Lysophosphatidic Acid Signaling Axis Mediates Ceramide 1-Phosphate-Induced Proliferation of C2C12 Myoblasts

 ,
,  ,
, 

Abstract
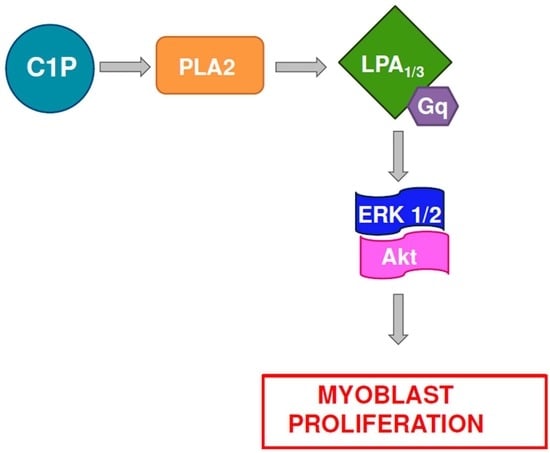
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
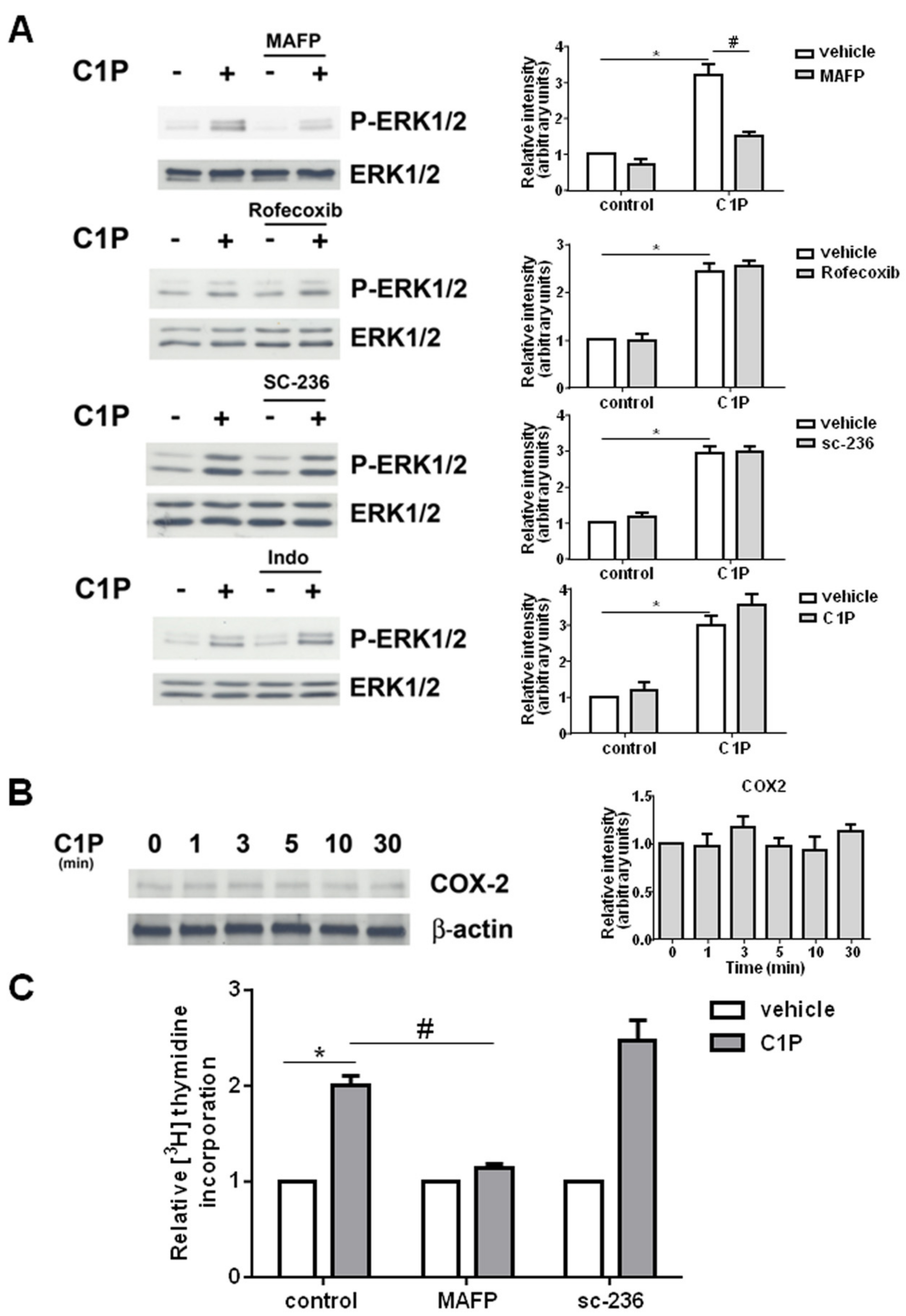
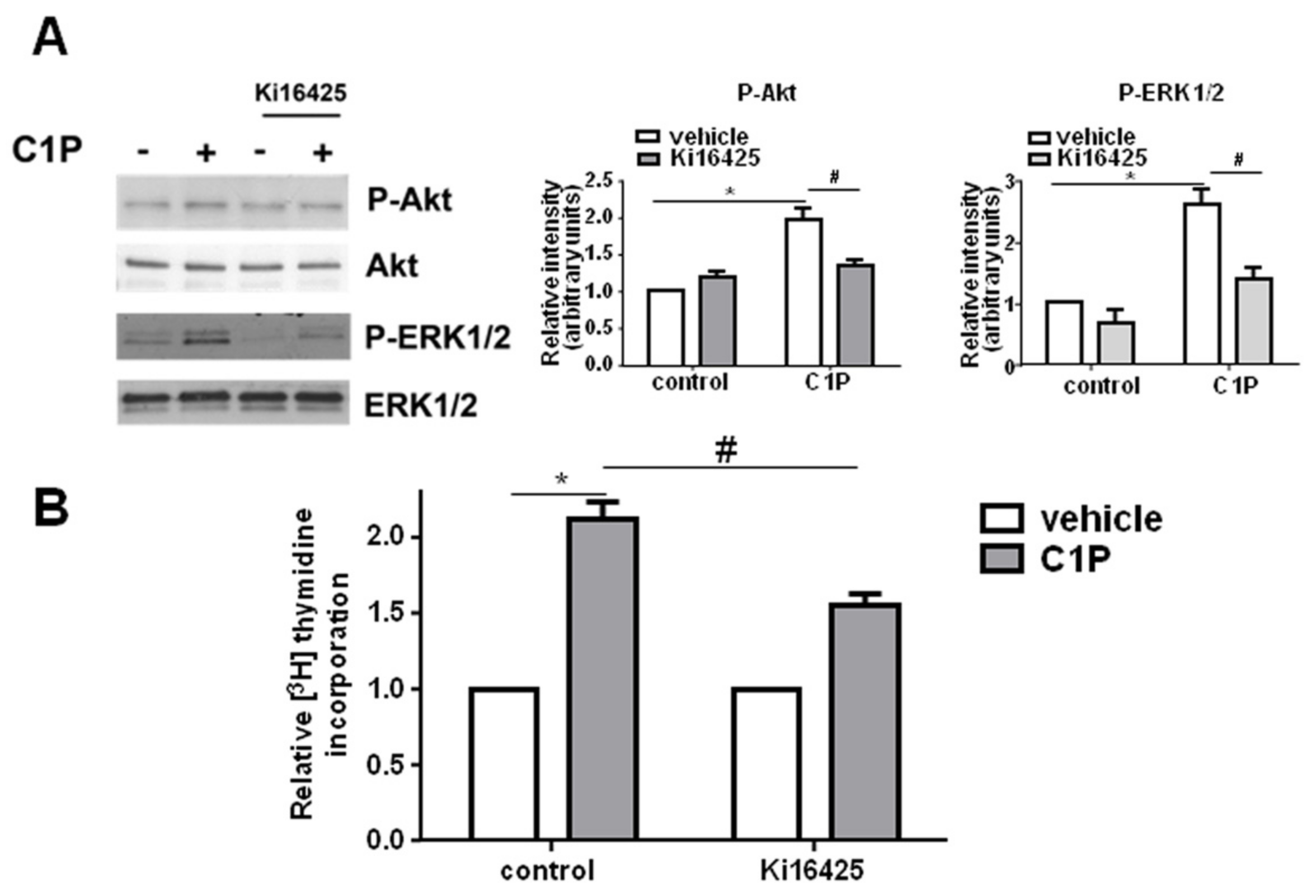
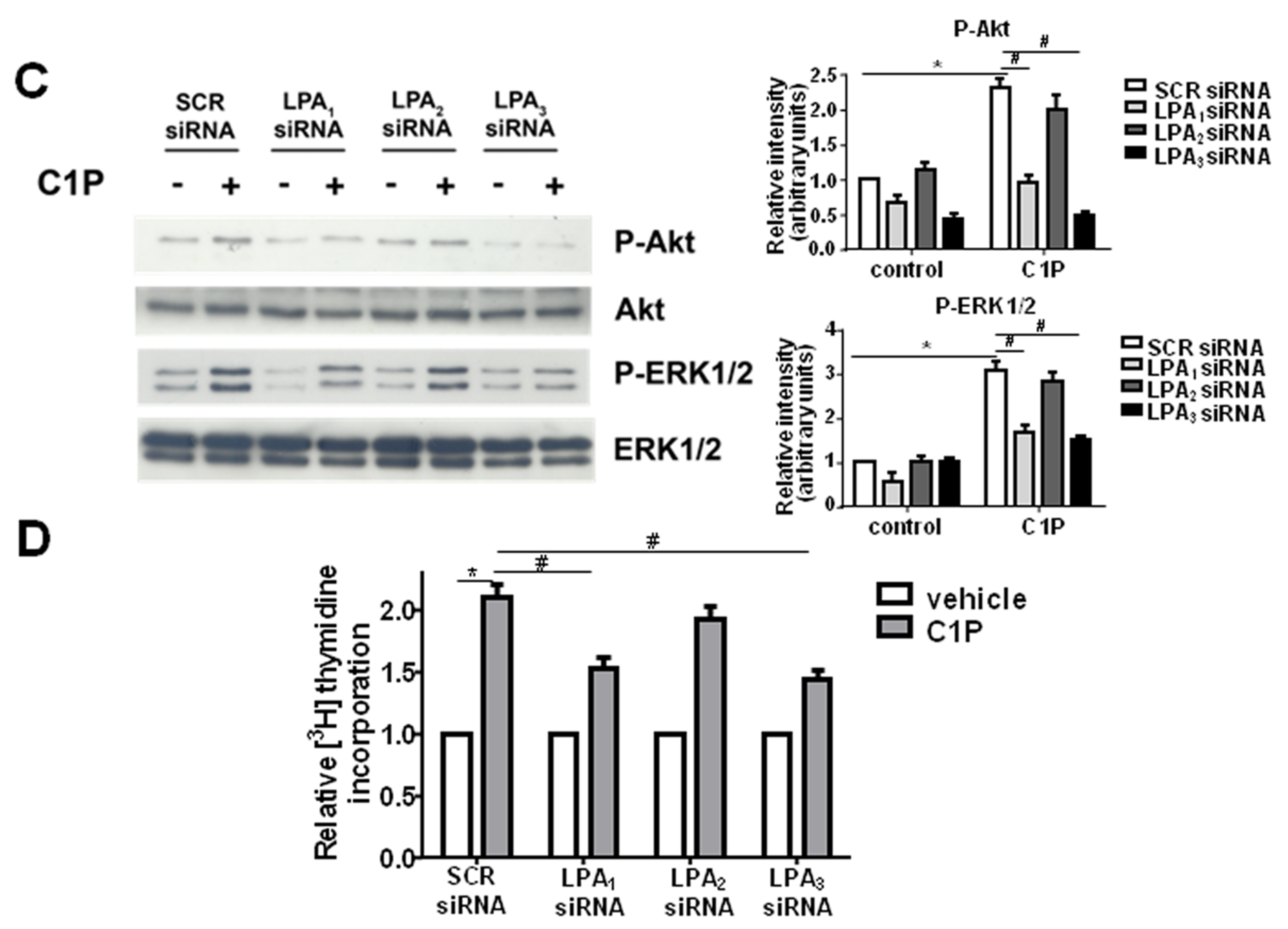
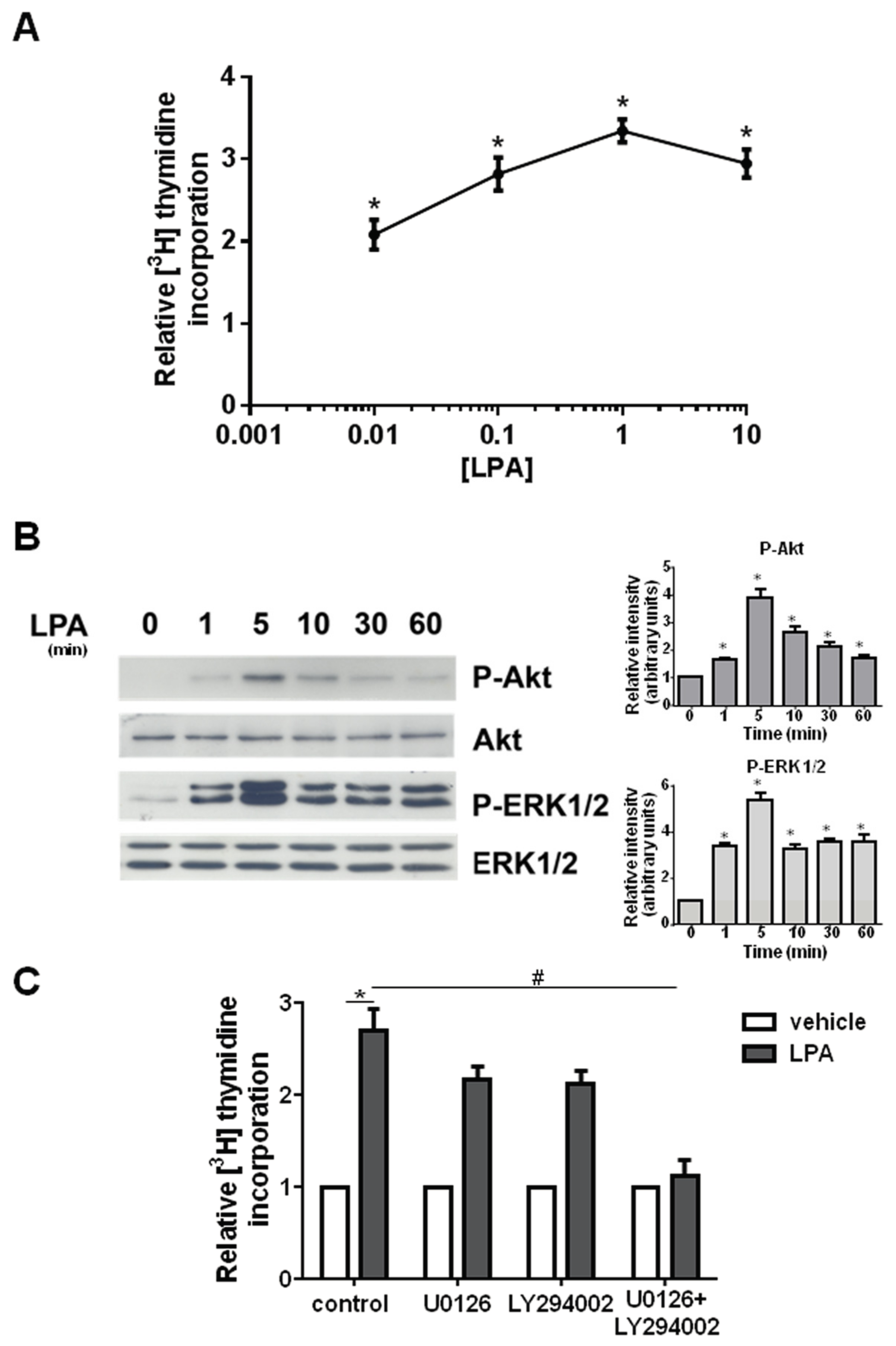
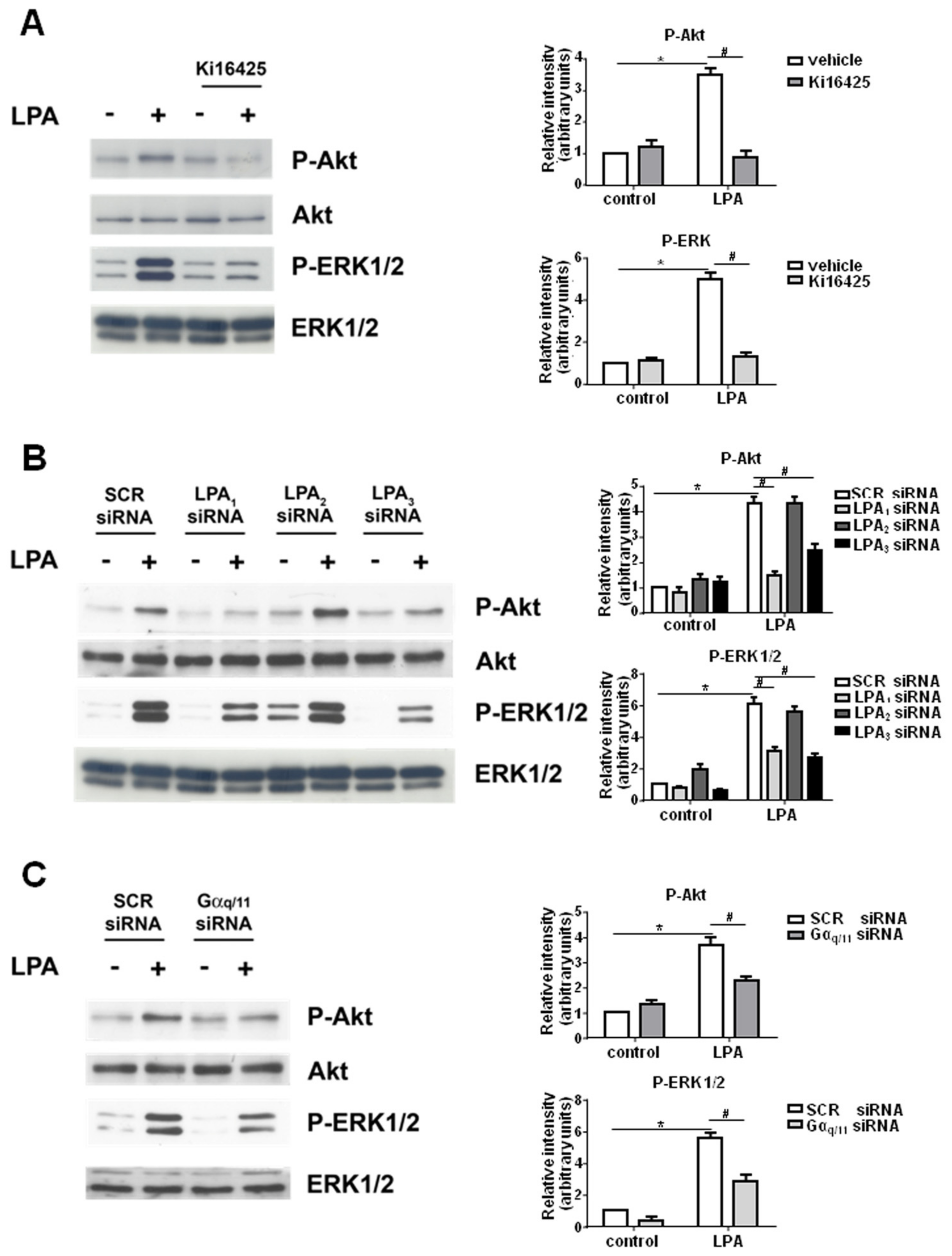
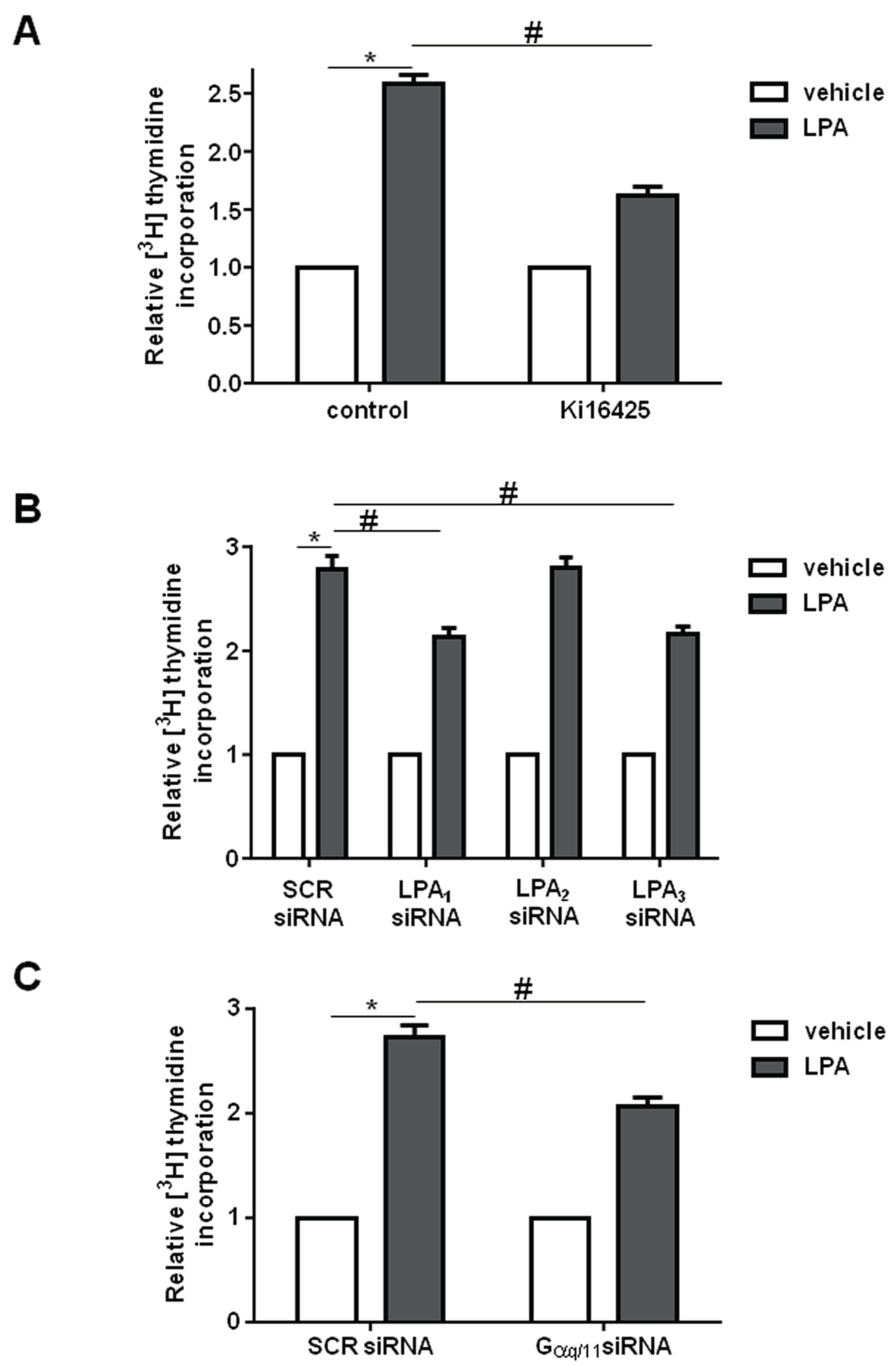
2. Results
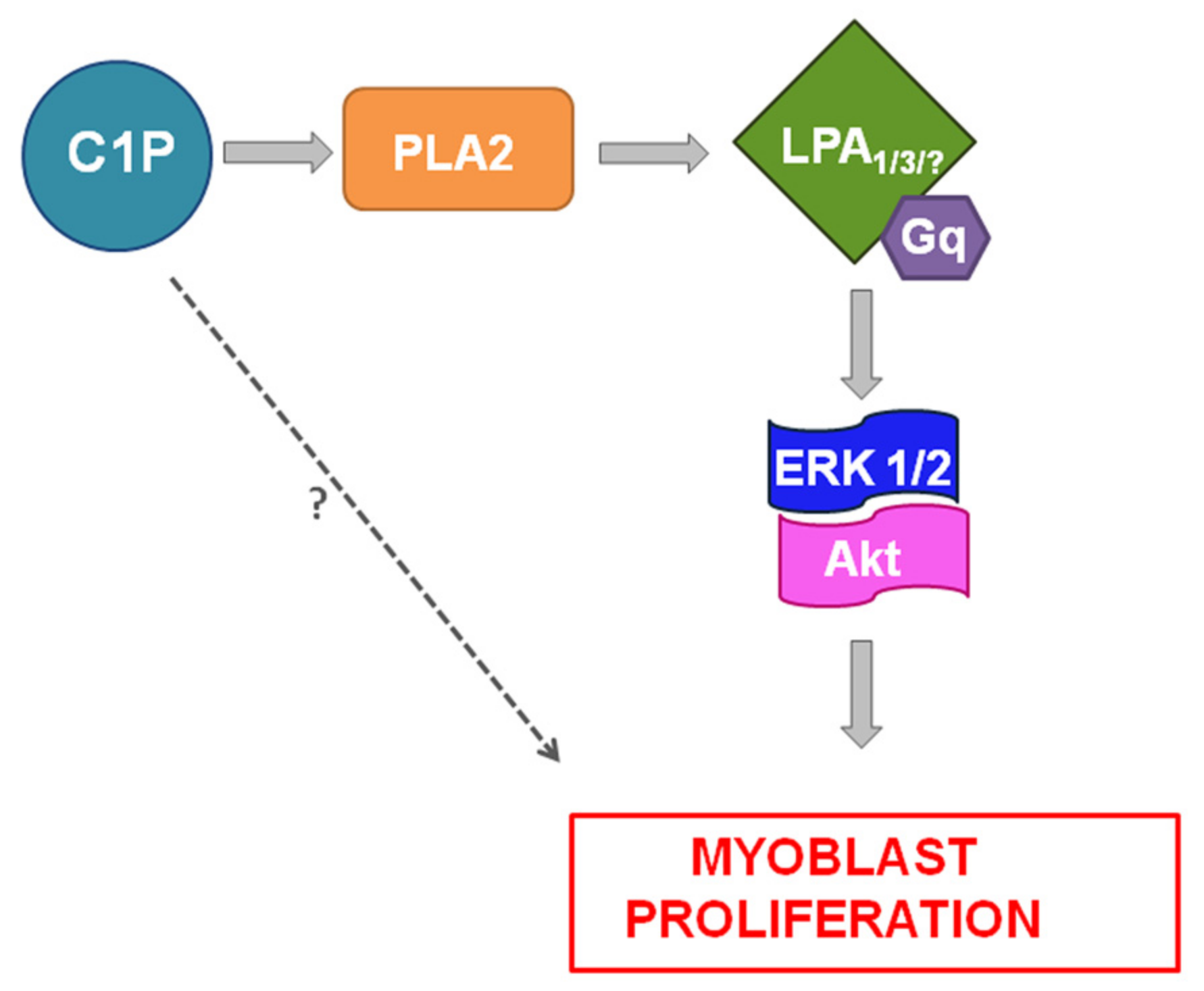
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Transfection
4.4. Cellular Fractionation
4.5. Western Blot Analysis
4.6. Cell Proliferation
4.7. Cell Membrane Preparation for C1P Radioligand-Binding Assay
4.8. Quantitative Real-Time Reverse Transcription PCR
4.9. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LPA | Lysophosphatidic acid |
| C1P | Ceramide 1-phosphate |
| COX | Cyclooxygenase |
| Cerk | Ceramide kinase |
| PLA2 | Phospholipase A2 |
| LPAR | Lysophosphatidic acid receptor |
References
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Desai, N.N.; Olivera, A.; Seki, T.; Brooker, G.; Spiegel, S. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell Biol. 1991, 114, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingosine-1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Arana, L.; Gangoiti, P.; Ouro, A.; Rivera, I.G.; Ordonez, M.; Trueba, M.; Lankalapalli, R.S.; Bittman, R.; Gomez-Munoz, A. Generation of reactive oxygen species (ROS) is a key factor for stimulation of macrophage proliferation by ceramide 1-phosphate. Exp. Cell Res. 2012, 318, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Gangoiti, P.; Bernacchioni, C.; Donati, C.; Cencetti, F.; Ouro, A.; Gomez-Munoz, A.; Bruni, P. Ceramide 1-phosphate stimulates proliferation of C2C12 myoblasts. Biochimie 2012, 94, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Gangoiti, P.; Granado, M.H.; Wang, S.W.; Kong, J.Y.; Steinbrecher, U.P.; Gomez-Munoz, A. Ceramide 1-phosphate stimulates macrophage proliferation through activation of the PI3-kinase/PKB, JNK and ERK1/2 pathways. Cell. Signal. 2008, 20, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Munoz, A.; Duffy, P.A.; Martin, A.; O’Brien, L.; Byun, H.S.; Bittman, R.; Brindley, D.N. Short-chain ceramide-1-phosphates are novel stimulators of DNA synthesis and cell division: Antagonism by cell-permeable ceramides. Mol. Pharmacol. 1995, 47, 833–839. [Google Scholar] [PubMed]
- Gomez-Munoz, A.; Frago, L.M.; Alvarez, L.; Varela-Nieto, I. Stimulation of DNA synthesis by natural ceramide 1-phosphate. Biochem. J. 1997, 325 Pt 2, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Kang, Y.J.; Lim, Y.; Lee, H.W.; Bae, K.; Lee, Y.S.; Yoo, J.M.; Yoo, H.S.; Yun, Y.P. Ceramide 1-phosphate induces neointimal formation via cell proliferation and cell cycle progression upstream of ERK1/2 in vascular smooth muscle cells. Exp. Cell Res. 2011, 317, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Arana, L.; Ordonez, M.; Ouro, A.; Rivera, I.G.; Gangoiti, P.; Trueba, M.; Gomez-Munoz, A. Ceramide 1-phosphate induces macrophage chemoattractant protein-1 release: Involvement in ceramide 1-phosphate-stimulated cell migration. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1213–E1226. [Google Scholar] [CrossRef] [PubMed]
- Granado, M.H.; Gangoiti, P.; Ouro, A.; Arana, L.; Gonzalez, M.; Trueba, M.; Gomez-Munoz, A. Ceramide 1-phosphate (C1P) promotes cell migration Involvement of a specific C1P receptor. Cell. Signal. 2009, 21, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Schneider, G.; Abdel-Latif, A.; Mierzejewska, K.; Sunkara, M.; Borkowska, S.; Ratajczak, J.; Morris, A.J.; Kucia, M.; Ratajczak, M.Z. Ceramide-1-phosphate regulates migration of multipotent stromal cells and endothelial progenitor cells—Implications for tissue regeneration. Stem Cells 2013, 31, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Hirabayashi, T.; Shimizu, M.; Murayama, T. Ceramide-1-phosphate activates cytosolic phospholipase A2α directly and by PKC pathway. Biochem. Pharmacol. 2006, 71, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Bielawska, A.; Subramanian, P.; Wijesinghe, D.S.; Maceyka, M.; Leslie, C.C.; Evans, J.H.; Freiberg, J.; Roddy, P.; Hannun, Y.A.; et al. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J. Biol. Chem. 2004, 279, 11320–11326. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, P.; Stahelin, R.V.; Szulc, Z.; Bielawska, A.; Cho, W.; Chalfant, C.E. Ceramide 1-phosphate acts as a positive allosteric activator of group IVA cytosolic phospholipase A2α and enhances the interaction of the enzyme with phosphatidylcholine. J. Biol. Chem. 2005, 280, 17601–17607. [Google Scholar] [CrossRef] [PubMed]
- Hammad, S.M.; Pierce, J.S.; Soodavar, F.; Smith, K.J.; al Gadban, M.M.; Rembiesa, B.; Klein, R.L.; Hannun, Y.A.; Bielawski, J.; Bielawska, A. Blood sphingolipidomics in healthy humans: Impact of sample collection methodology. J. Lipid Res. 2010, 51, 3074–3087. [Google Scholar] [CrossRef] [PubMed]
- Boath, A.; Graf, C.; Lidome, E.; Ullrich, T.; Nussbaumer, P.; Bornancin, F. Regulation and traffic of ceramide 1-phosphate produced by ceramide kinase: Comparative analysis to glucosylceramide and sphingomyelin. J. Biol. Chem. 2008, 283, 8517–8526. [Google Scholar] [CrossRef] [PubMed]
- Bruni, P.; Donati, C. Pleiotropic effects of sphingolipids in skeletal muscle. Cell. Mol. Life Sci. 2008, 65, 3725–3736. [Google Scholar] [CrossRef] [PubMed]
- Donati, C.; Cencetti, F.; Bruni, P. Sphingosine 1-phosphate axis: A new leader actor in skeletal muscle biology. Front. Physiol. 2013, 4, 338. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Almada, A.E.; Wagers, A.J. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease, Nature reviews. Mol. Cell Biol. 2016, 17, 267–279. [Google Scholar]
- Nagata, Y.; Kobayashi, H.; Umeda, M.; Ohta, N.; Kawashima, S.; Zammit, P.S.; Matsuda, R. Sphingomyelin levels in the plasma membrane correlate with the activation state of muscle satellite cells. J. Histochem. Cytochem. 2006, 54, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Partridge, T.A.; Matsuda, R.; Zammit, P.S. Entry of muscle satellite cells into the cell cycle requires sphingolipid signaling. J. Cell Biol. 2006, 174, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Donati, C.; Meacci, E.; Nuti, F.; Becciolini, L.; Farnararo, M.; Bruni, P. Sphingosine 1-phosphate regulates myogenic differentiation: A major role for S1P2 receptor. FASEB J. 2005, 19, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Meacci, E.; Nuti, F.; Donati, C.; Cencetti, F.; Farnararo, M.; Bruni, P. Sphingosine kinase activity is required for myogenic differentiation of C2C12 myoblasts. J. Cell. Physiol. 2008, 214, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Bernacchioni, C.; Cencetti, F.; Blescia, S.; Donati, C.; Bruni, P. Sphingosine kinase/sphingosine 1-phosphate axis: A new player for insulin-like growth factor-1-induced myoblast differentiation. Skelet. Muscle 2012, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Cencetti, F.; Bernacchioni, C.; Nincheri, P.; Donati, C.; Bruni, P. Transforming growth factor-β1 induces transdifferentiation of myoblasts into myofibroblasts via up-regulation of sphingosine kinase-1/S1P3 axis. Mol. Biol. Cell 2010, 21, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Donati, C.; Nincheri, P.; Cencetti, F.; Rapizzi, E.; Farnararo, M.; Bruni, P. Tumor necrosis factor-α exerts pro-myogenic action in C2C12 myoblasts via sphingosine kinase/S1P2 signaling. FEBS Lett. 2007, 581, 4384–4388. [Google Scholar] [CrossRef] [PubMed]
- Nincheri, P.; Bernacchioni, C.; Cencetti, F.; Donati, C.; Bruni, P. Sphingosine kinase-1/S1P1 signalling axis negatively regulates mitogenic response elicited by PDGF in mouse myoblasts. Cell. Signal. 2010, 22, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Cencetti, F.; Bruno, G.; Blescia, S.; Bernacchioni, C.; Bruni, P.; Donati, C. Lysophosphatidic acid stimulates cell migration of satellite cells. A role for the sphingosine kinase/sphingosine 1-phosphate axis. FEBS J. 2014, 281, 4467–4478. [Google Scholar] [CrossRef] [PubMed]
- Meadows, K.A.; Holly, J.M.; Stewart, C.E. Tumor necrosis factor-alpha-induced apoptosis is associated with suppression of insulin-like growth factor binding protein-5 secretion in differentiating murine skeletal myoblasts. J. Cell. Physiol. 2000, 183, 330–337. [Google Scholar] [CrossRef]
- Mebarek, S.; Komati, H.; Naro, F.; Zeiller, C.; Alvisi, M.; Lagarde, M.; Prigent, A.F.; Nemoz, G. Inhibition of de novo ceramide synthesis upregulates phospholipase D and enhances myogenic differentiation. J. Cell Sci. 2007, 120, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Dulin, N.O.; Sorokin, A.; Reed, E.; Elliott, S.; Kehrl, J.H.; Dunn, M.J. RGS3 inhibits G protein-mediated signaling via translocation to the membrane and binding to Gα11. Mol. Cell. Biol. 1999, 19, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Scheschonka, A.; Dessauer, C.W.; Sinnarajah, S.; Chidiac, P.; Shi, C.S.; Kehrl, J.H. RGS3 is a GTPase-activating protein for Giα and Gqα and a potent inhibitor of signaling by GTPase-deficient forms of Gqα and G11α. Mol. Pharmacol. 2000, 58, 719–728. [Google Scholar] [PubMed]
- Esposito, G.; Prasad, S.V.; Rapacciuolo, A.; Mao, L.; Koch, W.J.; Rockman, H.A. Cardiac overexpression of a Gq inhibitor blocks induction of extracellular signal-regulated kinase and c-Jun NH2-terminal kinase activity in in vivo pressure overload. Circulation 2001, 103, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Meacci, E.; Donati, C.; Cencetti, F.; Romiti, E.; Farnararo, M.; Bruni, P. Receptor-activated phospholipase D is present in caveolin-3-enriched light membranes of C2C12 myotubes. FEBS Lett. 2000, 473, 10–14. [Google Scholar] [CrossRef]
- Meacci, E.; Vasta, V.; Donati, C.; Farnararo, M.; Bruni, P. Receptor-mediated activation of phospholipase D by sphingosine 1-phosphate in skeletal muscle C2C12 cells. A role for protein kinase C. FEBS Lett. 1999, 457, 184–188. [Google Scholar] [CrossRef]
- Lamour, N.F.; Subramanian, P.; Wijesinghe, D.S.; Stahelin, R.V.; Bonventre, J.V.; Chalfant, C.E. Ceramide 1-phosphate is required for the translocation of group IVA cytosolic phospholipase A2 and prostaglandin synthesis. J. Biol. Chem. 2009, 284, 26897–26907. [Google Scholar] [CrossRef] [PubMed]
- Stahelin, R.V.; Subramanian, P.; Vora, M.; Cho, W.; Chalfant, C.E. Ceramide-1-phosphate binds group IVA cytosolic phospholipase A2 via a novel site in the C2 domain. J. Biol. Chem. 2007, 282, 20467–20474. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Ann. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Mitra, P.; Maceyka, M.; Payne, S.G.; Lamour, N.; Milstien, S.; Chalfant, C.E.; Spiegel, S. Ceramide kinase regulates growth and survival of A549 human lung adenocarcinoma cells. FEBS Lett. 2007, 581, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, I.H.; Jeon, Y.J.; Chung, C.H.; Ha, D.B. Sphingosine blocks both membrane fusion and calmodulin-dependent phosphorylation of the 100-kDa protein of chick embryonic myoblasts. Exp. Cell Res. 1993, 205, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Ordonez, M.; Trueba, M.; Gomez-Munoz, A. Regulation of cell migration and inflammation by ceramide 1-phosphate. Biochim. Biophys. Acta 2016, 1861, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N.; Lin, F.T.; Tigyi, G.J. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim. Biophys. Acta 2013, 1831, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Gaits, F.; Fourcade, O.; le Balle, F.; Gueguen, G.; Gaige, B.; Gassama-Diagne, A.; Fauvel, J.; Salles, J.P.; Mauco, G.; Simon, M.F.; et al. Lysophosphatidic acid as a phospholipid mediator: Pathways of synthesis. FEBS Lett. 1997, 410, 54–58. [Google Scholar] [CrossRef]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; Hilaire, A.S.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARγ agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Jean-Baptiste, G.; Yang, Z.; Khoury, C.; Greenwood, M.T. Lysophosphatidic acid mediates pleiotropic responses in skeletal muscle cells. Biochem. Biophys. Res. Commun. 2005, 335, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [PubMed]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; An, S. Diversity of cellular receptors and functions for the lysophospholipid growth factors lysophosphatidic acid and sphingosine 1-phosphate. FASEB J. 1998, 12, 1589–1598. [Google Scholar] [PubMed]
- Kim, R.H.; Takabe, K.; Milstien, S.; Spiegel, S. Export and functions of sphingosine-1-phosphate. Biochim. Biophys. Acta 2009, 1791, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Belinson, J.; Morton, R.E.; Xu, Y. Phorbol 12-myristate 13-acetate stimulates lysophosphatidic acid secretion from ovarian and cervical cancer cells but not from breast or leukemia cells. Gynecol. Oncol. 1998, 71, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Shida, D.; Nagahashi, M.; Fang, X.; Milstien, S.; Takabe, K.; Spiegel, S. Lysophosphatidic acid stimulates gastric cancer cell proliferation via ERK1-dependent upregulation of sphingosine kinase 1 transcription. FEBS Lett. 2010, 584, 4077–4082. [Google Scholar] [CrossRef] [PubMed]
- Bruno, G.; Cencetti, F.; Pertici, I.; Japtok, L.; Bernacchioni, C.; Donati, C.; Bruni, P. CTGF/CCN2 exerts profibrotic action in myoblasts via the up-regulation of sphingosine kinase-1/S1P3 signaling axis: Implications in the action mechanism of TGFβ. Biochim. Biophys. Acta 2015, 1851, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Bernacchioni, C.; Ghini, V.; Cencetti, F.; Japtok, L.; Donati, C.; Bruni, P.; Turano, P. NMR metabolomics highlights sphingosine kinase-1 as a new molecular switch in the orchestration of aberrant metabolic phenotype in cancer cells. Mol. Oncol. 2017, 11, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Donati, C.; Marseglia, G.; Magi, A.; Serrati, S.; Cencetti, F.; Bernacchioni, C.; Nannetti, G.; Benelli, M.; Brunelli, S.; Torricelli, F.; et al. Sphingosine 1-phosphate induces differentiation of mesoangioblasts towards smooth muscle. A role for GATA6. PLoS ONE 2011, 6, e20389. [Google Scholar] [CrossRef] [PubMed]
- Cencetti, F.; Bernacchioni, C.; Tonelli, F.; Roberts, E.; Donati, C.; Bruni, P. TGFβ1 evokes myoblast apoptotic response via a novel signaling pathway involving S1P4 transactivation upstream of Rho-kinase-2 activation. FASEB J. 2013, 27, 4532–4546. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernacchioni, C.; Cencetti, F.; Ouro, A.; Bruno, M.; Gomez-Muñoz, A.; Donati, C.; Bruni, P. Lysophosphatidic Acid Signaling Axis Mediates Ceramide 1-Phosphate-Induced Proliferation of C2C12 Myoblasts. Int. J. Mol. Sci. 2018, 19, 139. https://doi.org/10.3390/ijms19010139
Bernacchioni C, Cencetti F, Ouro A, Bruno M, Gomez-Muñoz A, Donati C, Bruni P. Lysophosphatidic Acid Signaling Axis Mediates Ceramide 1-Phosphate-Induced Proliferation of C2C12 Myoblasts. International Journal of Molecular Sciences. 2018; 19(1):139. https://doi.org/10.3390/ijms19010139
Chicago/Turabian StyleBernacchioni, Caterina, Francesca Cencetti, Alberto Ouro, Marina Bruno, Antonio Gomez-Muñoz, Chiara Donati, and Paola Bruni. 2018. "Lysophosphatidic Acid Signaling Axis Mediates Ceramide 1-Phosphate-Induced Proliferation of C2C12 Myoblasts" International Journal of Molecular Sciences 19, no. 1: 139. https://doi.org/10.3390/ijms19010139
APA StyleBernacchioni, C., Cencetti, F., Ouro, A., Bruno, M., Gomez-Muñoz, A., Donati, C., & Bruni, P. (2018). Lysophosphatidic Acid Signaling Axis Mediates Ceramide 1-Phosphate-Induced Proliferation of C2C12 Myoblasts. International Journal of Molecular Sciences, 19(1), 139. https://doi.org/10.3390/ijms19010139