Epigenetic Modifiers in Myeloid Malignancies: The Role of Histone Deacetylase Inhibitors

Abstract

1. Introduction to Myeloid Hematological Diseases and Their Treatment
2. Epigenetic Regulation of Normal and Malignant Hematopoiesis
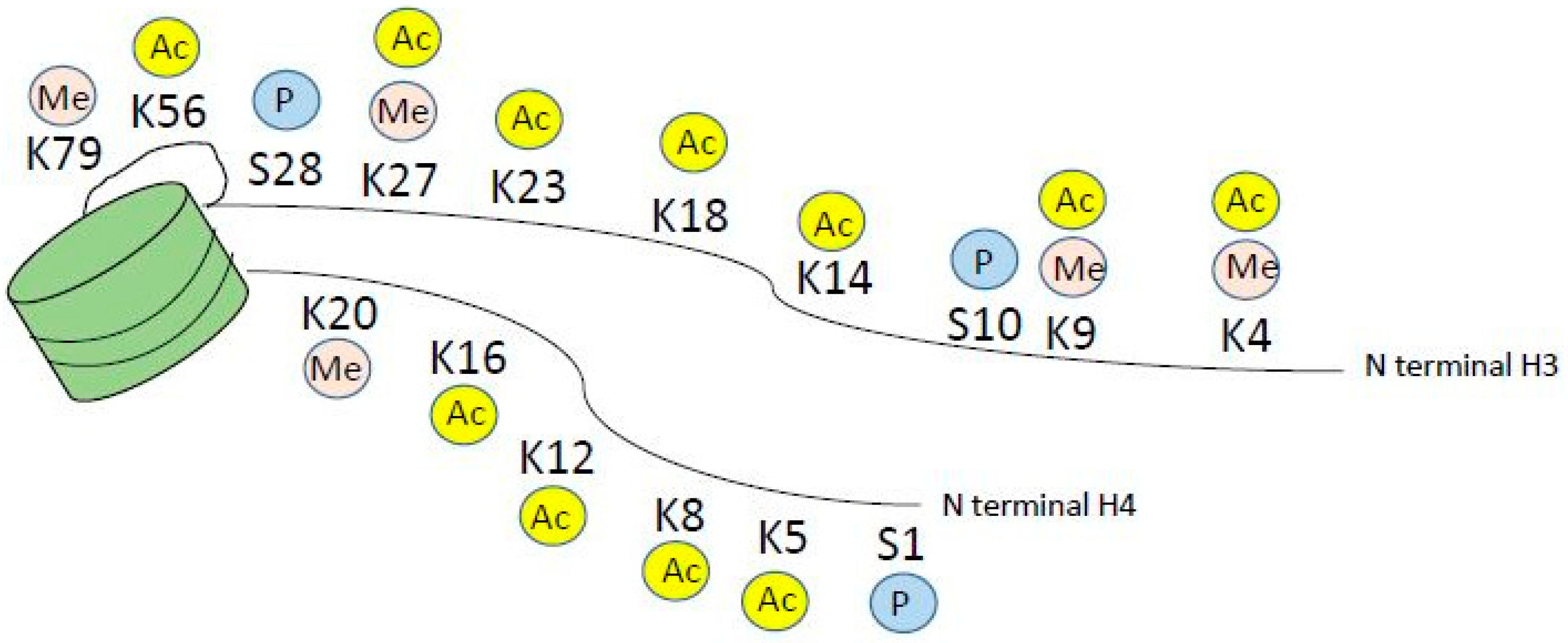
3. Dysregulation of Histone Acetylation and Methylation in Myeloid Malignancies
4. Preclinical Experience of HDACi in Myeloid Malignancies
5. Preclinical Rationale for Combination Therapy Including HDACi
6. Results from Clinical Studies of HDACi Monotherapy and Combination Therapy for Myeloid Malignancies
7. Why Have the Clinical Studies Failed?
8. Summary
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Madanat, Y.; Sekeres, M.A. Optimizing the use of hypomethylating agents in myelodysplastic syndromes: Selecting the candidate, predicting the response, and enhancing the activity. Semin. Hematol. 2017, 54, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, S.; Heil, O.; Lyko, F.; Brueckner, B. Azacytidine and decitabine induce gene-specific and non-random DNA demethylation in human cancer cell lines. PLoS ONE 2011, 6, e17388. [Google Scholar] [CrossRef] [PubMed]
- Daskalakis, M.; Nguyen, T.T.; Nguyen, C.; Guldberg, P.; Kohler, G.; Wijermans, P.; Jones, P.A.; Lubbert, M. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood 2002, 100, 2957–2964. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Schmidt-Mende, J.; Karimi, M.; Gogvadze, V.; Hassan, M.; Ekstrom, T.J.; Zhivotovsky, B.; Hellstrom-Lindberg, E. Hypomethylation and apoptosis in 5-azacytidine-treated myeloid cells. Exp. Hematol. 2008, 36, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Tobiasson, M.; Abdulkadir, H.; Lennartsson, A.; Katayama, S.; Marabita, F.; de Paepe, A.; Karimi, M.; Krjutskov, K.; Einarsdottir, E.; Grovdal, M.; et al. Comprehensive mapping of the effects of azacitidine on DNA methylation, repressive/permissive histone marks and gene expression in primary cells from patients with MDS and MDS-related disease. Oncotarget 2017, 8, 28812–28825. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.F.; Micklem, C.N.; Taguchi, M.; Itonaga, H.; Sawayama, Y.; Imanishi, D.; Nishikawa, S.; Miyazaki, Y.; Jakt, L.M. Longitudinal Analysis of DNA Methylation in CD34+ Hematopoietic Progenitors in Myelodysplastic Syndrome. Stem Cells Transl. Med. 2014, 3, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F. How I treat myelofibrosis. Blood 2014, 124, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Solary, E.; Itzykson, R. How I treat chronic myelomonocytic leukemia. Blood 2017, 130, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Chandesris, M.O.; Damaj, G.; Canioni, D.; Brouzes, C.; Lhermitte, L.; Hanssens, K.; Frenzel, L.; Cherquaoui, Z.; Durieu, I.; Durupt, S.; et al. Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2605–2607. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Reiter, A.; Scott, B.L.; Nakamura, R.; Damaj, G.; Kreil, S.; Shanley, R.; Hogan, W.J.; Perales, M.A.; Shore, T.; et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J. Clin. Oncol. 2014, 32, 3264–3274. [Google Scholar] [CrossRef] [PubMed]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [PubMed]
- Tenti, E.; Papayannidis, C.; Marconi, G.; Parisi, S.; Simonetti, G.; Paolini, S.; Sartor, C.; Ottaviani, E.; Testoni, N.; Martinelli, G. Efficacy of Azacitidine in the treatment of adult patients aged 65 years or older with AML. Expert Opin. Pharmacother. 2016, 17, 2479–2486. [Google Scholar] [CrossRef] [PubMed]
- Goyama, S.; Kitamura, T. Epigenetics in normal and malignant hematopoiesis: An overview and update 2017. Cancer Sci. 2017, 108, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Man, N.; Tan, Y.; Nimer, S.D.; Wang, L. The Role of Histone Acetyltransferases in Normal and Malignant Hematopoiesis. Front Oncol. 2015, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome, C. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Tefferi, A. Chronic myelomonocytic leukemia: 2018 update on diagnosis, risk stratification and management. Am. J. Hematol. 2018, 93, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Schischlik, F.; Kralovics, R. Mutations in myeloproliferative neoplasms-their significance and clinical use. Expert Rev. Hematol. 2017, 10, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.P.; et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013, 122, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Fenaux, P. Epigenetics of myelodysplastic syndromes. Leukemia 2014, 28, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug. Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, D.; Marschalek, R. How to effectively treat acute leukemia patients bearing MLL-rearrangements? Biochem. Pharmacol. 2018, 147, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Waters, N.J. Preclinical Pharmacokinetics and Pharmacodynamics of Pinometostat (EPZ-5676), a First-in-Class, Small Molecule S-Adenosyl Methionine Competitive Inhibitor of DOT1L. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Chaidos, A.; Caputo, V.; Karadimitris, A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: Emerging preclinical and clinical evidence. Ther. Adv. Hematol. 2015, 6, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Leaver, D.J.; Hermans, S.J.; Kelly, G.L.; Brennan, M.S.; Downer, N.L.; Nguyen, N.; Wichmann, J.; McRae, H.M.; Yang, Y.; et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 2018, 560, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Puda, A.; Milosevic, J.D.; Berg, T.; Klampfl, T.; Harutyunyan, A.S.; Gisslinger, B.; Rumi, E.; Pietra, D.; Malcovati, L.; Elena, C.; et al. Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. Am. J. Hematol. 2012, 87, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; McGirr, C.; Shakhbazov, K.; Barbier, V.; Krishnan, K.; Mukhopadhyay, P.; Hawthorne, P.; Trezise, A.; Ding, J.; Grimmond, S.M.; et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 2014, 123, 2682–2690. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Singh, S.; Hua, W.K.; Cai, Q.; Chao, S.W.; Li, L.; Liu, H.; Ho, Y.; McDonald, T.; Lin, A.; et al. HDAC8 Inhibition Specifically Targets Inv (16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation. Cell Stem Cell 2015, 17, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Maddipoti, S.; Quesada, A.; Bohannan, Z.; Cabrero Calvo, M.; Colla, S.; Wei, Y.; Estecio, M.; Wierda, W.; Bueso-Ramos, C.; et al. Analysis of class I and II histone deacetylase gene expression in human leukemia. Leuk. Lymphoma 2015, 56, 3426–3433. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Chen, C.; Dumlao, T.; Naik, S.; Chang, T.; Xiao, Y.Y.; Sominsky, I.; Burton, J. Enhanced histone deacetylase enzyme activity in primary myelofibrosis. Leuk. Lymphoma 2008, 49, 2321–2327. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Newbold, A.; Falkenberg, K.J.; Prince, H.M.; Johnstone, R.W. How do tumor cells respond to HDAC inhibition? FEBS J. 2016, 283, 4032–4046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef] [PubMed]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Marks, P.A. Histone deacetylase inhibitors in the therapy of cancer: Much to learn. Epigenomics 2010, 2, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, H.; Towatari, M.; Hatano, S.; Kitamura, K.; Kiyoi, H.; Kinoshita, T.; Tanimoto, M.; Murate, T.; Kawashima, K.; Saito, H.; et al. Histone deacetylase inhibitors are the potent inducer/enhancer of differentiation in acute myeloid leukemia: A new approach to anti-leukemia therapy. Leukemia 1999, 13, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Saunthararajah, Y.; Redner, R.L.; Liu, J.M. Inhibitors of histone deacetylase relieve ETO-mediated repression and induce differentiation of AML1-ETO leukemia cells. Cancer Res. 1999, 59, 2766–2769. [Google Scholar] [PubMed]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, G.; Medici, M.C.; Russo, G.L. Valproic acid sensitizes K562 erythroleukemia cells to TRAIL/Apo2L-induced apoptosis. Anticancer Res. 2008, 28, 855–864. [Google Scholar] [PubMed]
- Silva, G.; Cardoso, B.A.; Belo, H.; Almeida, A.M. Vorinostat induces apoptosis and differentiation in myeloid malignancies: Genetic and molecular mechanisms. PLoS ONE 2013, 8, e53766. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ruvolo, V.R.; McQueen, T.; Chen, W.; Samudio, I.J.; Conneely, O.; Konopleva, M.; Andreeff, M. HDAC inhibition by SNDX-275 (Entinostat) restores expression of silenced leukemia-associated transcription factors Nur77 and Nor1 and of key pro-apoptotic proteins in AML. Leukemia 2013, 27, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Klisovic, R.B.; Vukosavljevic, T.; Yu, J.; Paschka, P.; Huynh, L.; Pang, J.; Neviani, P.; Liu, Z.; Blum, W.; et al. Targeting AML1/ETO-histone deacetylase repressor complex: A novel mechanism for valproic acid-mediated gene expression and cellular differentiation in AML1/ETO-positive acute myeloid leukemia cells. J. Pharmacol. Exp. Ther. 2007, 321, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Akada, H.; Akada, S.; Gajra, A.; Bair, A.; Graziano, S.; Hutchison, R.E.; Mohi, G. Efficacy of vorinostat in a murine model of polycythemia vera. Blood 2012, 119, 3779–3789. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fiskus, W.; Chong, D.G.; Buckley, K.M.; Natarajan, K.; Rao, R.; Joshi, A.; Balusu, R.; Koul, S.; Chen, J.; et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 2009, 114, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Fenger, J.; Murahari, S.; Bear, M.D.; Kulp, S.K.; Wang, D.; Chen, C.S.; Kisseberth, W.C.; London, C.A. AR-42, a novel HDAC inhibitor, exhibits biologic activity against malignant mast cell lines via down-regulation of constitutively activated Kit. Blood 2010, 115, 4217–4225. [Google Scholar] [CrossRef] [PubMed]
- Lyberg, K.; Ali, H.A.; Grootens, J.; Kjellander, M.; Tirfing, M.; Arock, M.; Hagglund, H.; Nilsson, G.; Ungerstedt, J. Histone deacetylase inhibitor SAHA mediates mast cell death and epigenetic silencing of constitutively active D816V KIT in systemic mastocytosis. Oncotarget 2017, 8, 9647–9659. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, G.; Mancini, M.; De Benedittis, C.; Rondoni, M.; Papayannidis, C.; Manfrini, M.; Meggendorfer, M.; Calogero, R.; Guadagnuolo, V.; Fontana, M.C.; et al. SETD2 and histone H3 lysine 36 methylation deficiency in advanced systemic mastocytosis. Leukemia 2018, 32, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myohanen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Urbanavicius, D.; Tan, P.; Spencer, A.; Dear, A.E. Mechanisms and potential molecular markers of early response to combination epigenetic therapy in patients with myeloid malignancies. Int. J. Oncol. 2014, 45, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Shaker, S.; Bernstein, M.; Momparler, L.F.; Momparler, R.L. Preclinical evaluation of antineoplastic activity of inhibitors of DNA methylation (5-aza-2′-deoxycytidine) and histone deacetylation (trichostatin A, depsipeptide) in combination against myeloid leukemic cells. Leuk. Res. 2003, 27, 437–444. [Google Scholar] [CrossRef]
- Fiskus, W.; Buckley, K.; Rao, R.; Mandawat, A.; Yang, Y.; Joshi, R.; Wang, Y.; Balusu, R.; Chen, J.; Koul, S.; et al. Panobinostat treatment depletes EZH2 and DNMT1 levels and enhances decitabine mediated de-repression of JunB and loss of survival of human acute leukemia cells. Cancer Biol. Ther. 2009, 8, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Wei, A.; Mithraprabhu, S.; Cummings, N.; Liu, H.B.; Perugini, M.; Reed, K.; Avery, S.; Patil, S.; Walker, P.; et al. Dual epigenetic targeting with panobinostat and azacitidine in acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood Cancer J. 2014, 4, e170. [Google Scholar] [CrossRef] [PubMed]
- Morabito, F.; Voso, M.T.; Hohaus, S.; Gentile, M.; Vigna, E.; Recchia, A.G.; Iovino, L.; Benedetti, E.; Lo-Coco, F.; Galimberti, S. Panobinostat for the treatment of acute myelogenous leukemia. Expert Opin. Investig. Drugs 2016, 25, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Gore, S.D.; Vey, N.; Prebet, T. Lost in translation? Ten years of development of histone deacetylase inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Expert Opin. Investig. Drugs 2016, 25, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.; Gojo, I.; Goldberg, S.L.; Bredeson, C.; Kujawski, L.A.; Yang, A.; Marks, P.; Frankel, P.; Sun, X.; Tosolini, A.; et al. A phase 1 clinical trial of vorinostat in combination with decitabine in patients with acute myeloid leukaemia or myelodysplastic syndrome. Br. J. Haematol. 2014, 167, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Munakata, W.; Ogura, M.; Uchida, T.; Taniwaki, M.; Kobayashi, T.; Shimada, F.; Yonemura, M.; Matsuoka, F.; Tajima, T.; et al. Phase I study of panobinostat and 5-azacitidine in Japanese patients with myelodysplastic syndrome or chronic myelomonocytic leukemia. Int. J. Hematol. 2018, 107, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Qi, J.; You, T.; Yang, L.; Wu, D.; Han, Y.; Zhu, L. Addition of histone deacetylase inhibitors does not improve prognosis in patients with myelodysplastic syndrome and acute myeloid leukemia compared with hypomethylating agents alone: A systematic review and meta-analysis of seven prospective cohort studies. Leuk. Res. 2018, 71, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Garcia-Manero, G.; Huang, X.; Cortes, J.; Ravandi, F.; Jabbour, E.; Borthakur, G.; Brandt, M.; Pierce, S.; Kantarjian, H.M. Results of phase 2 randomized study of low-dose decitabine with or without valproic acid in patients with myelodysplastic syndrome and acute myelogenous leukemia. Cancer 2015, 121, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Othus, M.; List, A.F.; Odenike, O.; Stone, R.M.; Gore, S.D.; Litzow, M.R.; Buckstein, R.; Fang, M.; Roulston, D.; et al. Randomized Phase II Study of Azacitidine Alone or in Combination with Lenalidomide or With Vorinostat in Higher-Risk Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia: North American Intergroup Study SWOG S1117. J. Clin. Oncol. 2017, 35, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Sekeres, M.A.; Egyed, M.; Breccia, M.; Graux, C.; Cavenagh, J.D.; Salman, H.; Illes, A.; Fenaux, P.; DeAngelo, D.J.; et al. A phase 1b/2b multicenter study of oral panobinostat plus azacitidine in adults with MDS, CMML or AML with 30% blasts. Leukemia 2017, 31, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Montalban-Bravo, G.; Berdeja, J.G.; Abaza, Y.; Jabbour, E.; Essell, J.; Lyons, R.M.; Ravandi, F.; Maris, M.; Heller, B.; et al. Phase 2, randomized, double-blind study of pracinostat in combination with azacitidine in patients with untreated, higher-risk myelodysplastic syndromes. Cancer 2017, 123, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Sun, Z.; Figueroa, M.E.; Ketterling, R.; Melnick, A.; Greenberg, P.L.; Herman, J.; Juckett, M.; Smith, M.R.; Malick, L.; et al. Prolonged administration of azacitidine with or without entinostat for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia-related changes: Results of the US Leukemia Intergroup trial E1905. J. Clin. Oncol. 2014, 32, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Sun, Z.; Ketterling, R.P.; Zeidan, A.; Greenberg, P.; Herman, J.; Juckett, M.; Smith, M.R.; Malick, L.; Paietta, E.; et al. Eastern Cooperative Oncology G, North American Leukemia i: Azacitidine with or without Entinostat for the treatment of therapy-related myeloid neoplasm: Further results of the E1905 North American Leukemia Intergroup study. Br. J. Haematol. 2016, 172, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Duncavage, E.J.; Chang, G.S.; Jacoby, M.A.; Miller, C.A.; Shao, J.; Heath, S.; Elliott, K.; Reineck, T.; Fulton, R.S.; et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. Leukemia 2017, 31, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Fredly, H.; Gjertsen, B.T.; Bruserud, O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: The effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. Epigenetics 2013, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Grishina, O.; Schmoor, C.; Dohner, K.; Hackanson, B.; Lubrich, B.; May, A.M.; Cieslik, C.; Muller, M.J.; Lubbert, M. DECIDER: Prospective randomized multicenter phase II trial of low-dose decitabine (DAC) administered alone or in combination with the histone deacetylase inhibitor valproic acid (VPA) and all-trans retinoic acid (ATRA) in patients >60 years with acute myeloid leukemia who are ineligible for induction chemotherapy. BMC Cancer 2015, 15, 430. [Google Scholar]
- Bose, P.; Verstovsek, S. Investigational histone deacetylase inhibitors (HDACi) in myeloproliferative neoplasms. Expert Opin. Investig. Drugs 2016, 25, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; McMullin, M.F.; Ejerblad, E.; Zweegman, S.; Harrison, C.; Fernandes, S.; Bareford, D.; Knapper, S.; Samuelsson, J.; Lofvenberg, E.; et al. A phase II study of vorinostat (MK-0683) in patients with polycythaemia vera and essential thrombocythaemia. Br. J. Haematol. 2013, 162, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Vannucchi, A.M.; Martinelli, V.; Ruggeri, M.; Nobile, F.; Specchia, G.; Pogliani, E.M.; Olimpieri, O.M.; Fioritoni, G.; Musolino, C.; et al. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br. J. Haematol. 2013, 161, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Rambaldi, A.; Dellacasa, C.M.; Finazzi, G.; Carobbio, A.; Ferrari, M.L.; Guglielmelli, P.; Gattoni, E.; Salmoiraghi, S.; Finazzi, M.C.; Di Tollo, S.; et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br. J. Haematol. 2010, 150, 446–455. [Google Scholar] [PubMed]
- Quintas-Cardama, A.; Kantarjian, H.; Estrov, Z.; Borthakur, G.; Cortes, J.; Verstovsek, S. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk. Res. 2012, 36, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Sandy, L.; Lu, M.; Yoon, J.; Petersen, B.; Zhang, D.; Ye, F.; Newsom, C.; Najfeld, V.; Hochman, T.; et al. A phase II study of panobinostat in patients with primary myelofibrosis (PMF) and post-polycythemia vera/essential thrombocythemia myelofibrosis (post-PV/ET MF). Leuk. Res. 2017, 53, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ball, B.; Zeidan, A.; Gore, S.D.; Prebet, T. Hypomethylating agent combination strategies in myelodysplastic syndromes: Hopes and shortcomings. Leuk. Lymphoma 2017, 58, 1022–1036. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D. Epigenetic therapies in MDS and AML. Adv. Exp. Med. Biol. 2013, 754, 253–283. [Google Scholar] [PubMed]
- Stahl, M.; Zeidan, A.M. Hypomethylating agents in combination with histone deacetylase inhibitors in higher risk myelodysplastic syndromes: Is there a light at the end of the tunnel? Cancer 2017, 123, 911–914. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Gene | Loss/Gain of Function | Activity | Frequency in Myeloid Malignancies |
|---|---|---|---|---|
| DNA methylation | DNMT3A | loss | De novo DNA methylation | AML 12–22% |
| MDS 5–10% | ||||
| CMML 5% | ||||
| MPN 7–15% | ||||
| ASM 1% | ||||
| DNA methylation | TET2 | loss | 5-methyl-C to 5-hydroxy methyl-C | AML 7–23% |
| MDS 20–25% | ||||
| CMML 60% | ||||
| MPN 4–13% | ||||
| ASM 40% | ||||
| DNA methylation | IDH1/2 | gain | Cofactor for TET2 | AML 10–30% |
| MDS 3% | ||||
| CMML 1–10% | ||||
| MPN 2.5–5% | ||||
| Histone methylation | EZH2 | Loss | Trimethylation of H3K27, part of PRC2 complex | AML rare |
| MDS 6% | ||||
| CMML 5% | ||||
| MPN 3–13% | ||||
| ASM 3% | ||||
| Histone methylation | ASXL1 | loss | Associates with PRC1 and PRC2 | AML 5% |
| MDS 15–20% | ||||
| CMML 40–45% | ||||
| MPN 2–23% | ||||
| ASM 14% | ||||
| Histone methylation | SUZ12 | loss | Member of PRC2 | MDS rare, <1% |
| Histone methylation | EED | loss | Member of PRC2 | MDS rare, <1% |
| Histone methylation | KMT2A (MLL1) | gain | H3K4 lysine methyl transferase | AML 5% |
| MDS/AML 5% | ||||
| Histone methylation | MECOM (EVI1) | gain | H3K9(me1) lysine methyl transferase | MDS/AML rare |
| Histone methylation | PRDM16 | gain | H3K9(me1) lysine methyl transferase | MDS/AML rare |
| Histone methylation | SETD2 | loss | H3K36 lysine methyl transferase | AML 5% |
| Histone methylation | JARID2 | Recruits PRC2 to target | sAML(from MDS, MPN) 6.5% | |
| MDS, MPN 0.2% | ||||
| Histone methylation | UTX (=KDM6A) | loss | Counteracts PRC2 by removing di and trimethylated H3K27 | AML 3% |
| MDS 2.5% | ||||
| CMML 8% | ||||
| MDS/MPN 4.8% | ||||
| Histone acetylation | CREBBP (CBP) | gain | Lysine acetyl transferase | AML rare |
| Histone acetylation | P300 (EP300) | gain | Lysine acetyl transferase | AML rare |
| Histone deacetylation | HDAC2 | loss | Lysine deacetylase | AML rare |
| Histone deacetylation | HDAC3 | loss | Lysine deacetylase | AML rare |
| Study, Trial Number and Reference | Disease, Phase | Additive Clinical Effect of HDACi | Drugs | Clinical Response | Molecular Markers Analyzed |
|---|---|---|---|---|---|
| Tan [62], ACTRN12610000924055, Open label, phase Ib/II | Higher risk MDS, AML. n = 39 | NA | Azacitidine 1, Panobinostat 2 | ORR 31% in AML, 50% in MDS. Median OS 8 months in AML, 16 months in MDS. | Total PBMC histone H3 and H4 acetylation higher in responders. NUR77 and p21 markers of treatment efficacy [59] |
| Issa [68], NCT00414310, Randomized, Phase II | Higher risk MDS, AML. n = 149 | NO | Decitabine 3, valproic acid 4 | No improvement in CR or OS with adding valproic acid. | NO |
| Sekeres [69], NCT01522976, Randomized, Phase II | Higher risk MDS, CMML. n = 184 | NO | Azacitidine 5, Vorinostat 6 | ORR 38% monotherapy, 27% combination (p = 0.16). Study not powered for calculating OS. | NGS. ORR was higher in DNMT3A mutated patients. ORR lower for SRSF2 and ASXL1. Response duration low in TET2 and TP53 mutated patients. |
| Garcia-Manero [70], NCT00946647, Randomized phase Ib/II | MDS, CMML AML with 20–30% blasts. n = 113 | NO | Panobinostat 7, Azacitidine 5 | CR 27.5% in the combination arm, 14.3% in monotherapy. No difference in OS or time to progression. | NGS data on 24 myeloid mutations, no clear correlation between mutation pattern and response. |
| Garcia-Manero [71], NCT01873703, Randomized phase II, double blinded | MDS (up to 30% blasts). n = 102 | NO | Azacitidine 5, Pracinostat 8 | CR 18% in the combination group, 33% in monotherapy group (p = 0.07). No difference in OS (16 vs. 19 months). | NO |
| Prebet [72], NCT00313586, Prebet [73], Open label phase II | MDS, CMML, MDS/AML. n = 149 | NO | Azacitidine 9, entinostat 10 | OS 18 months for monotherapy, 13 for combination. | No correlation between overall methylation decrease and clinical response, or with treatment arm. Possible correlation of SOCS1 methylation and response. |
| Uy [74], NCT00691938, Open label observational phase I/II | AML, MDS. n = 52 | NA | Decitabine 3, panobinostat 11 | ORR 11/37 AML and 7/14 MDS, total 36% ORR. Median OS 6.4 months. | Extensive sequencing, complex patterns. Mutations persist during complete remission. |
| Drug Type | Compound | Name | Selectivity | Clinical Status | Used in Myeloid Disease |
|---|---|---|---|---|---|
| Hydroxamates | MK0653 (SAHA) | Vorinostat | Pan HDACi | Phase II/III. Approved. | Yes. Single and combination |
| LBH589 | Panobinostat | Pan HDACi | Phase II/III. Approved. | Yes. Single and combination | |
| PXD101 | Belinostat | Pan HDACi | Phase I/ II/III. Approved. | Yes. Combination therapy | |
| JNJ-26481585 | Quisinostat | HDAC1,3,5,8 | Phase I/II | MDS and AML. Single therapy | |
| ITF2357 | Givinostat | Class I and II | Phase I/II | MPN. Single and combination | |
| SB939 | Pracinostat | Class I, II, IV | Phase II | Yes. Single and combination | |
| SHP141 | Remetinostat | Phase II/III | No | ||
| 4SC201 | Resminostat | Pan HDACi | Phase I/II | No | |
| 4SC202 | Domatinostat | HDAC1,2,3 | Phase I/II Approved in melanoma (combination) | Yes. Single therapy | |
| ACY1215 | Ricolinostat | HDAC6 | Phase I/II | No | |
| Cyclic tetrapeptides | FK228 | Romidepsin | Class I | Phase I/II/III. Approved. | Yes. Single and combination |
| Benzamides | MS275 | Entinostat | HDAC1,2,3 | Phase I/II | Yes Combination therapy |
| MGCD0103 | Mocetinostat | Class I | Phase I/II | Yes Single and combination | |
| Fatty acids | Valproic acid | Valproate | Class I and IIa | Phase I/ II | Yes Combination therapy |
| Sodium Butyrate | Butyrate | Class I and IIa | Phase I/II | Mostly non cancer diseases |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungerstedt, J.S. Epigenetic Modifiers in Myeloid Malignancies: The Role of Histone Deacetylase Inhibitors. Int. J. Mol. Sci. 2018, 19, 3091. https://doi.org/10.3390/ijms19103091
Ungerstedt JS. Epigenetic Modifiers in Myeloid Malignancies: The Role of Histone Deacetylase Inhibitors. International Journal of Molecular Sciences. 2018; 19(10):3091. https://doi.org/10.3390/ijms19103091
Chicago/Turabian StyleUngerstedt, Johanna S. 2018. "Epigenetic Modifiers in Myeloid Malignancies: The Role of Histone Deacetylase Inhibitors" International Journal of Molecular Sciences 19, no. 10: 3091. https://doi.org/10.3390/ijms19103091
APA StyleUngerstedt, J. S. (2018). Epigenetic Modifiers in Myeloid Malignancies: The Role of Histone Deacetylase Inhibitors. International Journal of Molecular Sciences, 19(10), 3091. https://doi.org/10.3390/ijms19103091