A New Synthetic Histone Deacetylase Inhibitor, MHY2256, Induces Apoptosis and Autophagy Cell Death in Endometrial Cancer Cells via p53 Acetylation


 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. MHY2256 Is Highly Cytotoxicity to Ishikawa Endometrial Cancer Cells
2.2. MHY2256 Reduces Both SIRT1 Enzyme Activity and SIRT Protein Levels in Ishikawa Cells
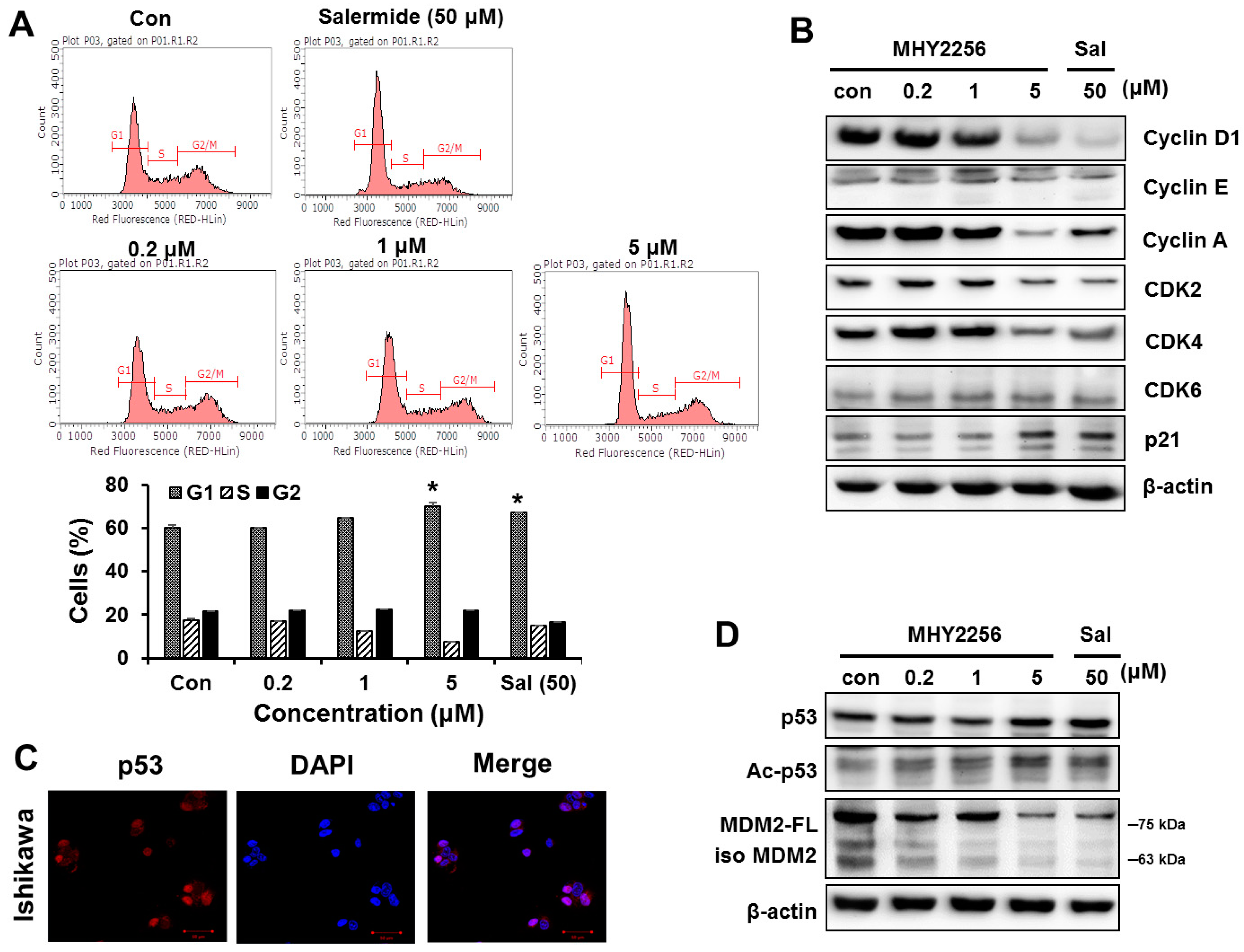
2.3. MHY2256 Inhibits Cell Cycle Distribution
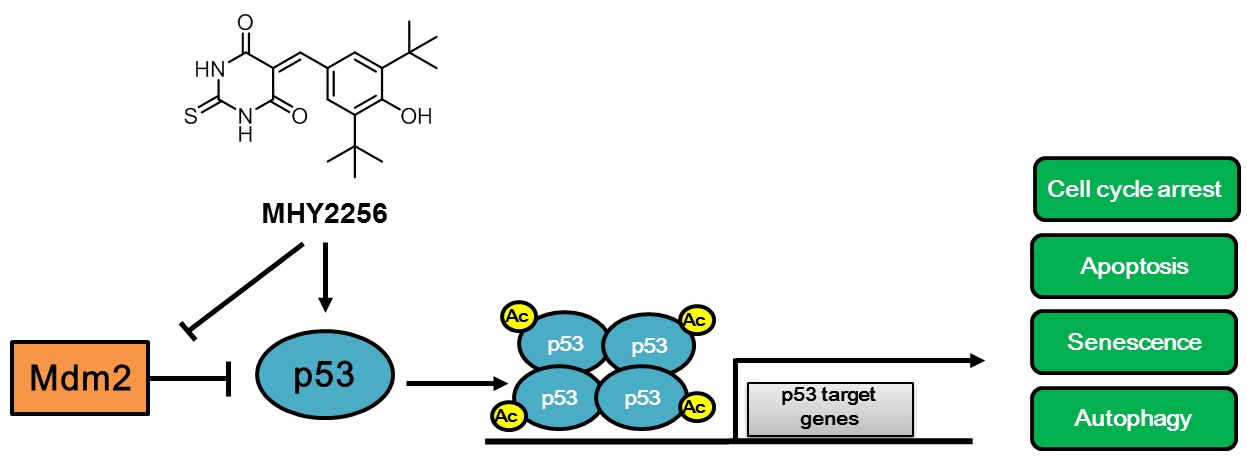
2.4. MHY2256 the Steady State Level of p53 Protein in Increase via Downregulation of the MDM2 Expression
2.5. MHY2256 Induces Apoptotic Cellular Death in Ishikawa Cells
2.6. MHY2256 Induces Autophagic Cell Death
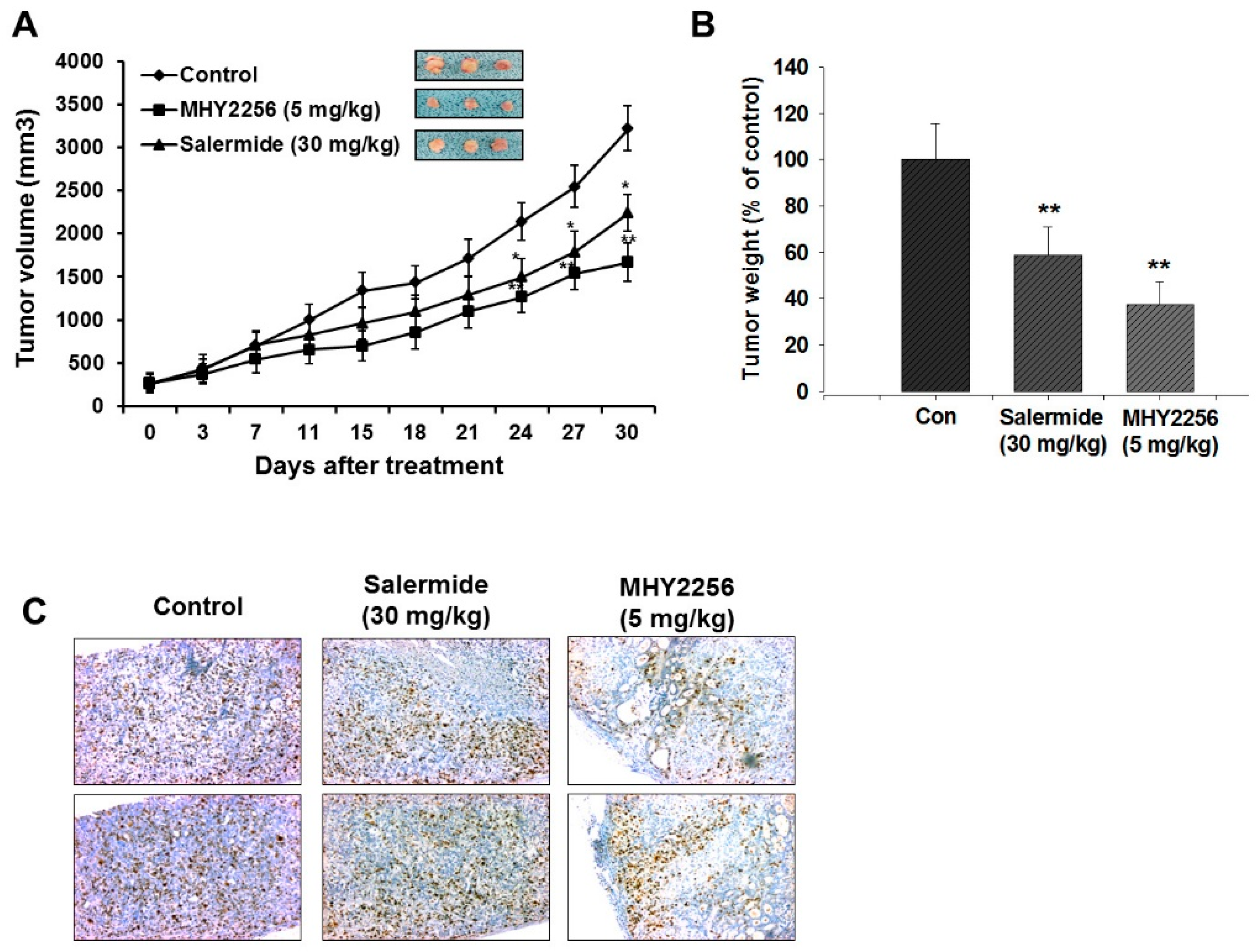
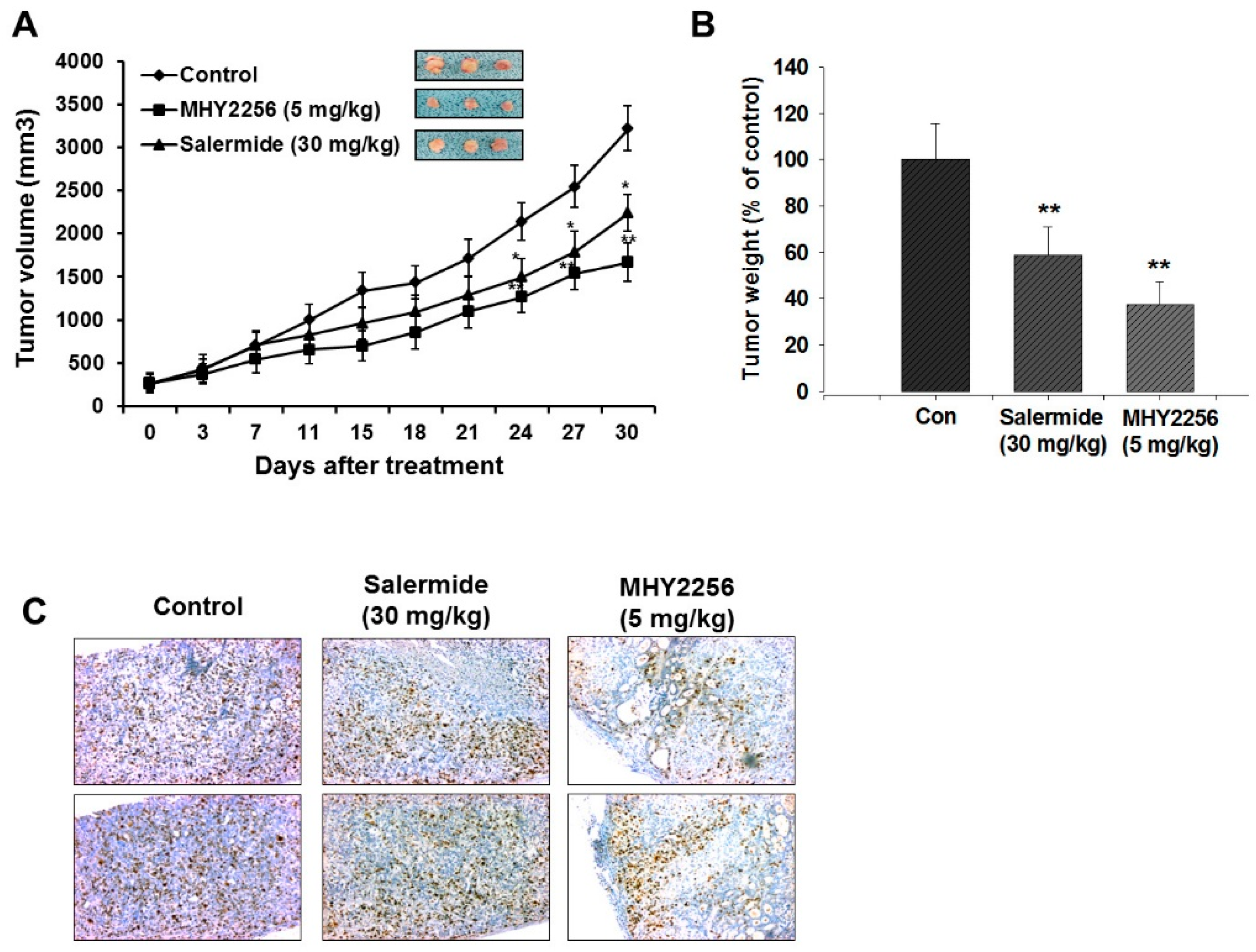
2.7. MHY2256 Inhibits Ishikawa Endometric Cell Tumors in a Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. Synthesis of MHY2256
4.3. Cell Culture
4.4. SIRT1 Activity Assay
4.5. Cytotoxicity Assay
4.6. Western Blot Analysis
4.7. Flow Cytometry Analysis
4.8. Annexin V-FITC/PI Binding Assay
4.9. Detection of Acidic Vesicular Organelles (AVOs)
4.10. In Vivo Tumor Xenograft Model
4.11. Statistical Methods
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Amant, F.; Moerman, P.; Neven, P.; Timmerman, D.; Van Limbergen, E.; Vergote, I. Endometrial cancer. Lancet 2005, 366, 491–505. [Google Scholar] [CrossRef]
- Tran, A.Q.; Gehrig, P. Recent Advances in Endometrial Cancer. F1000Research 2017, 6, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arend, R.C.; Jones, B.A.; Martinez, A.; Goodfellow, P. Endometrial cancer: Molecular markers and management of advanced stage disease. Gynecol. Oncol. 2018, 150, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Matias-Guiu, X.; Prat, J. Molecular pathology of endometrial carcinoma. Histopathology 2013, 62, 111–123. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, J.T.; Farrington, W.J.; Tollefsbol, T.O. An overview of epigenetic assays. Mol. Biotechnol. 2008, 38, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Garmpi, A.; Spartalis, E.; Kalampokas, E.; Kalampokas, T.; Margonis, G.A.; Schizas, D.; Andreatos, N.; Angelou, A.; Lavaris, A.; et al. Targeting histone deacetylases in endometrial cancer: A paradigm-shifting therapeutic strategy? Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 950–960. [Google Scholar]
- Baek, M.H.; Park, J.Y.; Rhim, C.C.; Kim, J.H.; Park, Y.; Kim, K.R.; Nam, J.H. Investigation of new therapeutic targets in undifferentiated endometrial sarcoma. Gynecol. Obstet. Investig. 2017, 82, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Desmond, J.C.; Kumagai, T.; Gui, D.; Said, J.W.; Whittaker, S.; Miyakawa, I.; Koeffler, H.P. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin. Cancer Res. 2004, 10, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tsai, Y.H.; Tseng, S.H. HDAC inhibitors and RECK modulate endoplasmic reticulum stress in tumor cells. Int. J. Mol. Sci. 2017, 18, 258. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef] [PubMed]
- Li, L.H.; Zhang, P.R.; Cai, P.Y.; Li, Z.C. Histone deacetylase inhibitor, Romidepsin (FK228) inhibits endometrial cancer cell growth through augmentation of p53–p21 pathway. Biomed. Pharmacother. 2016, 82, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Gonfloni, S.; Iannizzotto, V.; Maiani, E.; Bellusci, G.; Ciccone, S.; Diederich, M. P53 and Sirt1: Routes of metabolism and genome stability. Biochem. Pharmacol. 2014, 92, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S. Human SIRT1: A potential biomarker for tumorigenesis? Cell Biol. Int. 2007, 31, 636–637. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.L.; Dai, Y.; Faller, D.V. Sirtuin 1 (SIRT1) and steroid hormone receptor activity in cancer. J. Endocrinol. 2012, 213, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.L.; Faller, D.V. SIRT1 represses estrogen-signaling, ligand-independent ERα-mediated transcription, and cell proliferation in estrogen-responsive breast cells. J. Endocrinol. 2013, 216, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Park, E.Y.; Woo, Y.; Kim, S.J.; Kim, D.H.; Lee, E.K.; De, U.; Kim, K.S.; Lee, J.; Jung, J.H.; Ha, K.T.; et al. Anticancer effects of a new SIRT inhibitor, MHY2256, against human breast cancer MCF-7 Cells via regulation of MDM2-p53 binding. Int. J. Biol. Sci. 2016, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, B.; Gao, W.; Huang, S.; Liu, Z.; Li, W.; Jia, J. SIRT1 is downregulated in gastric cancer and leads to G1-phase arrest via NF-κB/Cyclin D1 signaling. Mol. Cancer Res. 2013, 11, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Chen, C.Y.; Ho, K.K.; Di Fruscia, P.; Myatt, S.S.; Coombes, R.C.; Fuchter, M.J.; Hsiao, C.D.; Lam, E.W. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 2010, 9, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.; Breitenbücher, F.; Schuler, M.; Ehrenhofer-Murray, A.E. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J. Biol. Chem. 2014, 289, 5208–5216. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kim, T.H.; Ahn, M.Y.; Lee, J.; Jung, J.H.; Choi, W.S.; Lee, B.M.; Yoon, K.S.; Yoon, S.; Kim, H.S. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Lara, E.; Mai, A.; Calvanese, V.; Altucci, L.; Lopez-Nieva, P.; Martinez-Chantar, M.L.; Varela-Rey, M.; Rotili, D.; Nebbioso, A.; Ropero, S.; et al. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 2009, 28, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Heckman, C.A.; Boxer, L.M. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol. Cell. Biol. 2005, 25, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Ahn, S.H.; Kim, Y.K.; Bae, G.U.; Yoon, J.W.; Hong, S.; Lee, H.Y.; Lee, Y.W.; Lee, H.W.; Han, J.W. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 2002, 277, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Librizzi, M.; Spencer, J.; Luparello, C. Biological effect of a hybrid anticancer agent based on kinase and histone deacetylase inhibitors on triple-negative (MDA-MB231) breast cancer cells. Int. J. Mol. Sci. 2016, 17, 1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Xu, M.; Wu, B.; Chen, L. Histone deacetylases in hearing loss: Current perspectives for therapy. J. Otol. 2017, 12, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Won, A.J.; Lee, J.; Jung, J.H.; Yoon, S.; Lee, B.M.; Kim, H.S. Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells. Int. J. Med. Sci. 2012, 9, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Jung, J.H.; Na, Y.J.; Kim, H.S. A natural histone deacetylase inhibitor, Psammaplin A, induces cell cycle arrest and apoptosis in human endometrial cancer cells. Gynecol. Oncol. 2008, 108, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Chung, H.Y.; Choi, W.S.; Lee, B.M.; Yoon, S.; Kim, H.S. Anti-tumor effect of apicidin on Ishikawa human endometrial cancer cells both in vitro and in vivo by blocking histone deacetylase 3 and 4. Int. J. Oncol. 2010, 36, 125–131. [Google Scholar] [PubMed]
- Lin, P.-C.; Hsieh, H.-Y.; Chu, P.-C.; Chen, C.S. Therapeutic opportunities of targeting histone deacetylase isoforms to eradicate cancer stem cells. Int. J. Mol. Sci. 2018, 19, 1939. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, C.; Monteiro-Reis, S.; Almeida-Rios, D.; Vieira, R.; Castro, A.; Moutinho, M.; Rodrigues, M.; Graça, I.; Lopes, J.M.; Jerónimo, C. Assessing sirtuin expression in endometrial carcinoma and non-neoplastic endometrium. Oncotarget 2016, 7, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Luo, J.; Brooks, C.L.; Gu, W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 2002, 277, 50607–50611. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De, U.; Son, J.Y.; Sachan, R.; Park, Y.J.; Kang, D.; Yoon, K.; Lee, B.M.; Kim, I.S.; Moon, H.R.; Kim, H.S. A New Synthetic Histone Deacetylase Inhibitor, MHY2256, Induces Apoptosis and Autophagy Cell Death in Endometrial Cancer Cells via p53 Acetylation. Int. J. Mol. Sci. 2018, 19, 2743. https://doi.org/10.3390/ijms19092743
De U, Son JY, Sachan R, Park YJ, Kang D, Yoon K, Lee BM, Kim IS, Moon HR, Kim HS. A New Synthetic Histone Deacetylase Inhibitor, MHY2256, Induces Apoptosis and Autophagy Cell Death in Endometrial Cancer Cells via p53 Acetylation. International Journal of Molecular Sciences. 2018; 19(9):2743. https://doi.org/10.3390/ijms19092743
Chicago/Turabian StyleDe, Umasankar, Ji Yeon Son, Richa Sachan, Yu Jin Park, Dongwan Kang, Kyungsil Yoon, Byung Mu Lee, In Su Kim, Hyung Ryong Moon, and Hyung Sik Kim. 2018. "A New Synthetic Histone Deacetylase Inhibitor, MHY2256, Induces Apoptosis and Autophagy Cell Death in Endometrial Cancer Cells via p53 Acetylation" International Journal of Molecular Sciences 19, no. 9: 2743. https://doi.org/10.3390/ijms19092743
APA StyleDe, U., Son, J. Y., Sachan, R., Park, Y. J., Kang, D., Yoon, K., Lee, B. M., Kim, I. S., Moon, H. R., & Kim, H. S. (2018). A New Synthetic Histone Deacetylase Inhibitor, MHY2256, Induces Apoptosis and Autophagy Cell Death in Endometrial Cancer Cells via p53 Acetylation. International Journal of Molecular Sciences, 19(9), 2743. https://doi.org/10.3390/ijms19092743