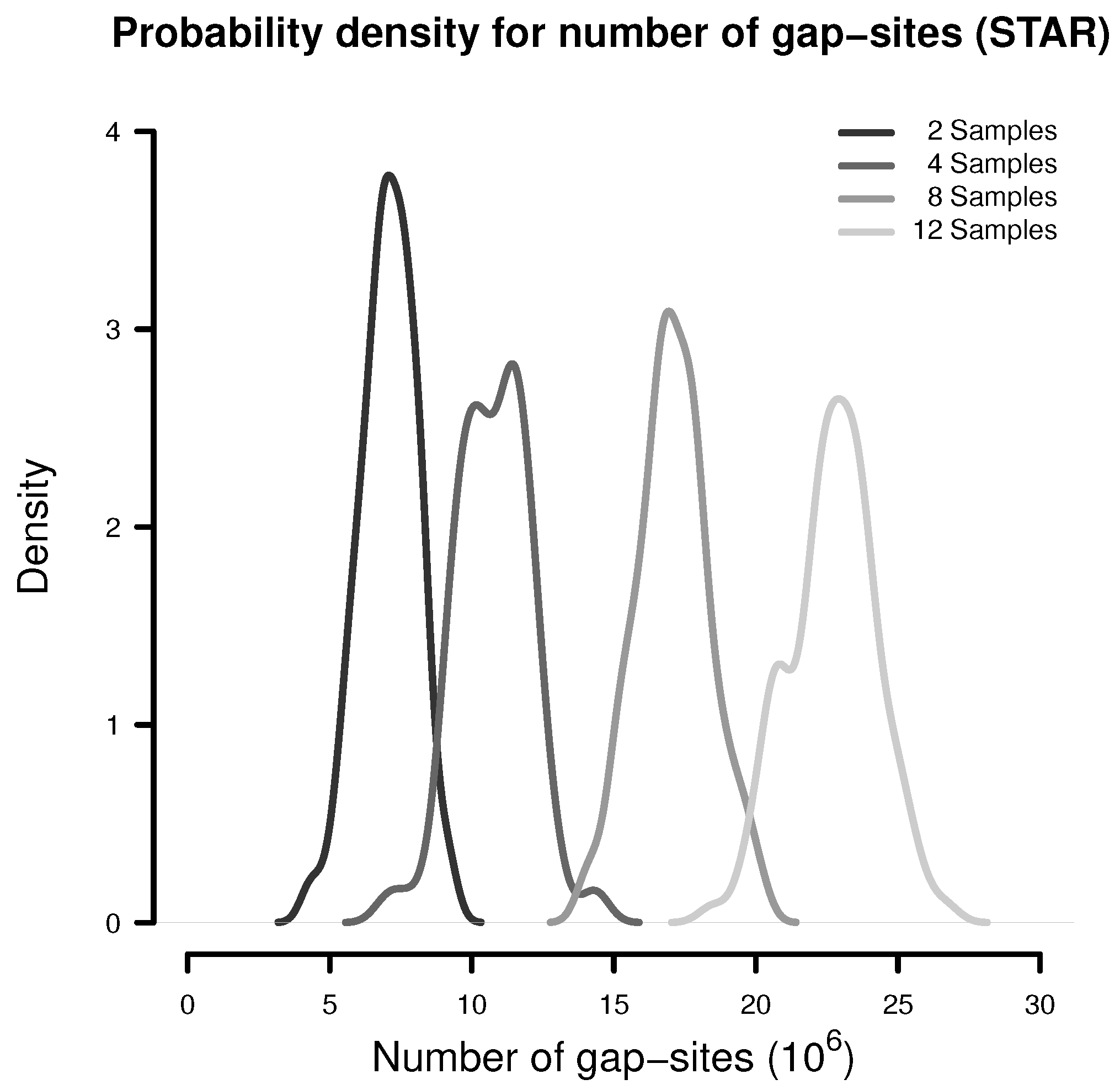
Number of Gap-Sites Observed in Small Samples
2.2. The Probabilistic Model for Prediction of Event Numbers
- random events
- independent from each other
2.2.1. Definition of Two Probabilities
2.2.2. Calculation of Expected Values
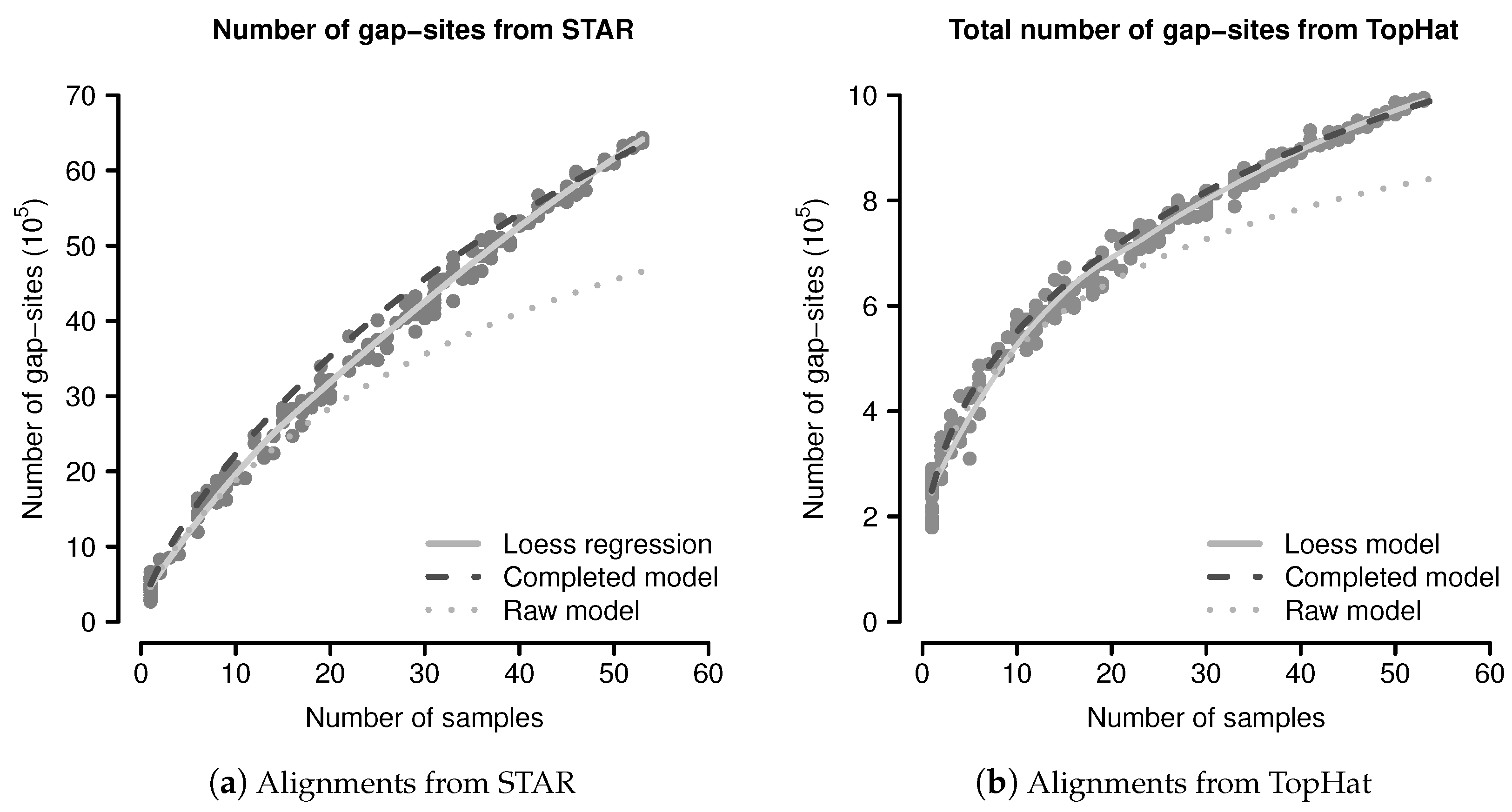
2.3. Evaluation of the Probabilistic Model
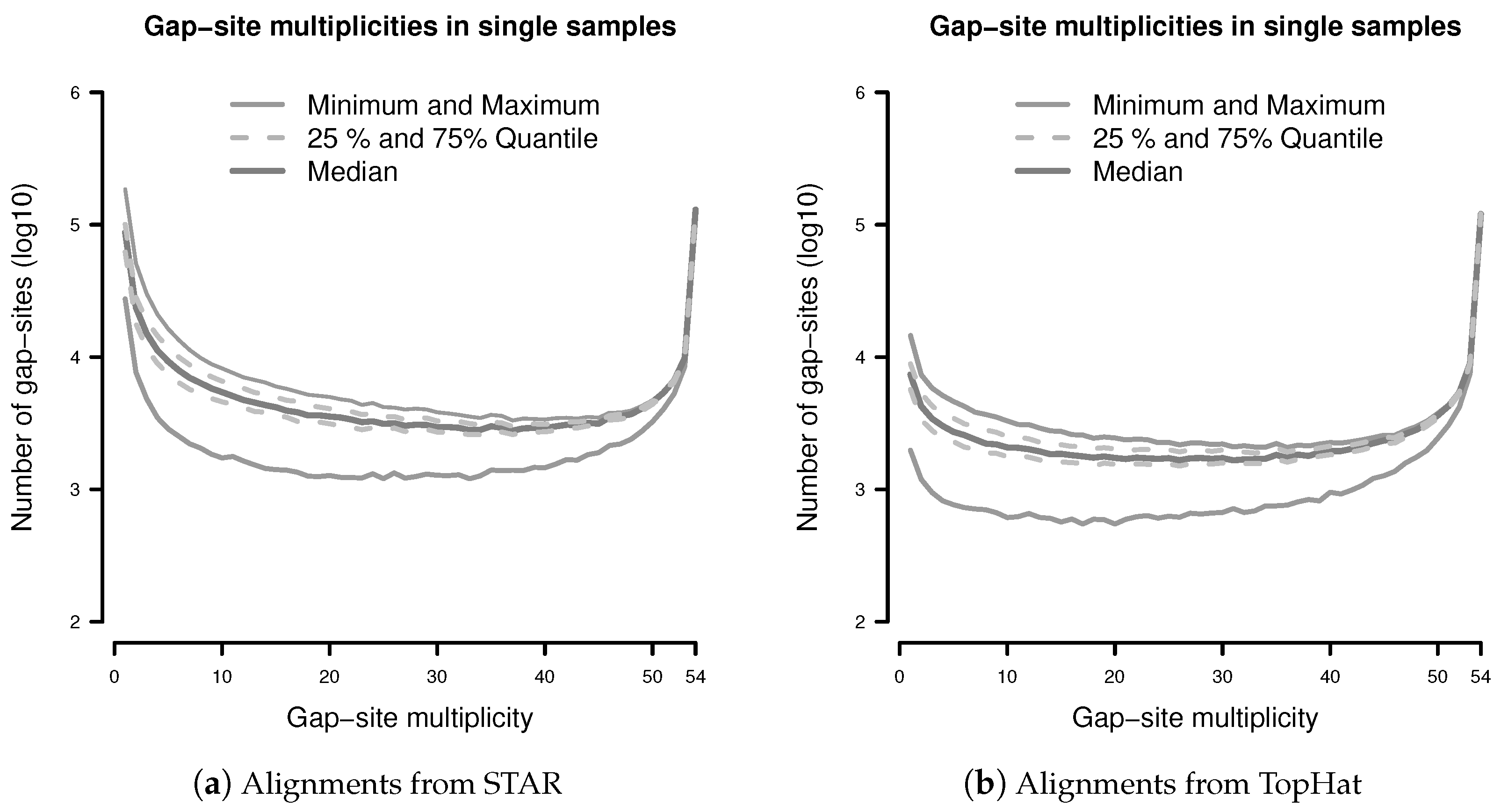
Limitations Arising from Finite Samples
2.4. Correction for Estimation Inaccuracies
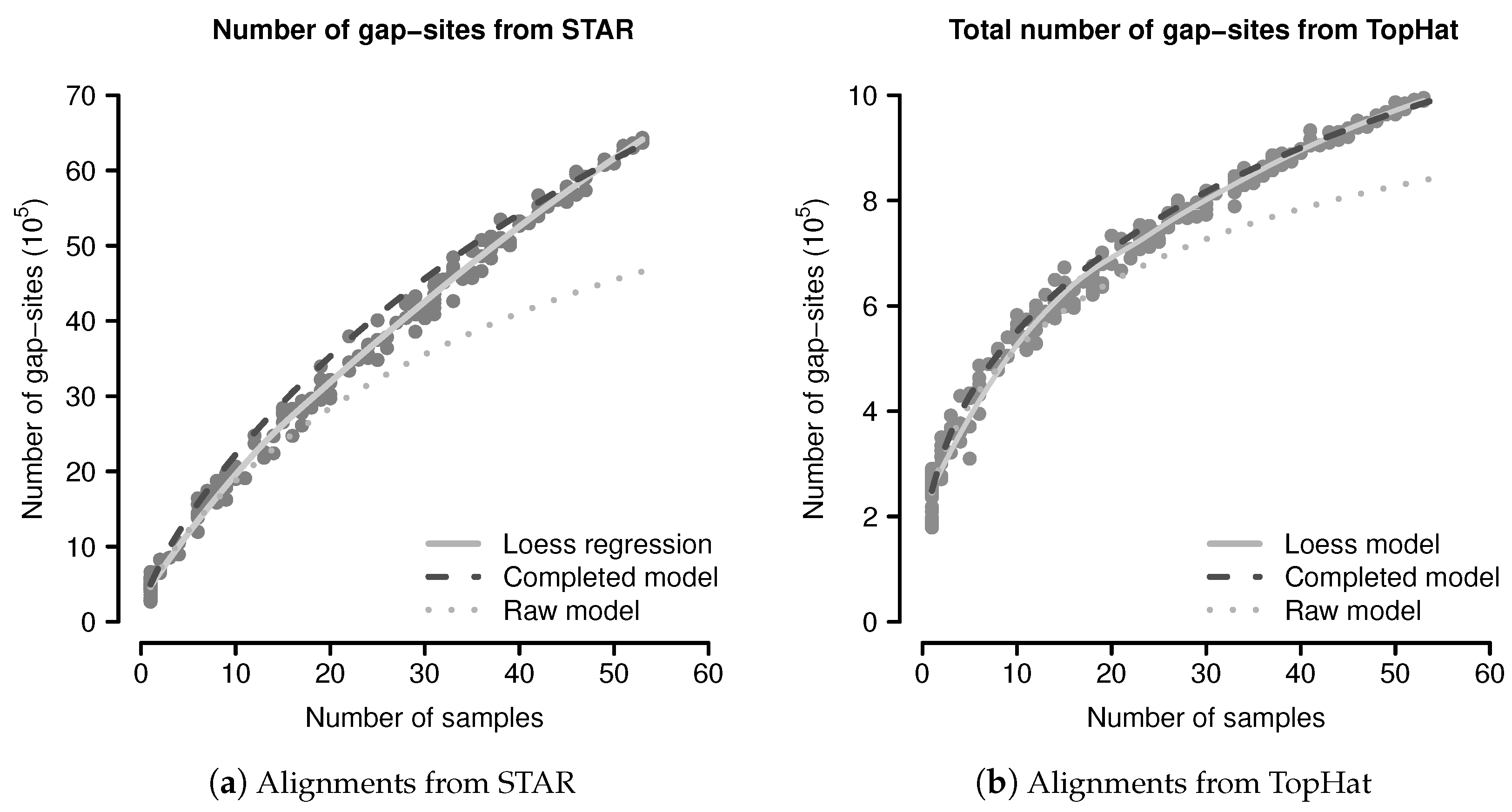
Predictions by the Completed Probabilistic Model
2.5. Basal Rate for Observation of Gap-Sites
2.6. Sample Size Estimation
3. Discussion
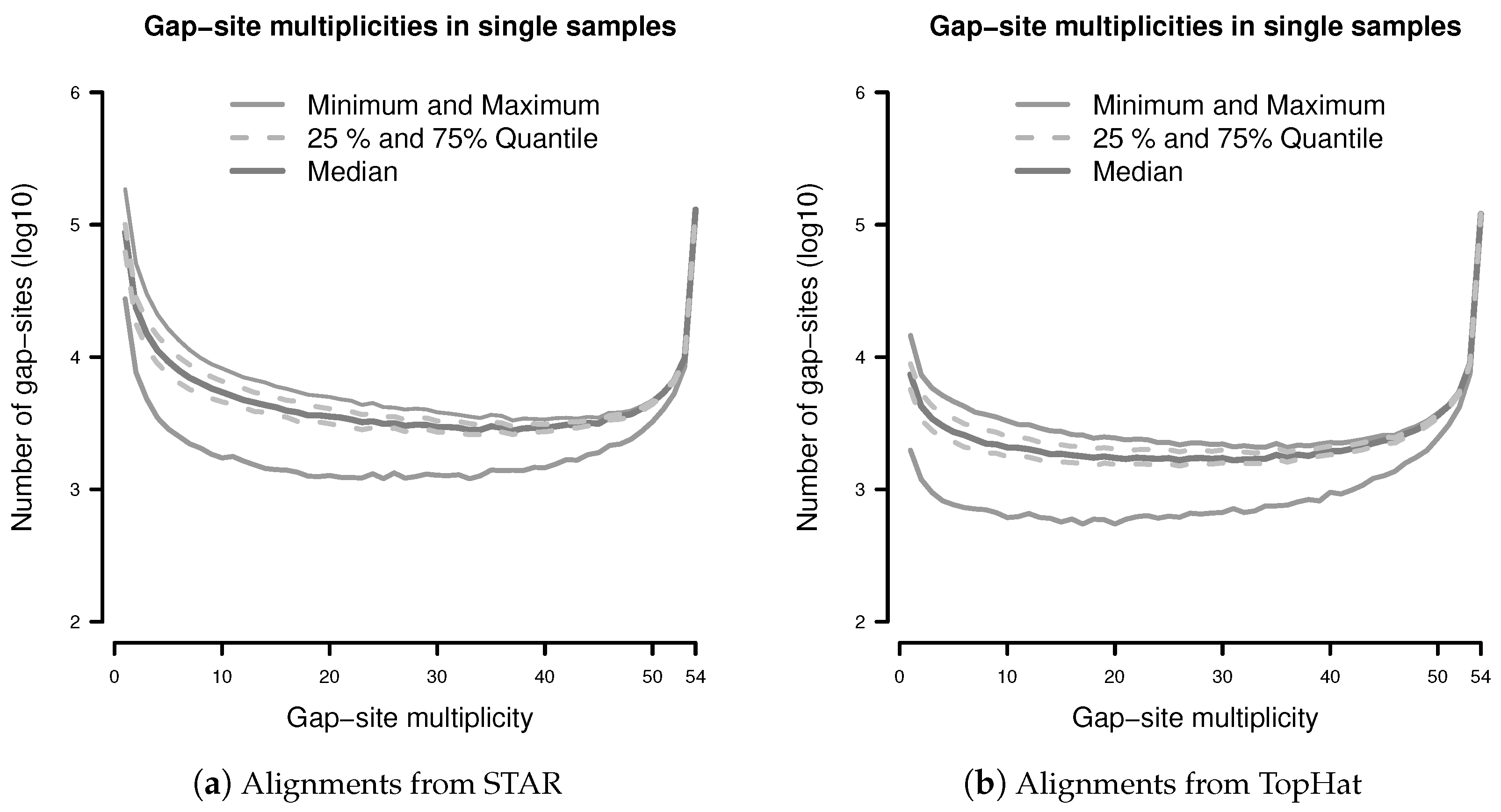
3.1. Independency of Gap-Site Observations
3.2. Observations of Gap-Sites Are Random Events
Stochastic Noise in the Splicing Machinery
3.3. Consequences of Basal Rates for Observation of Gap-Sites
3.4. Observation of Gap-Sites under Different Experimental Conditions
3.4.1. Influence of Read-Length and Alignment Depth
3.4.2. Influence of Sequencing Technology
3.4.3. Influence of Different Species and Tissues
3.5. Comparison with Other Analysis Strategies
4. Materials and Methods
Transcriptome Sequencing Data
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| FDR | False Discovery Rate |
| nAligns | Number of supporting alignments (for a gap-site) |
| nProbes | Number of samples in which a gap-site is identified |
| gqs | Gap quality score |
| wgis | Weighted Gap Information Score |
| mcl | Minimum CIGAR length |
| qsm | Quartet sum of MCL |
| nlstart | Number of left start (positions) |
| SD | Standard deviation |
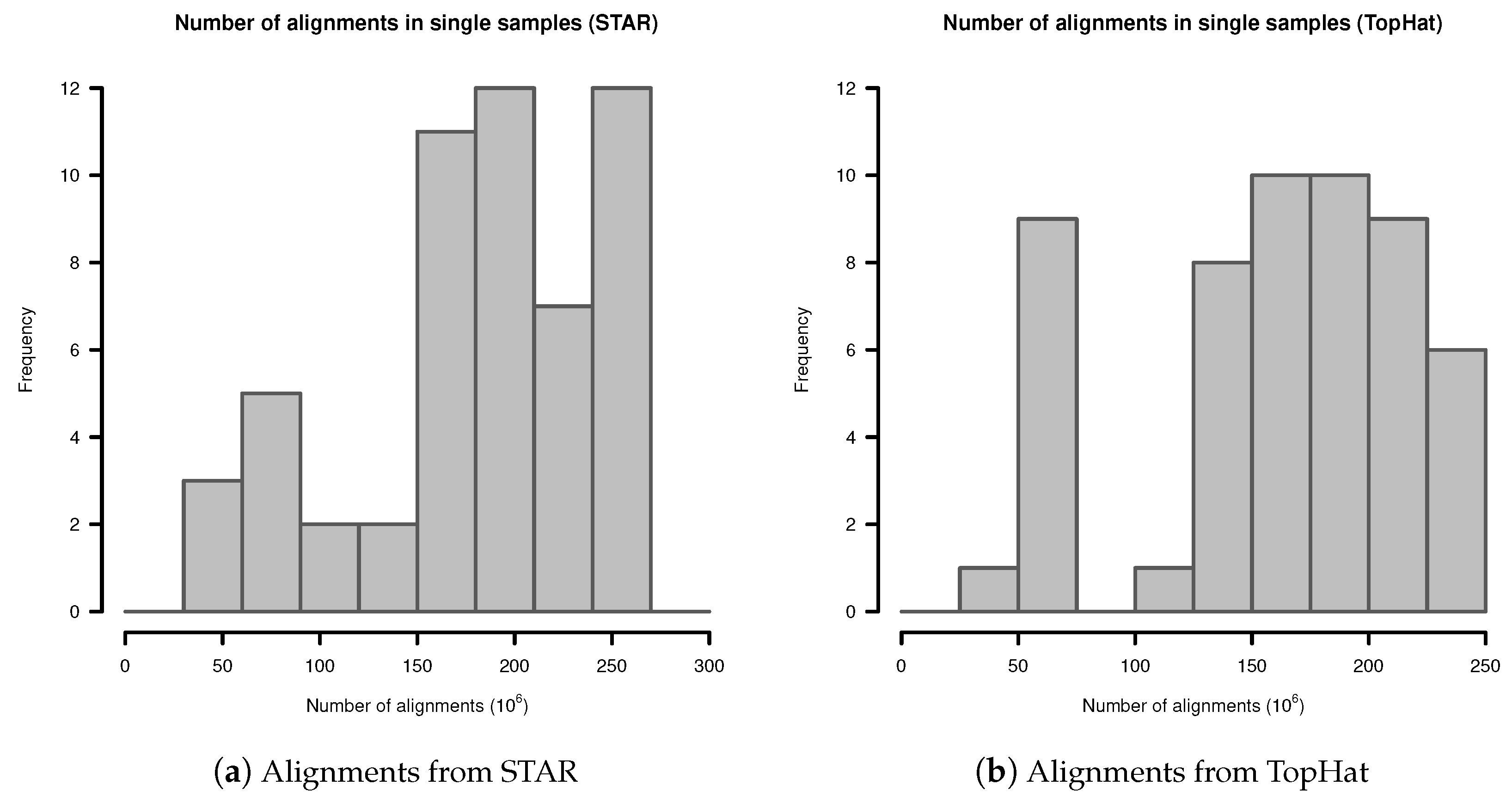
Appendix A. Number of Alignments in Fibroblast Transcriptome Samples
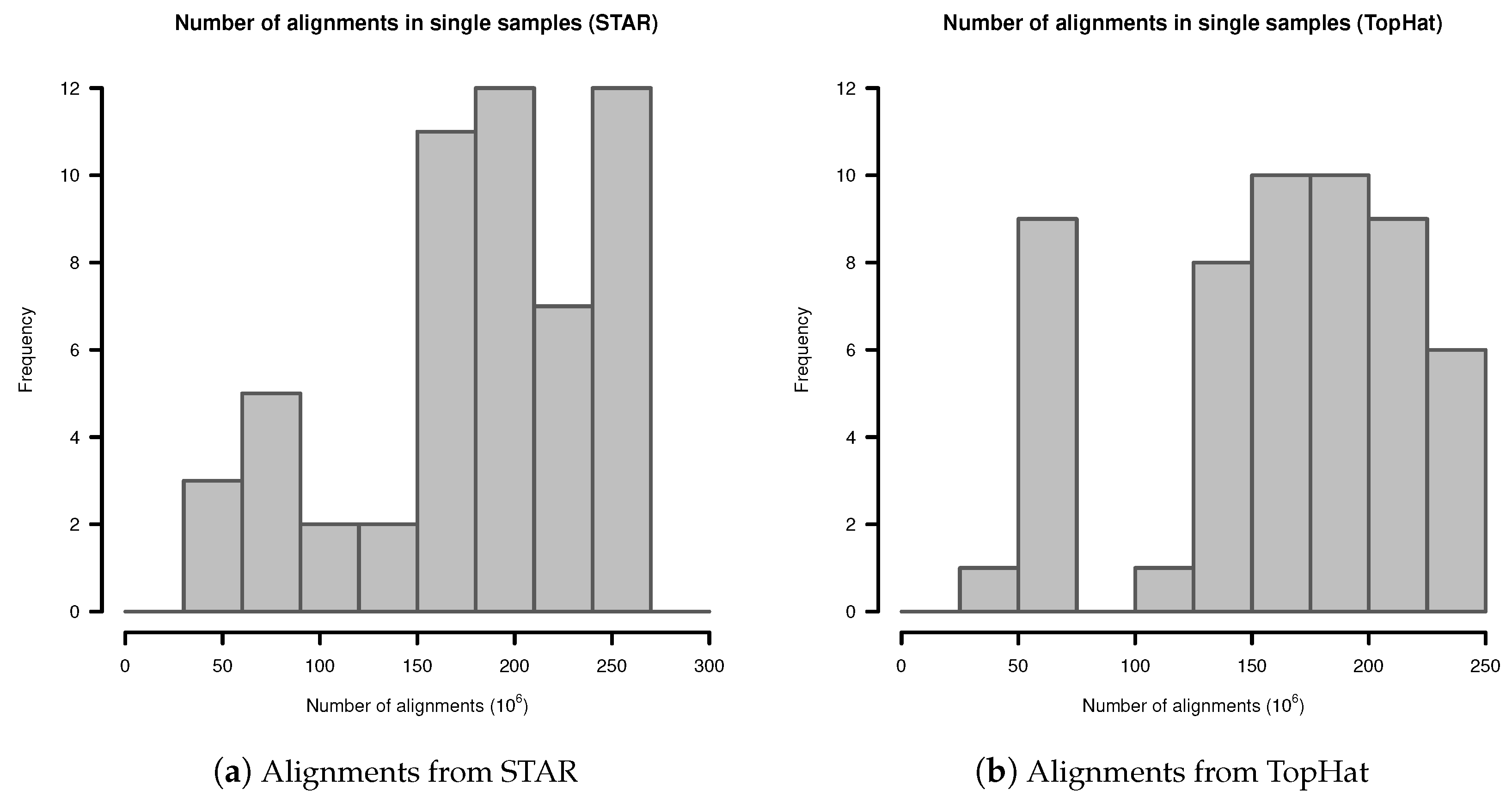
Appendix B. Probabilistic Model for Observation of Events
Appendix B.1. Definitions
Appendix B.1.1. Observation Probabilities
Appendix B.1.2. Observation of Events in Merged Samples
- First, observation probabilities () are drawn from an observation prior .
- Second, the actual observation of events are iid (independent identical distributed) drawn from a Bernoulli distribution .
Appendix B.1.3. Estimation of Observation Prior from Real Samples
Appendix B.2. Evaluation of Model Predictions for Special Cases
Appendix B.2.1. Equal Observation Probabilities
Appendix B.2.2. Events Observed in All Samples
Appendix B.2.3. Observation of Rare Events
Appendix B.2.4. Rare Events Observed with Equal Probability:
Appendix C. Gap-Sites Identified in Only One Sample
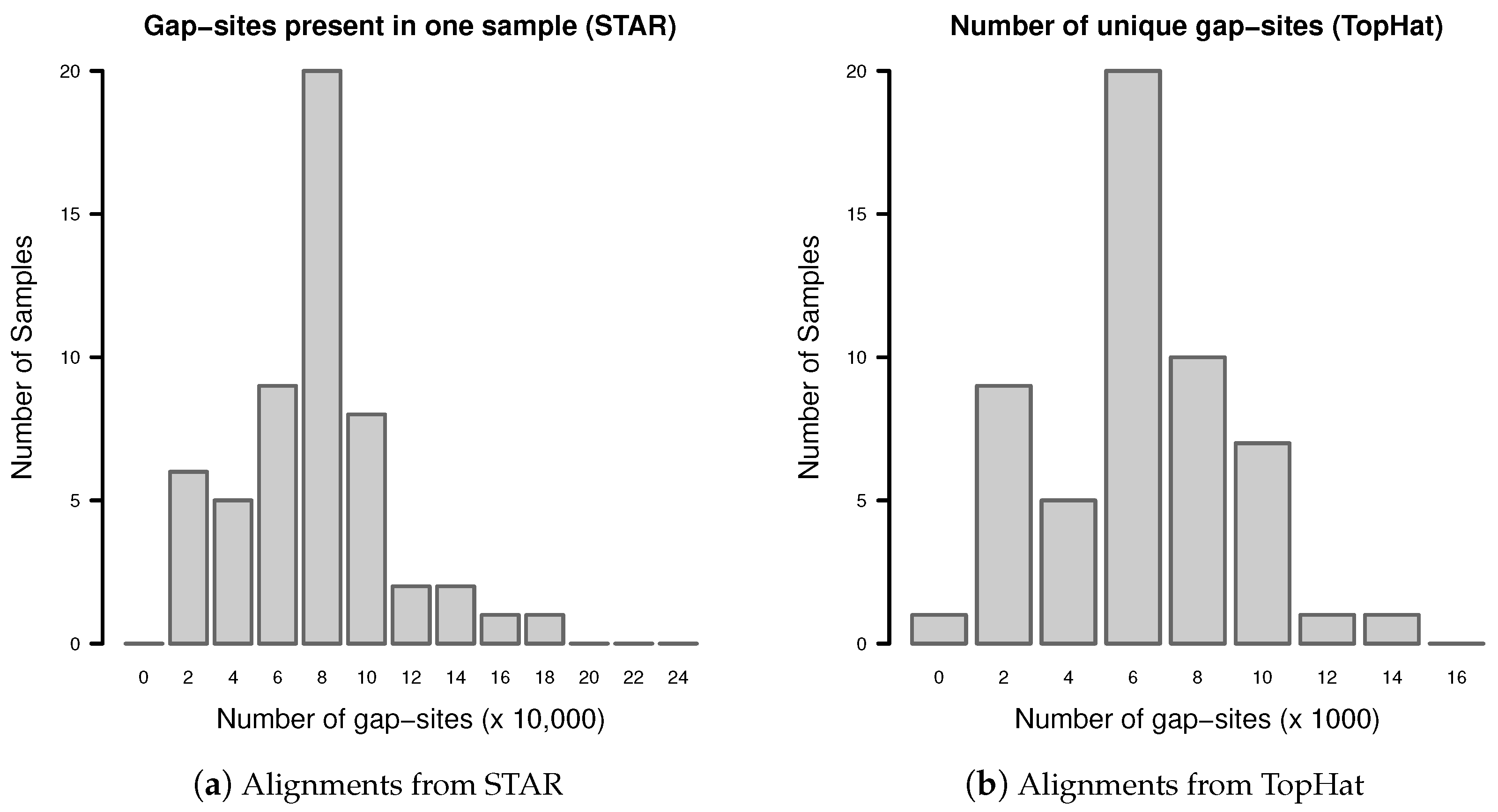
Appendix D. Derivation of of the Formula for Sample Size Estimation
References
- Shen, S.; Park, J.W.; Huang, J.; Dittmar, K.A.; Lu, Z.X.; Zhou, Q.; Carstens, R.P.; Xing, Y. MATS: A Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012, 40, e61. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed]
- Drewe, P.; Stegle, O.; Hartmann, L.; Kahles, A.; Bohnert, R.; Wachter, A.; Borgwardt, K.; Ratsch, G. Accurate detection of differential RNA processing. Nucleic Acids Res. 2013, 41, 5189–5198. [Google Scholar] [CrossRef] [PubMed]
- Steijger, T.; Abril, J.F.; Engstrom, P.G.; Kokocinski, F.; Hubbard, T.J.; Guigo, R.; Harrow, J.; Bertone, P.; Abril, J.F.; Akerman, M.; et al. Assessment of transcript reconstruction methods for RNA-Seq. Nat. Methods 2013, 10, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, S.A.; Chen, W.Y.; Wong, T.; Deveson, I.W.; Blackburn, J.; Andersen, S.B.; Nielsen, L.K.; Mattick, J.S.; Mercer, T.R. Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat. Methods 2016, 13, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.E. A survey of software for genome-wide discovery of differential splicing in RNA-Seq data. Hum. Genom. 2014, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Kaisers, W.; Ptok, J.; Schwender, H.; Schaal, H. Validation of Splicing Events in Transcriptome Sequencing Data. Int. J. Mol. Sci. 2017, 18, 1110. [Google Scholar] [CrossRef] [PubMed]
- Kaisers, W.; Schaal, H.; Schwender, H. rbamtools: An R interface to samtools enabling fast accumulative tabulation of splicing events over multiple RNA-seq samples. Bioinformatics 2015, 31, 1663–1664. [Google Scholar] [CrossRef] [PubMed]
- Loguinov, A.V.; Mian, I.S.; Vulpe, C.D. Exploratory differential gene expression analysis in microarray experiments with no or limited replication. Genome Biol. 2004, 5, R18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, W.; Koonin, E.V. Introns and the origin of nucleus-cytosol compartmentalization. Nature 2006, 440, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Stamm, S.; Ben-Ari, S.; Rafalska, I.; Tang, Y.; Zhang, Z.; Toiber, D.; Thanaraj, T.A.; Soreq, H. Function of alternative splicing. Gene 2005, 344, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W. Why genes in pieces? Nature 1978, 271, 501. [Google Scholar] [CrossRef] [PubMed]
- Ast, G. How did alternative splicing evolve? Nat. Rev. Genet. 2004, 5, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Modrek, B.; Lee, C.J. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat. Genet. 2003, 34, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Tress, M.L.; Abascal, F.; Valencia, A. Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem. Sci. 2016, 42, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Tress, M.L.; Martelli, P.L.; Frankish, A.; Reeves, G.A.; Wesselink, J.J.; Yeats, C.; Olason, P.I.; Albrecht, M.; Hegyi, H.; Giorgetti, A.; et al. The implications of alternative splicing in the ENCODE protein complement. Proc. Natl. Acad. Sci. USA 2007, 104, 5495–5500. [Google Scholar] [CrossRef] [PubMed]
- Ezkurdia, I.; Rodriguez, J.M.; Carrillo-de Santa Pau, E.; Vazquez, J.; Valencia, A.; Tress, M.L. Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res. 2015, 14, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Brogna, S.; McLeod, T.; Petric, M. The Meaning of NMD: Translate or Perish. Trends Genet. 2016, 32, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J.; Bennett, E.J. Protecting the proteome: Eukaryotic cotranslational quality control pathways. J. Cell Biol. 2014, 204, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Pai, A.A.; Gilad, Y.; Pritchard, J.K. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet. 2010, 6, e1001236. [Google Scholar] [CrossRef] [PubMed]
- Melamud, E.; Moult, J. Stochastic noise in splicing machinery. Nucleic Acids Res. 2009, 37, 4873–4886. [Google Scholar] [CrossRef] [PubMed]
- Melamud, E.; Moult, J. Structural implication of splicing stochastics. Nucleic Acids Res. 2009, 37, 4862–4872. [Google Scholar] [CrossRef] [PubMed]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Cardenas, J.A.; Garcia-Maroto, F.; Alvarez-Bermejo, J.A.; Manzano-Agugliaro, F. DNA Sequencing Sensors: An Overview. Sensors 2017, 17, 588. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Q.; Wang, Z. The evolution of nanopore sequencing. Front. Genet. 2014, 5, 449. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ferguson, J.F.; Xue, C.; Silverman, I.M.; Gregory, B.; Reilly, M.P.; Li, M. Evaluating the impact of sequencing depth on transcriptome profiling in human adipose. PLoS ONE 2013, 8, e66883. [Google Scholar] [CrossRef] [PubMed]
- Kaisers, W.; Boukamp, P.; Stark, H.J.; Schwender, H.; Tigges, J.; Krutmann, J.; Schaal, H. Age, gender and UV-exposition related effects on gene expression in in vivo aged short term cultivated human dermal fibroblasts. PLoS ONE 2017, 12, e0175657. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: The raw FASTQ files are available under ArrayExpress accession E-MTAB-4652 (ENA study ERP015294). The software is available in R packages: rbamtools and refGenome on CRAN and spliceSites (including algorithm for wgis) on Bioconductor. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| nFiles | Total | gql = 0 | gql = 3 |
|---|---|---|---|
| 2 | 706 | 378 | 92 |
| 4 | 1076 | 666 | 105 |
| 8 | 1708 | 1179 | 124 |
| 12 | 2270 | 1659 | 137 |
| Sample Size | |
|---|---|
| 0.1 | 16 |
| 0.15 | 10 |
| 0.2 | 8 |
| 0.5 | 3 |
| 0.8 | 1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaisers, W.; Schwender, H.; Schaal, H. Sample Size Estimation for Detection of Splicing Events in Transcriptome Sequencing Data. Int. J. Mol. Sci. 2017, 18, 1900. https://doi.org/10.3390/ijms18091900
Kaisers W, Schwender H, Schaal H. Sample Size Estimation for Detection of Splicing Events in Transcriptome Sequencing Data. International Journal of Molecular Sciences. 2017; 18(9):1900. https://doi.org/10.3390/ijms18091900
Chicago/Turabian StyleKaisers, Wolfgang, Holger Schwender, and Heiner Schaal. 2017. "Sample Size Estimation for Detection of Splicing Events in Transcriptome Sequencing Data" International Journal of Molecular Sciences 18, no. 9: 1900. https://doi.org/10.3390/ijms18091900