Studies on Anticonvulsant Effects of Novel Histamine H3R Antagonists in Electrically and Chemically Induced Seizures in Rats

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Material and Methods
2.1. In Vitro Pharmacology
2.1.1. Human Histamine H3 Receptor (hH3R) Binding Affinity for Tested Compounds 1–16
2.1.2. Human Histamine H1 Receptor (hH1R) Binding Affinity for Selected Compounds 4, 7 and 13
2.1.3. Human Histamine H4 Receptor (hH4R) Binding Affinity for Selected Compounds 4, 7 and 13
2.2. In Vivo Pharmacology
2.2.1. Animals
2.2.2. Drugs
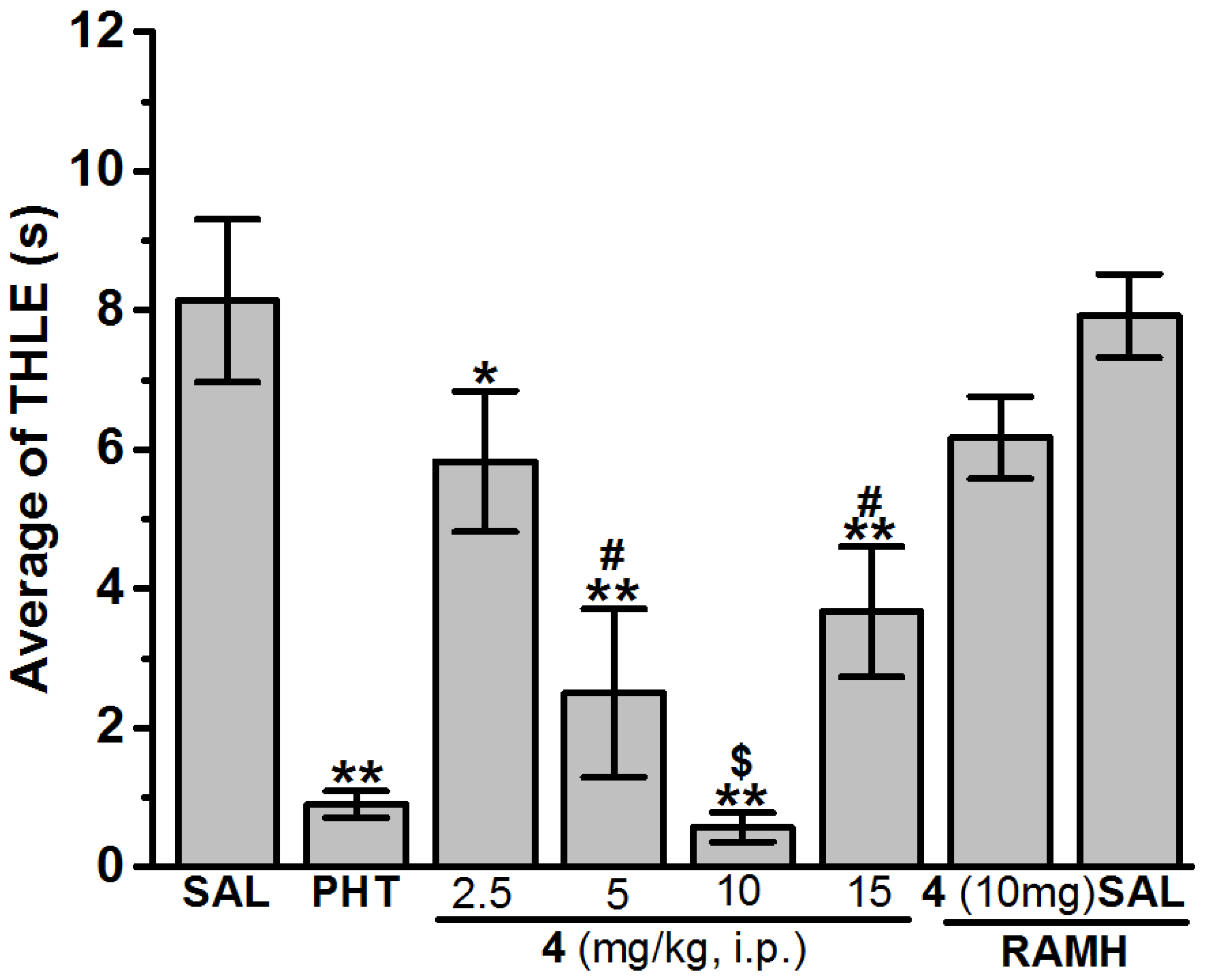
2.2.3. Maximal Electroshock (MES)-Induced Seizure
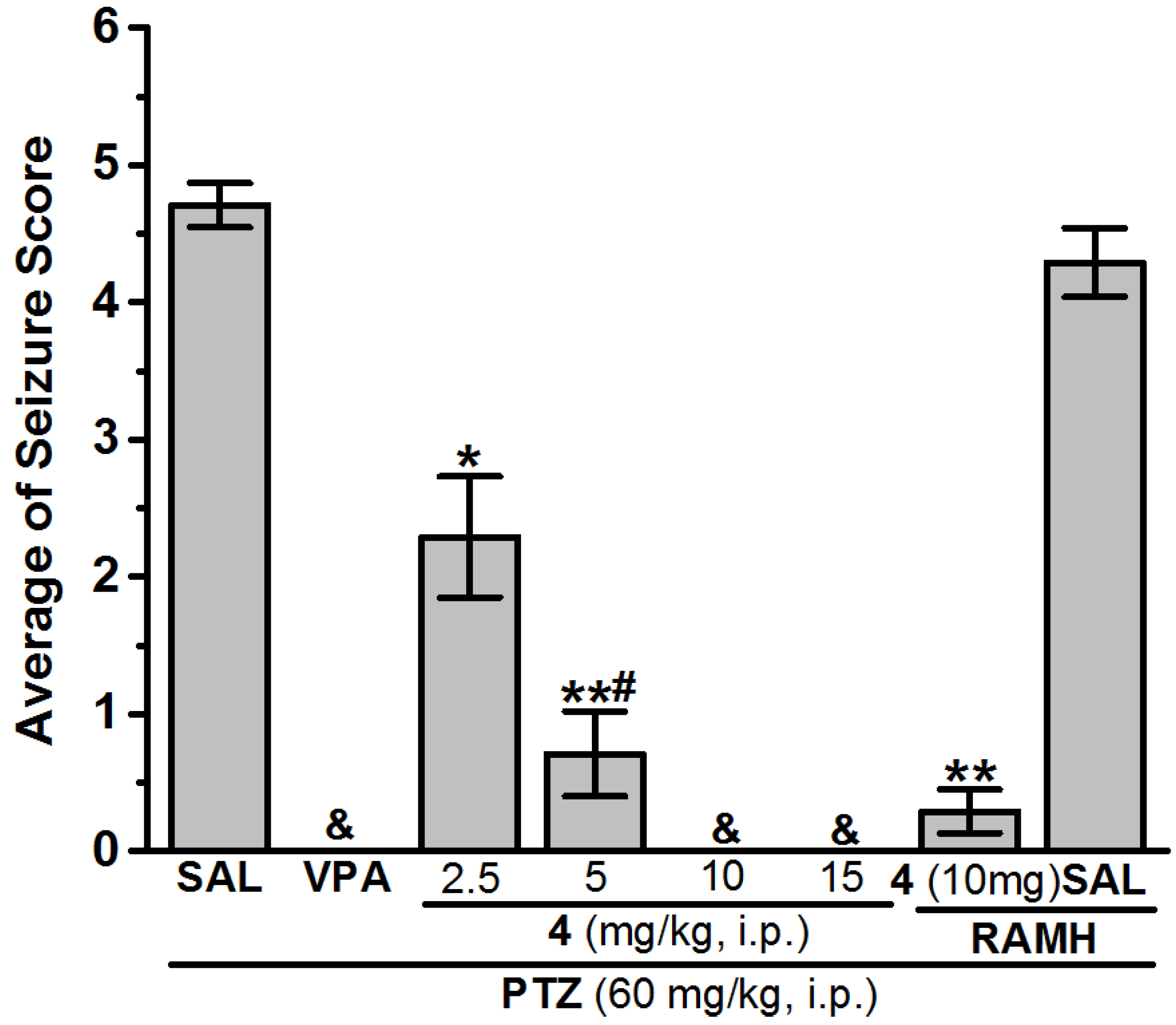
2.2.4. Chemically-Induced Seizures
2.2.5. Statistical Analysis
2.3. ADME-Tox Properties
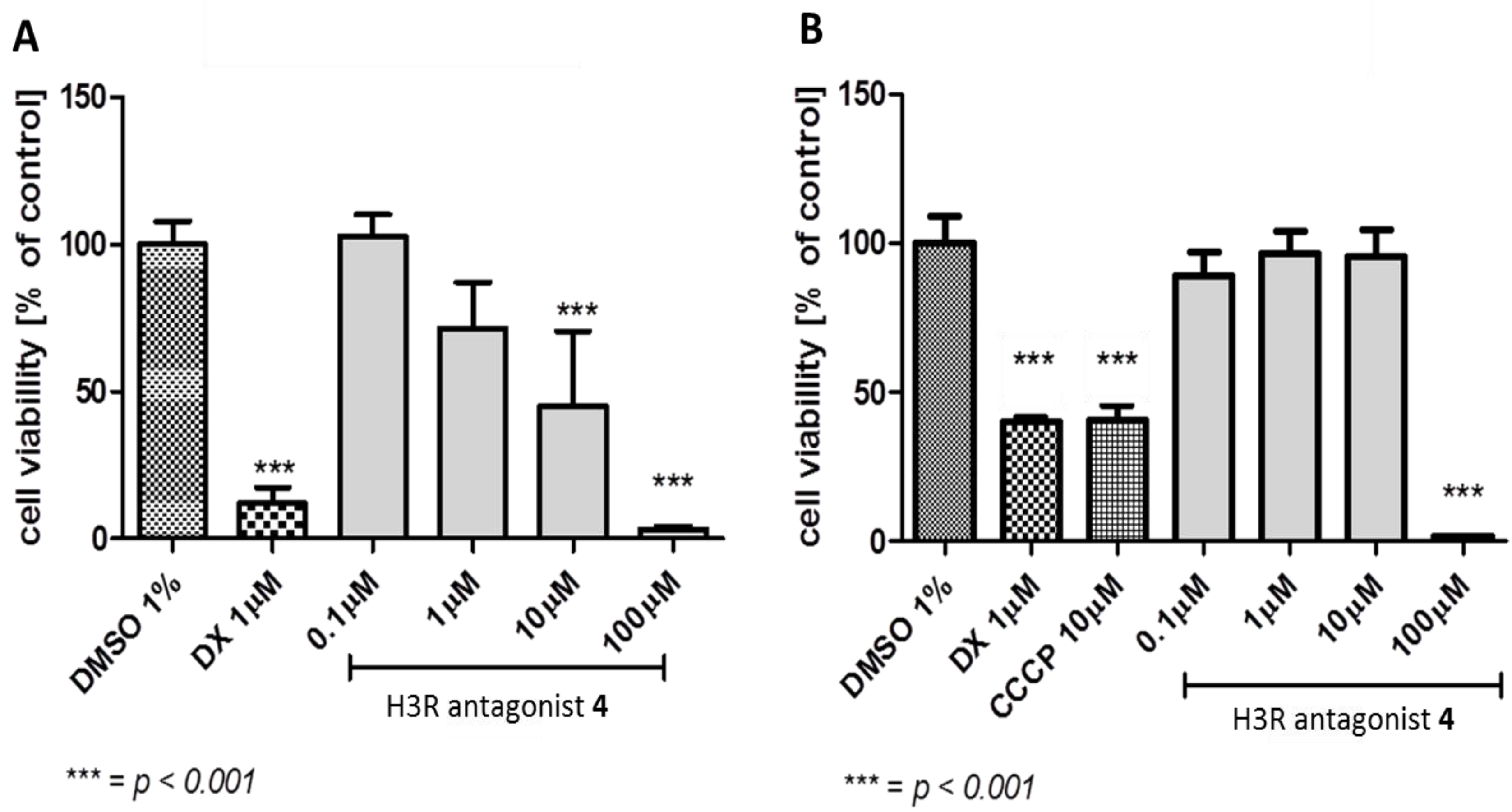
2.3.1. Antiproliferative Activity
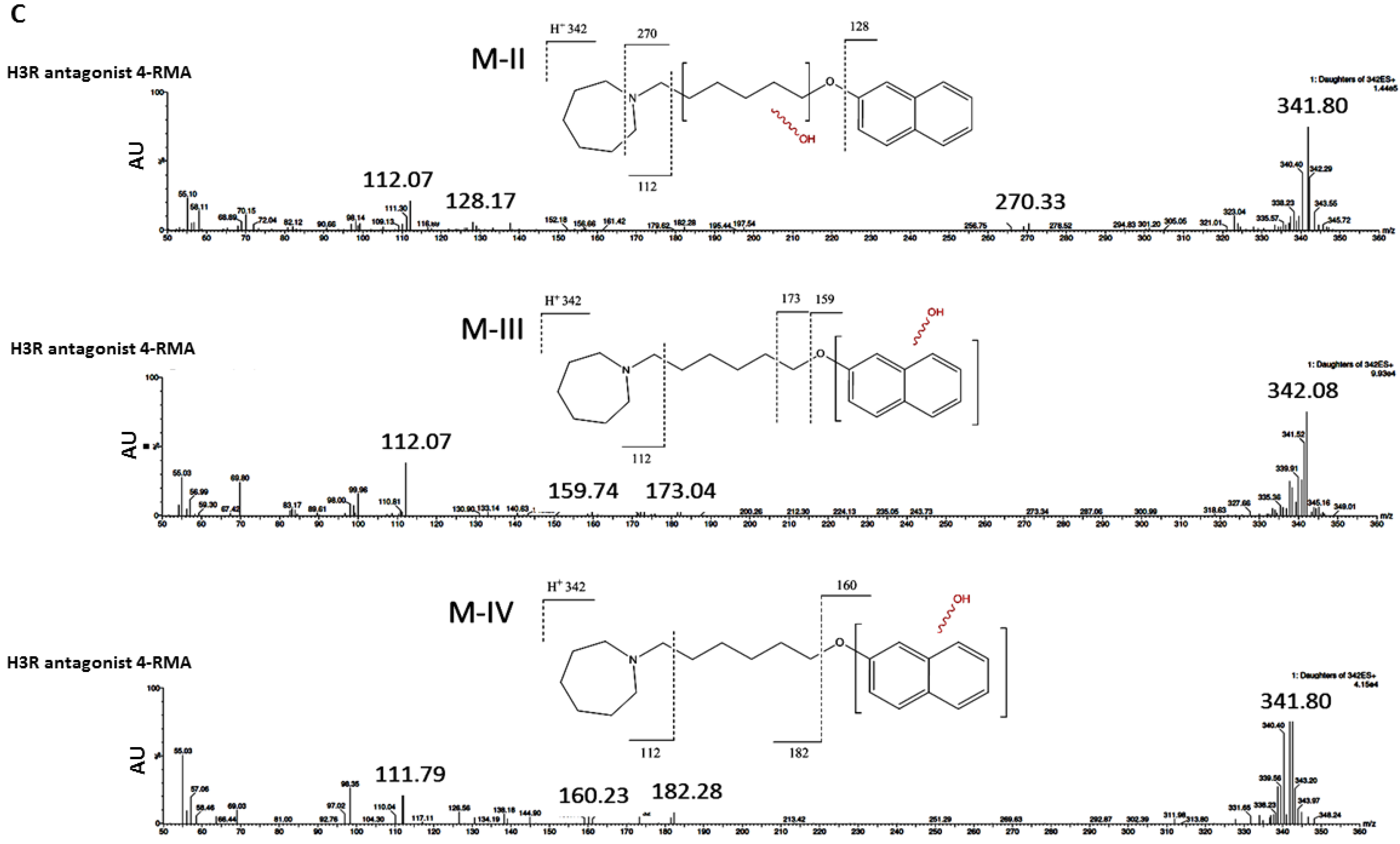
2.3.2. Prediction of In Silico Metabolism
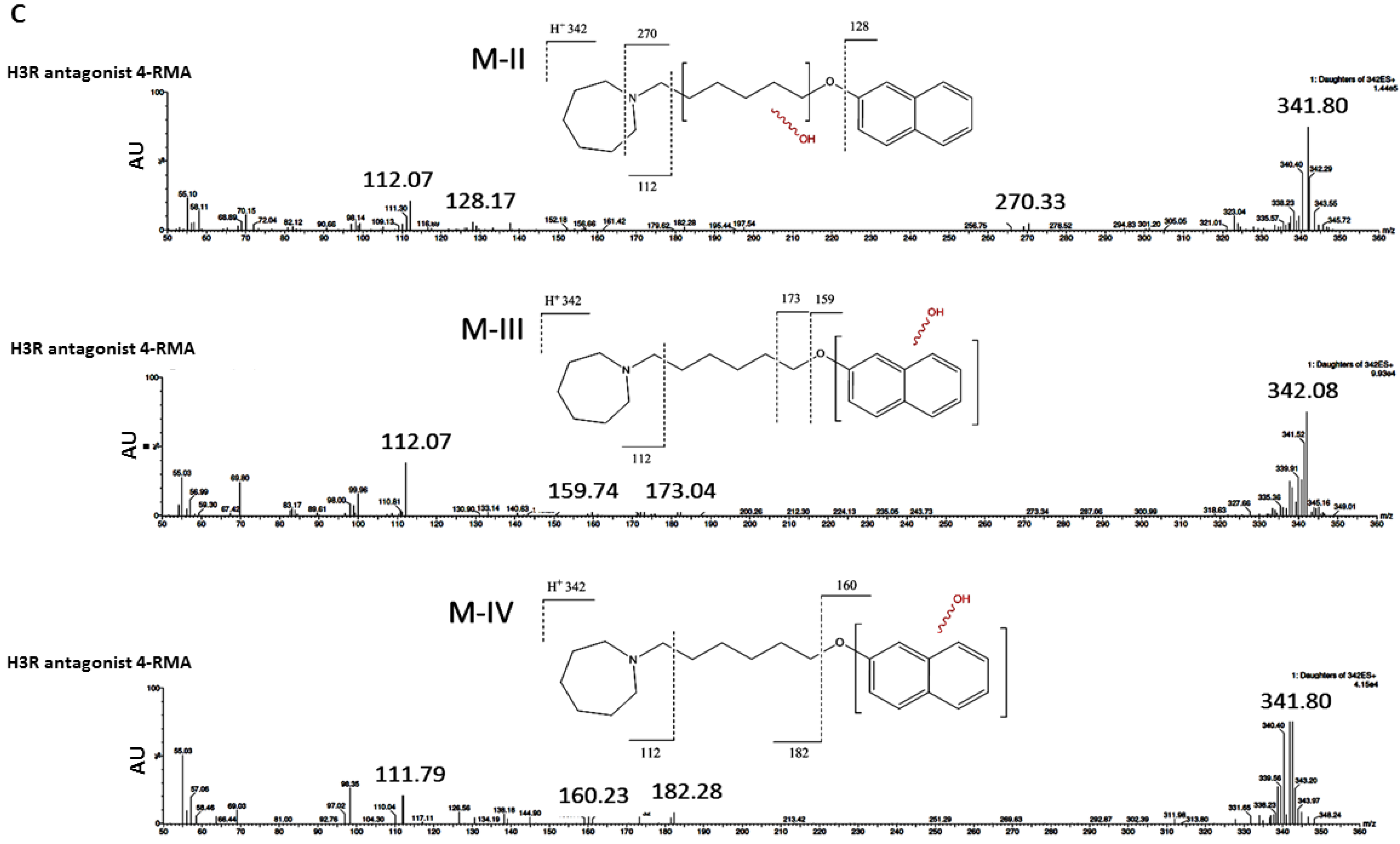
2.3.3. Metabolic Stability
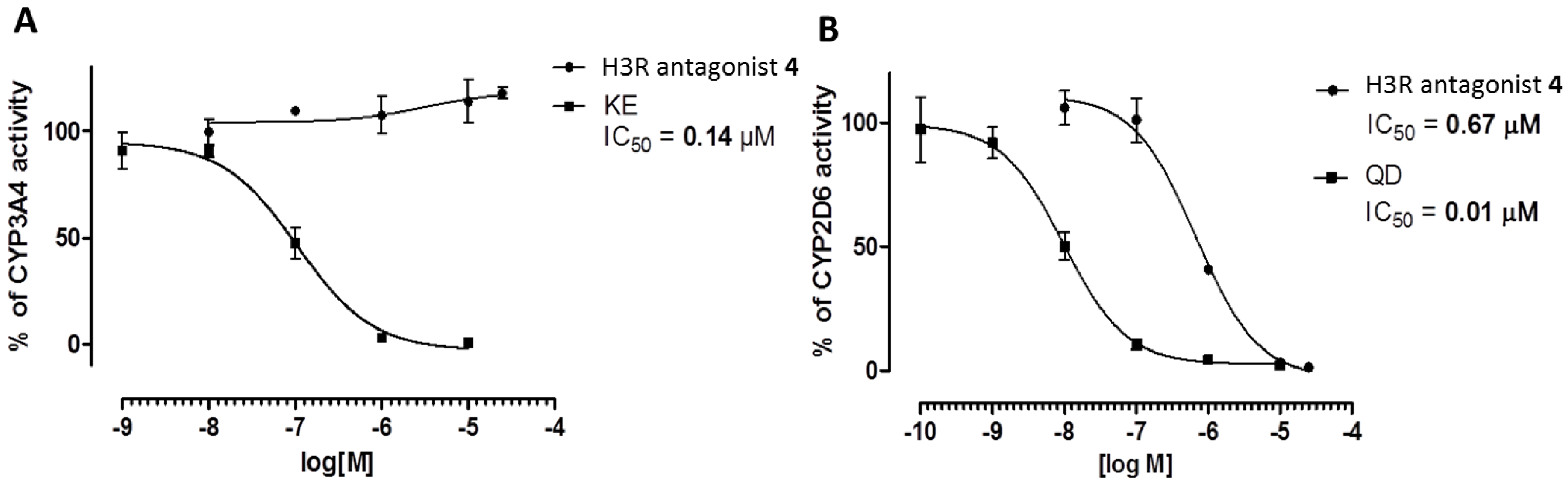
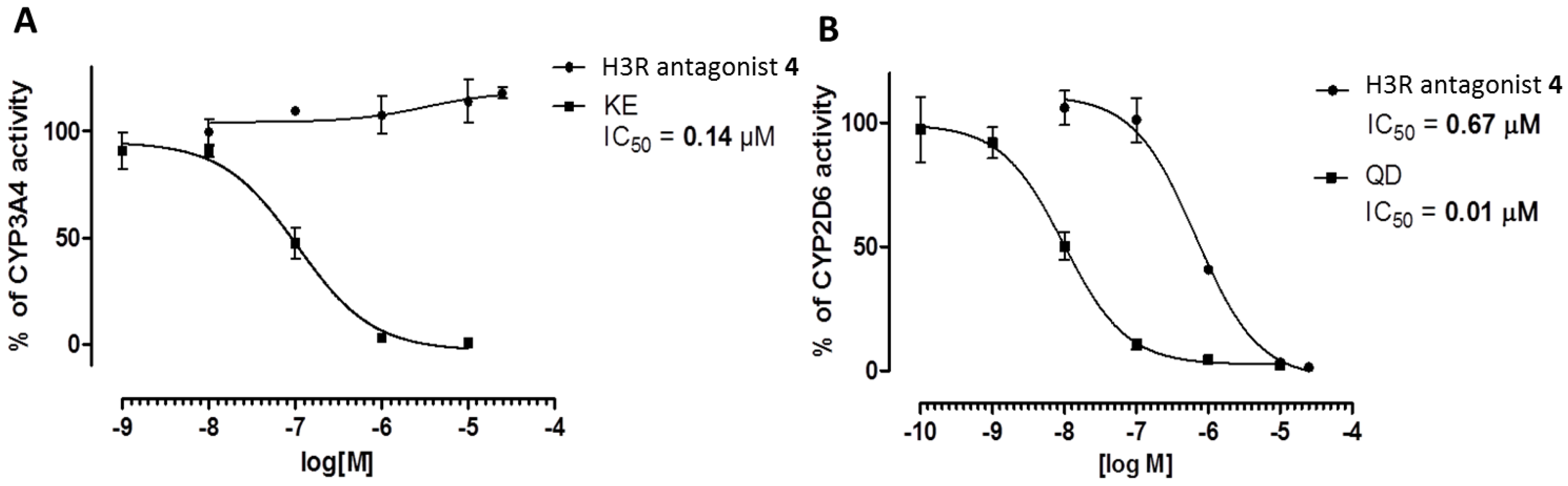
2.3.4. Metabolic Interactions
3. Results
3.1. Pharmacology
3.1.1. In Vitro Affinities at hH1Rs, hH3Rs, and hH4Rs
3.1.2. In Vivo Seizure Models
Anticonvulsant Screening of H3R Antagonists 1–16 in MES-Induced Seizure
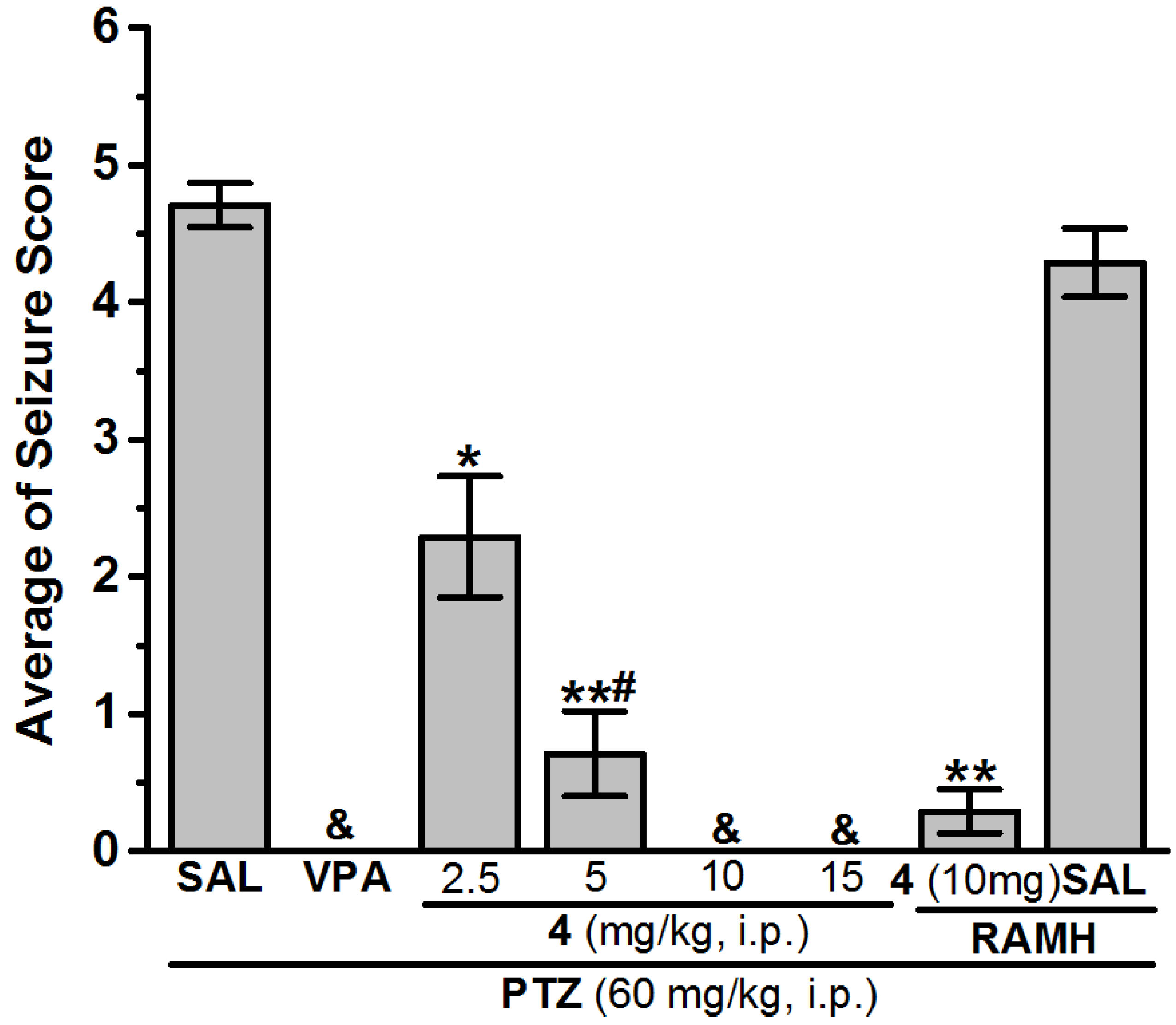
Anticonvulsant Screening for H3R Antagonists 1–16 in PTZ-Induced Seizures
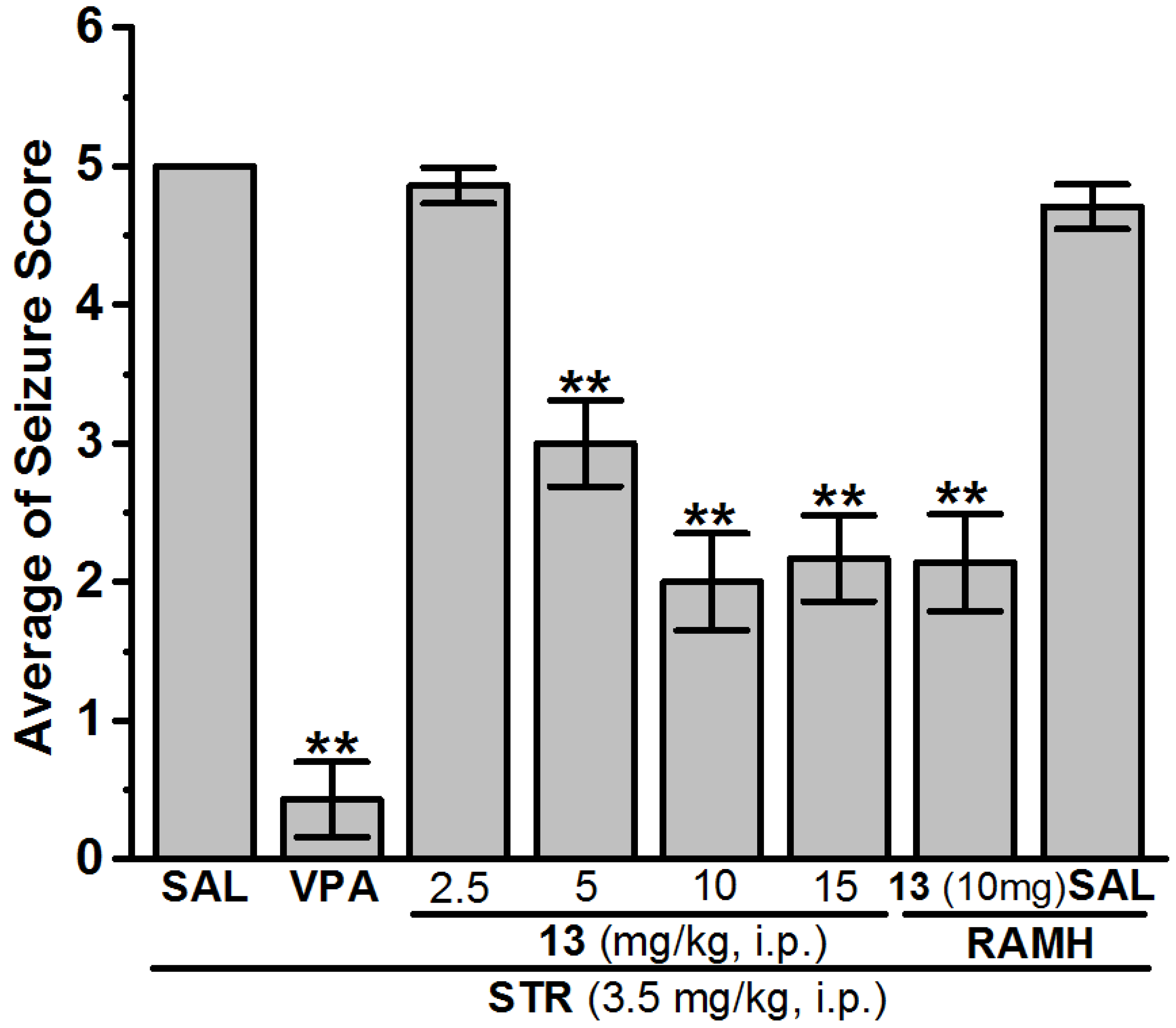
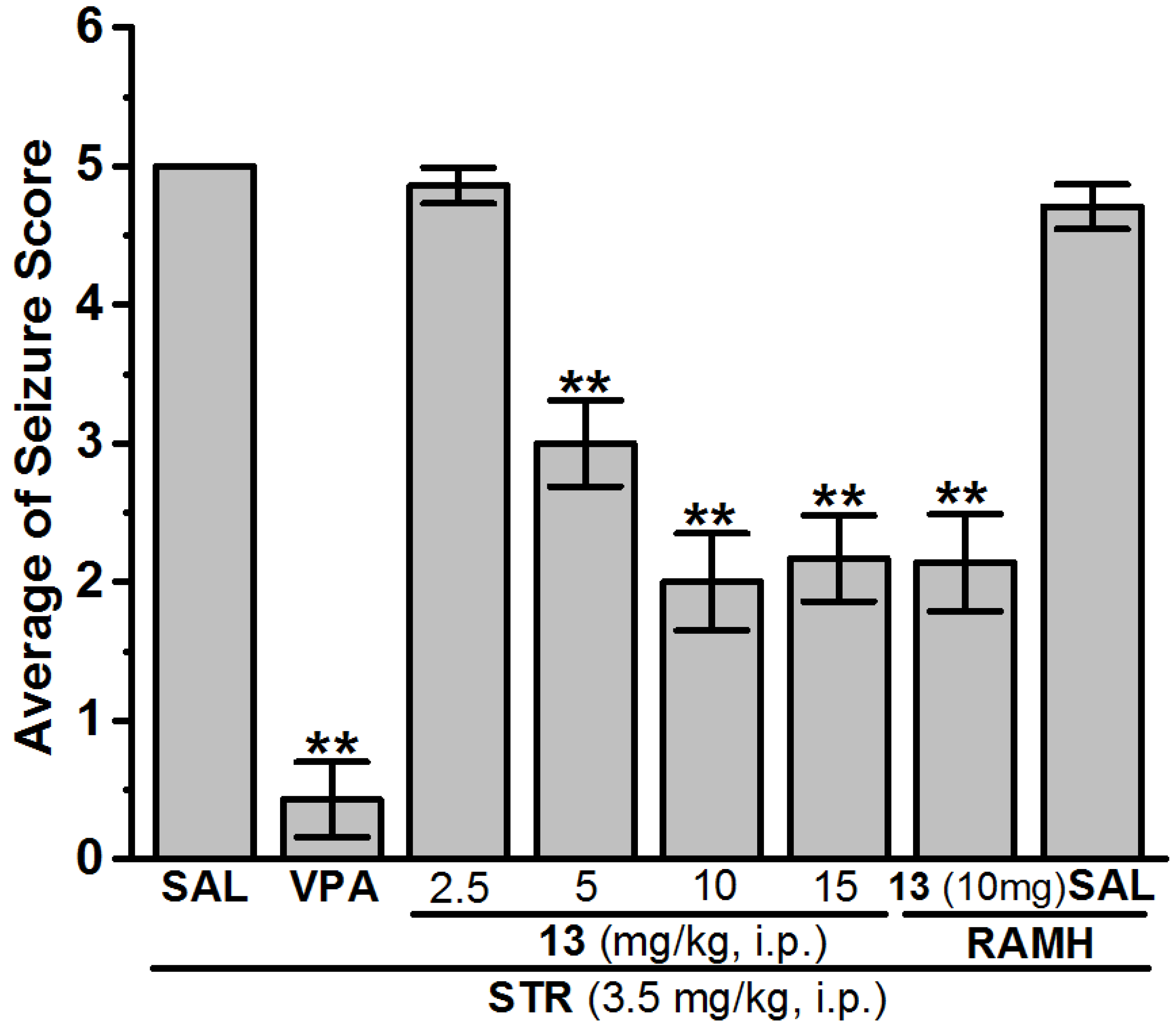
Anticonvulsant Screening for H3R Antagonists 1–16 in STR-Induced Seizures
3.2. ADME-Tox Properties
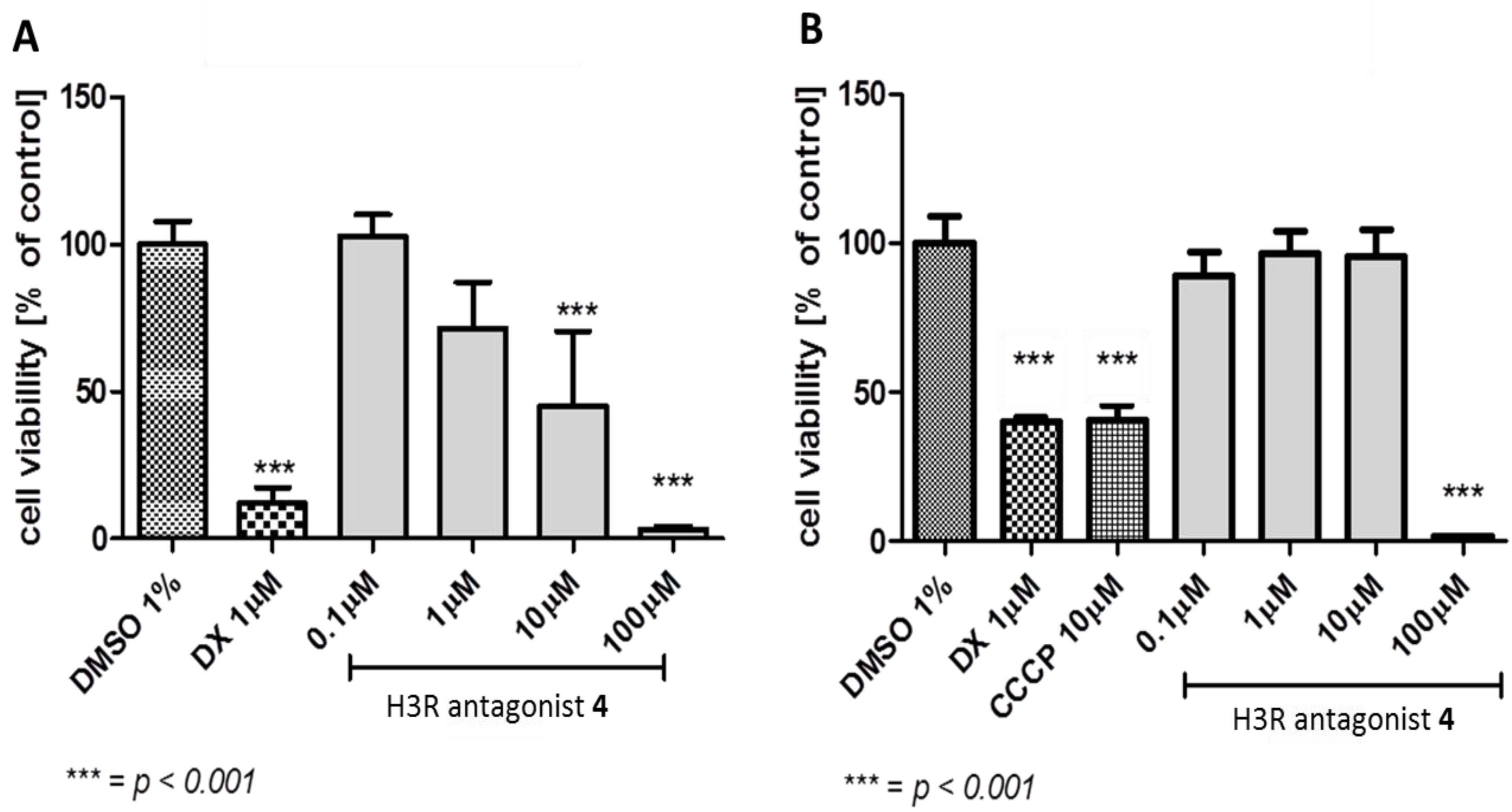
3.2.1. Antiproliferative Assay
3.2.2. In Silico Metabolic Stability
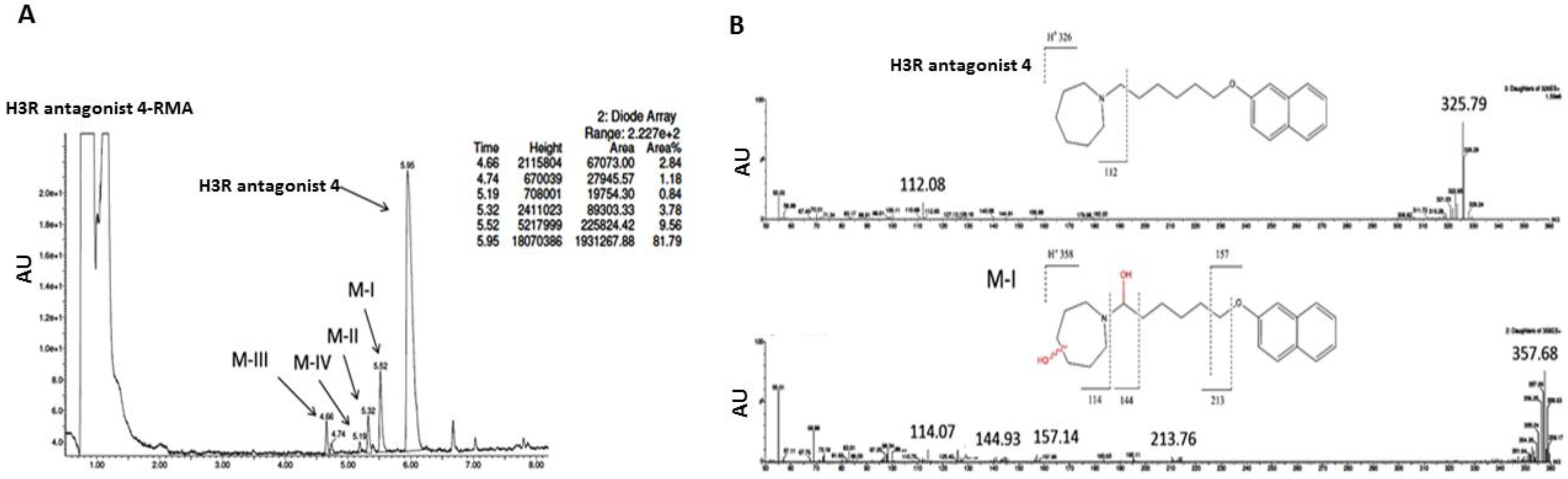
3.2.3. In Vitro Metabolic Stability
3.2.4. Metabolic Interactions
4. Discussion
4.1. In Vitro Histamine H3 Receptor Affinity of Test Compounds 1–16
4.2. Selectivity of Selected H3R Antagonists towards Other Histamine Receptors (H1 and H4)
4.2.1. Histamine H1 Receptor Affinity
4.2.2. Histamine H4 Receptor Affinity
4.3. In Vivo Anticonvulsant Activity
4.4. ADME-Tox Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Baulac, M.; de Boer, H.; Elger, C.; Glynn, M.; Kalviainen, R.; Little, A.; Mifsud, J.; Perucca, E.; Pitkanen, A.; Ryvlin, P. Epilepsy priorities in Europe: A report of the ILAE-IBE Epilepsy Advocacy Europe Task Force. Epilepsia 2015, 56, 1687–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobow, K.; Blumcke, I. Epigenetics in epilepsy. Neurosci. Lett. 2018, 667, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Mula, M.; Hermann, B.P. Uncovering the neurobehavioural comorbidities of epilepsy over the lifespan. Lancet 2012, 380, 1180–1192. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, D. The clinical impact of new antiepileptic drugs after a decade of use in epilepsy. Epilepsy Res. 2002, 50, 21–32. [Google Scholar] [CrossRef]
- Franco, V.; French, J.A.; Perucca, E. Challenges in the clinical development of new antiepileptic drugs. Pharmacol. Res. 2016, 103, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Burakgazi, E.; French, J.A. Treatment of epilepsy in adults. Epileptic Disord. 2016, 18, 228–239. [Google Scholar] [PubMed]
- Di Giovanni, G.; Svob Strac, D.; Sole, M.; Unzeta, M.; Tipton, K.F.; Muck-Seler, D.; Bolea, I.; Della Corte, L.; Nikolac Perkovic, M.; Pivac, N.; et al. Monoaminergic and Histaminergic Strategies and Treatments in Brain Diseases. Front. Neurosci. 2016, 10, 541. [Google Scholar] [CrossRef] [PubMed]
- Svob Strac, D.; Pivac, N.; Smolders, I.J.; Fogel, W.A.; De Deurwaerdere, P.; Di Giovanni, G. Monoaminergic Mechanisms in Epilepsy May Offer Innovative Therapeutic Opportunity for Monoaminergic Multi-Target Drugs. Front. Neurosci. 2016, 10, 492. [Google Scholar] [CrossRef] [PubMed]
- Kamei, C. Involvement of central histamine in amygdaloid kindled seizures in rats. Behav. Brain Res. 2001, 124, 243–250. [Google Scholar] [CrossRef]
- Kamei, C.; Ishizawa, K.; Kakinoki, H.; Fukunaga, M. Histaminergic mechanisms in amygdaloid-kindled seizures in rats. Epilepsy Res. 1998, 30, 187–194. [Google Scholar] [CrossRef]
- Vohora, D.; Pal, S.N.; Pillai, K.K. Histamine and selective H3-receptor ligands: A possible role in the mechanism and management of epilepsy. Pharmacol. Biochem. Behav. 2001, 68, 735–741. [Google Scholar] [CrossRef]
- Miyata, I.; Saegusa, H.; Sakurai, M. Seizure-modifying potential of histamine H1 antagonists: A clinical observation. Pediatr. Int. 2011, 53, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Ago, J.; Ishikawa, T.; Matsumoto, N.; Ashequr Rahman, M.; Kamei, C. Mechanism of imipramine-induced seizures in amygdala-kindled rats. Epilepsy Res. 2006, 72, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kakinoki, H.; Ishizawa, K.; Fukunaga, M.; Fujii, Y.; Kamei, C. The effects of histamine H3-receptor antagonists on amygdaloid kindled seizures in rats. Brain Res. Bull. 1998, 46, 461–465. [Google Scholar] [CrossRef]
- Yawata, I.; Tanaka, K.; Nakagawa, Y.; Watanabe, Y.; Murashima, Y.L.; Nakano, K. Role of histaminergic neurons in development of epileptic seizures in EL mice. Brain Res. Mol. Brain Res. 2004, 132, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Onodera, K.; Yamatodani, A.; Watanabe, T. Effects of alpha-fluoromethylhistidine on locomotor activity, brain histamine and catecholamine contents in rats. Methods Find Exp. Clin. Pharmacol. 1992, 14, 97–105. [Google Scholar] [PubMed]
- Tuomisto, L.; Tacke, U. Is histamine an anticonvulsive inhibitory transmitter? Neuropharmacology 1986, 25, 955–958. [Google Scholar] [CrossRef]
- Zhang, L.S.; Chen, Z.; Huang, Y.W.; Hu, W.W.; Wei, E.Q.; Yanai, K. Effects of endogenous histamine on seizure development of pentylenetetrazole-induced kindling in rats. Pharmacology 2003, 69, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.; Stark, H.; Thurmond, R.L.; Haas, H.L. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.H.; Seifert, R. The histamine H-receptor and the central and peripheral nervous system: A critical analysis of the literature. Neuropharmacology 2015, 106, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Arrang, J.M.; Garbarg, M.; Schwartz, J.C. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 1983, 302, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Arrang, J.M.; Garbarg, M.; Schwartz, J.C. Autoinhibition of histamine synthesis mediated by presynaptic H3-receptors. Neuroscience 1987, 23, 149–157. [Google Scholar] [CrossRef]
- Brown, R.E.; Stevens, D.R.; Haas, H.L. The physiology of brain histamine. Prog. Neurobiol. 2001, 63, 637–672. [Google Scholar] [CrossRef]
- Kuder, K.; Lazewska, D.; Latacz, G.; Schwed, J.S.; Karcz, T.; Stark, H.; Karolak-Wojciechowska, J.; Kiec-Kononowicz, K. Chlorophenoxy aminoalkyl derivatives as histamine H(3)R ligands and antiseizure agents. Bioorg. Med. Chem. 2016, 24, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Kuder, K.; Subramanian, D.; Shafiullah, M.; Stark, H.; Lazewska, D.; Adem, A.; Kiec-Kononowicz, K. Anticonvulsive effect of nonimidazole histamine H3 receptor antagonists. Behav. Pharmacol. 2014, 25, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Saad, A.; Latacz, G.; Kuder, K.; Olejarz, A.; Karcz, T.; Stark, H.; Kiec-Kononowicz, K. Non-imidazole-based histamine H3 receptor antagonists with anticonvulsant activity in different seizure models in male adult rats. Drug Des. Dev. Ther. 2016, 10, 3879–3898. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Saad, A.; Schwed, J.S.; Weizel, L.; Walter, M.; Stark, H. Anticonvulsant effects of isomeric nonimidazole histamine H3 receptor antagonists. Drug Des. Dev. Ther. 2016, 10, 3633–3651. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Saad, A.; Subramanian, D.; Shafiullah, M.; Lazewska, D.; Kiec-Kononowiczc, K. Anticonvulsant and procognitive properties of the non-imidazole histamine H3 receptor antagonist DL77 in male adult rats. Neuropharmacology 2016, 106, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Schwed, J.S.; Subramanian, D.; Weizel, L.; Walter, M.; Adem, A.; Stark, H. Non-imidazole histamine H3 receptor ligands incorporating antiepileptic moieties. Eur. J. Med. Chem. 2014, 77, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Shehab, S.; Wiecek, M.; Subramanian, D.; Shafiullah, M.; Kiec-Kononowicz, K.; Adem, A. Anticonvulsant properties of histamine H3 receptor ligands belonging to N-substituted carbamates of imidazopropanol. Bioorg. Med. Chem. Lett. 2013, 23, 4886–4891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, Z.; Ren, K.; Leurs, R.; Chen, J.; Zhang, W.; Ye, B.; Wei, E.; Timmerman, H. Effects of clobenpropit on pentylenetetrazole-kindled seizures in rats. Eur. J. Pharmacol. 2003, 482, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.S.; Chen, J.F.; Chen, G.F.; Hu, X.Y.; Ding, M.P. Effects of thioperamide on seizure development and memory impairment induced by pentylenetetrazole-kindling epilepsy in rats. Chin. Med. J. 2013, 126, 95–100. [Google Scholar] [PubMed]
- Kasteleijn-Nolst Trenite, D.; Parain, D.; Genton, P.; Masnou, P.; Schwartz, J.C.; Hirsch, E. Efficacy of the histamine 3 receptor (H3R) antagonist pitolisant (formerly known as tiprolisant; BF2.649) in epilepsy: Dose-dependent effects in the human photosensitivity model. Epilepsy Behav. 2013, 28, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Collart Dutilleul, P.; Ryvlin, P.; Kahane, P.; Vercueil, L.; Semah, F.; Biraben, A.; Schwartz, J.C.; De Seze, J.; Hirsch, E.; Collongues, N. Exploratory Phase II Trial to Evaluate the Safety and the Antiepileptic Effect of Pitolisant (BF2.649) in Refractory Partial Seizures, Given as Adjunctive Treatment During 3 Months. Clin. Neuropharmacol. 2016, 39, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Arrang, J.M.; Garbarg, M.; Lancelot, J.C.; Lecomte, J.M.; Pollard, H.; Robba, M.; Schunack, W.; Schwartz, J.C. Highly potent and selective ligands for histamine H3-receptors. Nature 1987, 327, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ligneau, X.; Morisset, S.; Tardivel-Lacombe, J.; Gbahou, F.; Ganellin, C.R.; Stark, H.; Schunack, W.; Schwartz, J.C.; Arrang, J.M. Distinct pharmacology of rat and human histamine H(3) receptors: Role of two amino acids in the third transmembrane domain. Br. J. Pharmacol. 2000, 131, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Kollb-Sielecka, M.; Demolis, P.; Emmerich, J.; Markey, G.; Salmonson, T.; Haas, M. The European Medicines Agency review of pitolisant for treatment of narcolepsy: Summary of the scientific assessment by the Committee for Medicinal Products for Human Use. Sleep Med. 2017, 33, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Ligneau, X.; Perrin, D.; Landais, L.; Camelin, J.C.; Calmels, T.P.; Berrebi-Bertrand, I.; Lecomte, J.M.; Parmentier, R.; Anaclet, C.; Lin, J.S.; et al. BF2.649 [1-{3-[3-(4-Chlorophenyl)propoxy]propyl}piperidine, hydrochloride], a nonimidazole inverse agonist/antagonist at the human histamine H3 receptor: Preclinical pharmacology. J. Pharmacol. Exp. Ther. 2007, 320, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Stark, H. Cherry-picked ligands at histamine receptor subtypes. Neuropharmacology 2015, 106, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Saad, A.; Sadeq, A.; Jalal, F.; Stark, H. Histamine H3 receptor as a potential target for cognitive symptoms in neuropsychiatric diseases. Behav. Brain Res. 2016, 312, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, M.; Khanam, R.; Vohora, D. Histamine H3 receptor antagonists in relation to epilepsy and neurodegeneration: A systemic consideration of recent progress and perspectives. Br. J. Pharmacol. 2012, 167, 1398–1414. [Google Scholar] [CrossRef] [PubMed]
- Sander, K.; Kottke, T.; Weizel, L.; Stark, H. Kojic acid derivatives as histamine H(3) receptor ligands. Chem. Pharm. Bull. 2010, 58, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Schreeb, A.; Schwed, J.S.; Weizel, L.; Stark, H. Drug-likeness approach of 2-aminopyrimidines as histamine H3 receptor ligands. Drug Des. Dev. Ther. 2014, 8, 1499–1513. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Lazewska, D.; Kaleta, M.; Hagenow, S.; Mogilski, S.; Latacz, G.; Karcz, T.; Lubelska, A.; Honkisz, E.; Handzlik, J.; Reiner, D.; et al. Novel naphthyloxy derivatives—Potent histamine H3 receptor ligands. Synthesis and pharmacological evaluation. Bioorg. Med. Chem. 2018, 26, 2573–2585. [Google Scholar] [CrossRef] [PubMed]
- Alachkar, A.; Latacz, G.; Siwek, A.; Lubelska, A.; Honkisz, E.; Grybos, A.; Lazewska, D.; Handzlik, J.; Stark, H.; Kiec-Kononowicz, K.; et al. Anticonvulsant evaluation of novel non-imidazole histamine H3R antagonists in different convulsion models in rats. Pharmacol. Biochem. Behav. 2018, 170, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Khanian, S.S.; Ashoor, A.; Prytkova, T.; Ghattas, M.A.; Atatreh, N.; Nurulain, S.M.; Yang, K.H.; Howarth, F.C.; Oz, M. Effects of antihistamines on the function of human alpha7-nicotinic acetylcholine receptors. Eur. J. Pharmacol. 2014, 746, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Loscher, W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 2011, 20, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Serdiuk, S.E.; Gmiro, V.E. Phenylephrine potentiates antidepressive and eliminates sedative action of amitriptyline in rats. Ross Fiziol. Zh Im I M Sechenova 2014, 100, 18–26. [Google Scholar] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Sadek, B.; Oz, M.; Nurulain, S.M.; Jayaprakash, P.; Latacz, G.; Kiec-Kononowicz, K.; Szymanska, E. Phenylalanine derivatives with modulating effects on human alpha1-glycine receptors and anticonvulsant activity in strychnine-induced seizure model in male adult rats. Epilepsy Res. 2017, 138, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, G.; Carosati, E.; De Boeck, B.; Ethirajulu, K.; Mackie, C.; Howe, T.; Vianello, R. MetaSite: Understanding metabolism in human cytochromes from the perspective of the chemist. J. Med. Chem. 2005, 48, 6970–6979. [Google Scholar] [CrossRef] [PubMed]
- Loscher, W.; Honack, D.; Fassbender, C.P.; Nolting, B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. III. Pentylenetetrazole seizure models. Epilepsy Res. 1991, 8, 171–189. [Google Scholar] [CrossRef]
- Vohora, D.; Pal, S.N.; Pillai, K.K. Thioperamide, a selective histamine H3 receptor antagonist, protects against PTZ-induced seizures in mice. Life Sci. 2000, 66, PL297–PL301. [Google Scholar] [CrossRef]
- Sowemimo, A.A.; Adio, O.; Fageyinbo, S. Anticonvulsant activity of the methanolic extract of Justicia extensa T. Anders. J. Ethnopharmacol. 2011, 138, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Takei, H.; Yamamoto, K.; Bae, Y.C.; Shirakawa, T.; Kobayashi, M. Histamine H3 Heteroreceptors Suppress Glutamatergic and GABAergic Synaptic Transmission in the Rat Insular Cortex. Front Neural Circuits. 2017, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Swinyard, E.A.; Sofia, R.D.; Kupferberg, H.J. Comparative anticonvulsant activity and neurotoxicity of felbamate and four prototype antiepileptic drugs in mice and rats. Epilepsia 1986, 27, 27–34. [Google Scholar] [CrossRef] [PubMed]
- White, H.S. Comparative anticonvulsant and mechanistic profile of the established and newer antiepileptic drugs. Epilepsia 1999, 40, S2–S10. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Structure | In Vitro Affinity Ki (hH3R) a in nM [CI] |
|---|---|---|
| 1 | 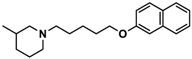 | 42.3 b [18.3; 97.4] |
| 2 | 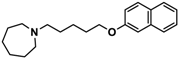 | 57.3 b [47.2; 69.7] |
| 3 | 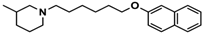 | 55.9 b [44.8; 69.7] |
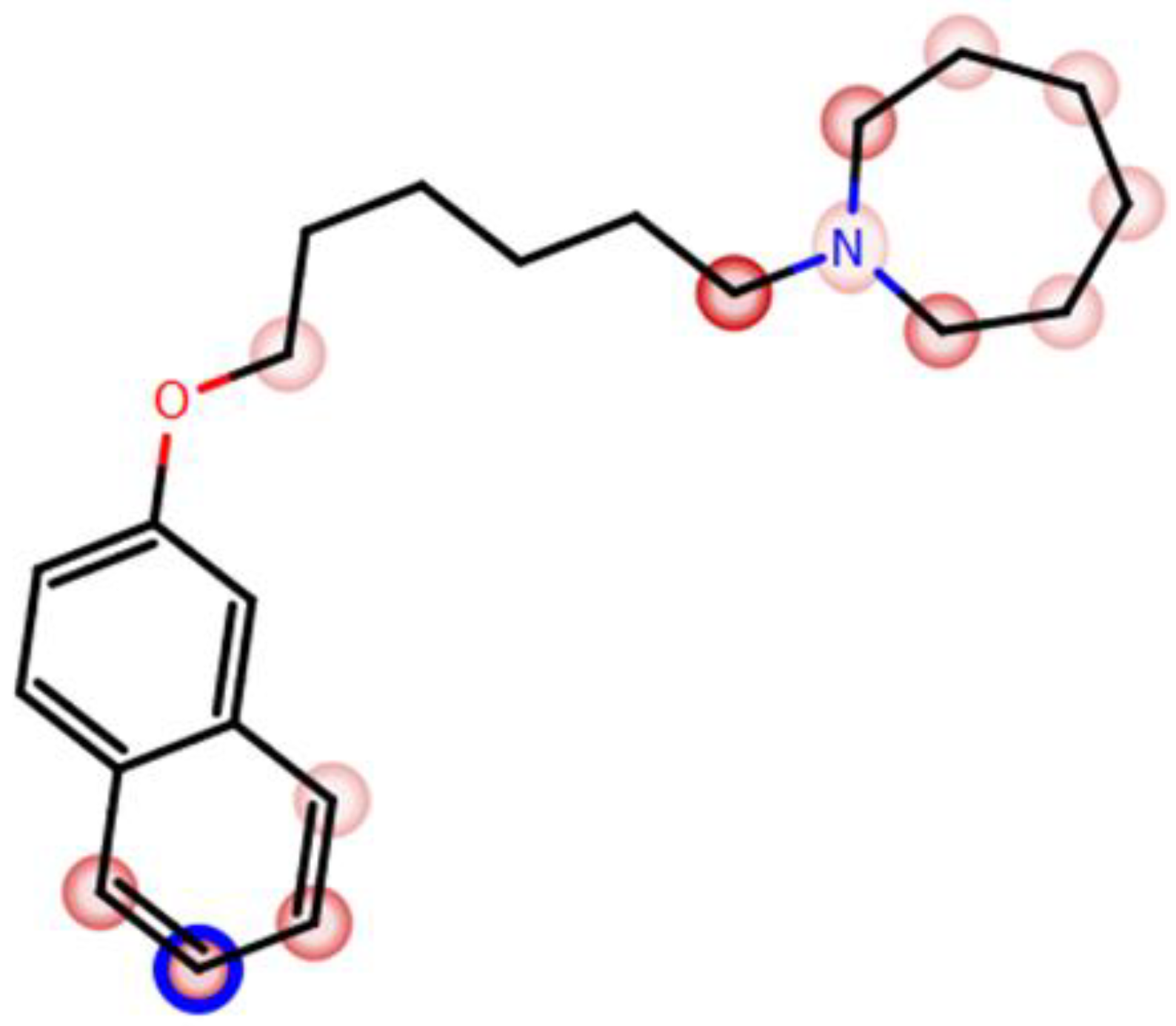
| 4 | 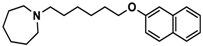 | 69.3 b [59.2; 81.1] |
| 5 | 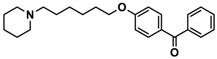 | 41.1 [25.6; 66.2] |
| 6 | 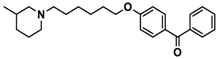 | 52.8 [31.4; 88.8] |
| 7 | 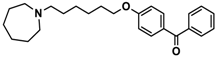 | 40.5 [32.9; 50.0] |
| 8 | 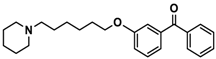 | 76.1 [53.5; 108.3] |
| 9 | 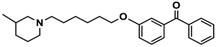 | 110.2 [61.8; 196.4] |
| 10 |  | 69.5 [44.4; 108.8] |
| 11 | 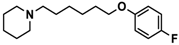 | 115.2 [78.4; 169.5] |
| 12 |  | 83.6 [65.8; 106.4] |
| 13 | 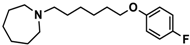 | 137.2 [60.0; 313.9] |
| 14 | 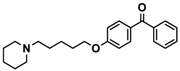 | 36.2 [10.0; 130.3] |
| 15 | 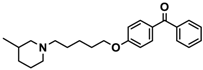 | 40.2 [13.5; 119.4] |
| 16 | 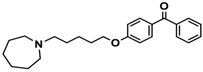 | 38.5 [10.5; 141.6] |
| Pitolisant (PIT) |  | 11.69 c |
| Ligand | Structure | MES a-Induced Seizure | PTZ b-Induced Seizure | STR c-Induced Seizure |
|---|---|---|---|---|
| 1 |  | − | − | − |
| 2 | 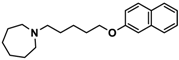 | − | − | − |
| 3 | 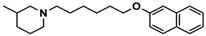 | ++ | − | − |
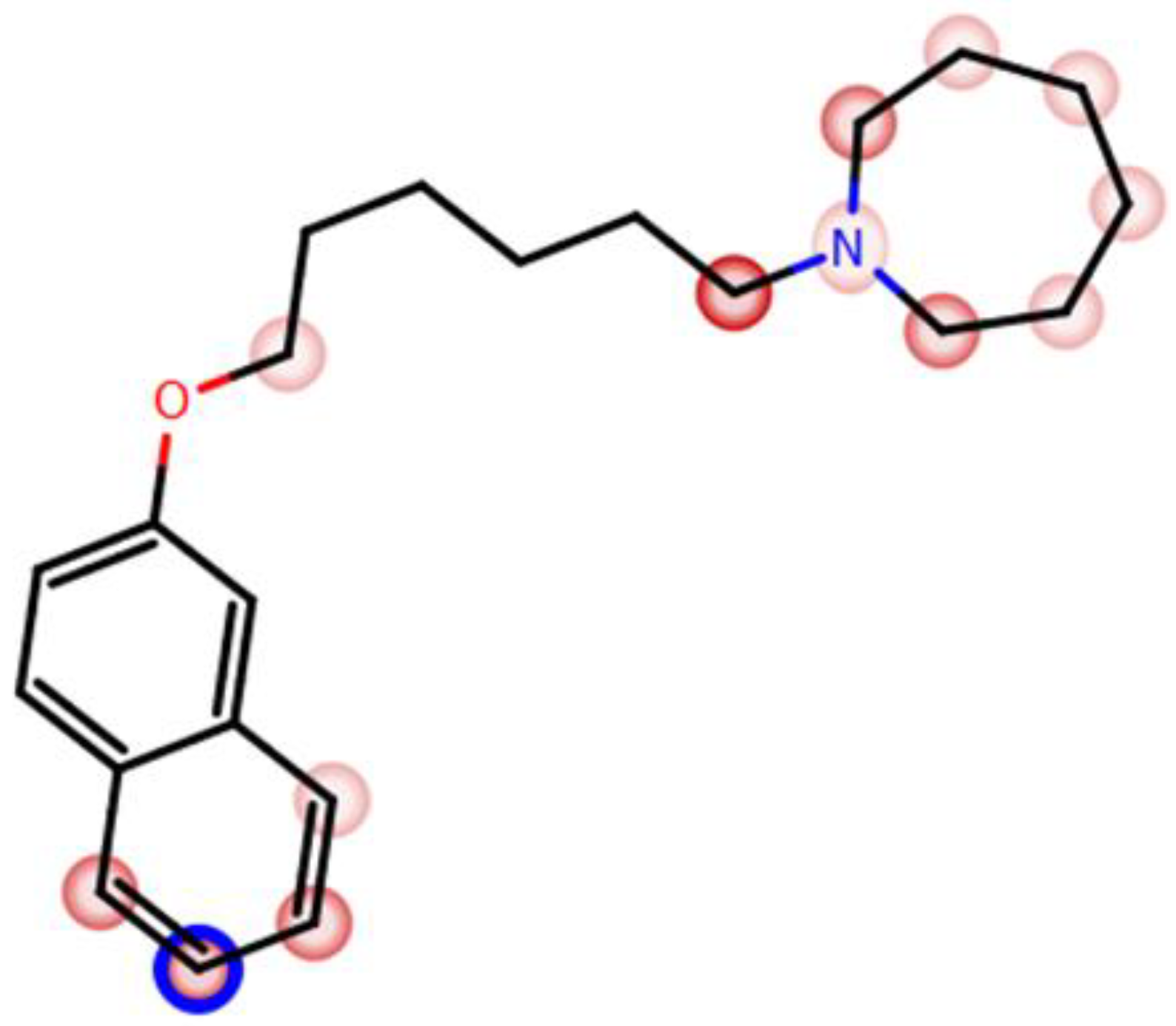
| 4 | 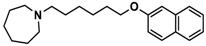 | ++ | ++ | − |
| 5 | 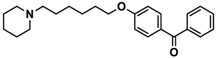 | ++ | − | − |
| 6 | 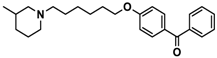 | + | − | − |
| 7 | 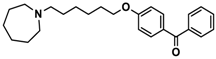 | + | ++ | − |
| 8 |  | + | − | − |
| 9 | 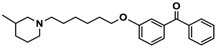 | − | − | − |
| 10 | 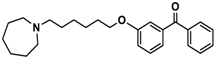 | − | − | − |
| 11 | 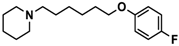 | − | ++ | − |
| 12 | 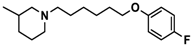 | − | + | − |
| 13 | 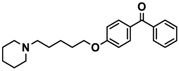 | − | + | + |
| 14 |  | + | + | − |
| 15 | 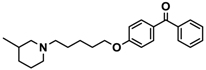 | − | + | − |
| 16 | 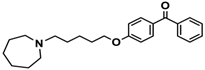 | − | − | − |
| Group | MES a-Induced Seizure | Group | PTZ b-Induced Seizure | Group | STR c-Induced Seizure | ||
|---|---|---|---|---|---|---|---|
| Average THLE (s) | Average Seizure Score | % Protection against GTCS | Average Seizure Score | % Protection against GTCS | |||
| SAL | 8.14 ± 1.17 | SAL | 4.71 ± 0.16 | 28.57 | SAL | 5 | 0 |
| PHT (10 mg) | 0.90 ± 0.19 ** | VPA (300 mg) | 0.00 ± 0.00 | 100 | VPA (300 mg) | 0 | 100 |
| 4 (2.5 mg) | 5.83 ± 1.01 * | 4 (2.5 mg) | 2.29 ± 0.44 * | 100 | 4 (2.5 mg) | − | − |
| 4 (5 mg) | 2.50 ± 1.21 **,# | 4 (5 mg) | 0.71 ± 0.31 **,# | 100 | 4 (5 mg) | − | − |
| 4 (10 mg) | 0.57 ± 0.21 **,$ | 4 (10 mg) | 0.00 ± 0.00 & | 100 | 4 (10 mg) | 4.57 ± 0.19 | 28.57 |
| 4 (15 mg) | 3.67 ± 0.94 **,# | 4 (15 mg) | 0.00 ± 0.00 & | 100 | 4 (15 mg) | − | − |
| 4 (10 mg) + RAMH | 6.17 ± 0.59 | 4 (10 mg) + RAMH | 0.29 ± 0.16 ** | 100 | 4 (10 mg) + RAMH | − | − |
| SAL + RAMH | 7.92 ± 0.60 | SAL + RAMH | 4.29 ± 0.25 | 14.29 | SAL + RAMH | − | − |
| 13 (2.5 mg) | − | 13 (2.5 mg) | − | − | 13 (2.5 mg) | 4.86 ± 0.13 | 14.29 |
| 13 (5 mg) | − | 13 (5 mg) | − | − | 13 (5 mg) | 3.00 ± 0.31 ** | 85.71 |
| 13 (10 mg) | 6.43 ± 1.14 | 13 (10 mg) | 2.14 ± 0.47 * | 85.71 | 13 (10 mg) | 2.00 ± 0.35 ** | 100 |
| 13 (15 mg) | − | 13 (15 mg) | − | − | 13 (15 mg) | 2.17 ± 0.31 ** | 100 |
| 13 (10 mg) + RAMH | − | 13 (10 mg) + RAMH | − | − | 13 (10 mg) + RAMH | 2.14 ± 0.35 ** | 100 |
| SAL + RAMH | − | SAL + RAMH | − | − | SAL + RAMH | 4.71 ± 0.16 | 28.57 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alachkar, A.; Łażewska, D.; Latacz, G.; Frank, A.; Siwek, A.; Lubelska, A.; Honkisz-Orzechowska, E.; Handzlik, J.; Stark, H.; Kieć-Kononowicz, K.; et al. Studies on Anticonvulsant Effects of Novel Histamine H3R Antagonists in Electrically and Chemically Induced Seizures in Rats. Int. J. Mol. Sci. 2018, 19, 3386. https://doi.org/10.3390/ijms19113386
Alachkar A, Łażewska D, Latacz G, Frank A, Siwek A, Lubelska A, Honkisz-Orzechowska E, Handzlik J, Stark H, Kieć-Kononowicz K, et al. Studies on Anticonvulsant Effects of Novel Histamine H3R Antagonists in Electrically and Chemically Induced Seizures in Rats. International Journal of Molecular Sciences. 2018; 19(11):3386. https://doi.org/10.3390/ijms19113386
Chicago/Turabian StyleAlachkar, Alaa, Dorota Łażewska, Gniewomir Latacz, Annika Frank, Agata Siwek, Annamaria Lubelska, Ewelina Honkisz-Orzechowska, Jadwiga Handzlik, Holger Stark, Katarzyna Kieć-Kononowicz, and et al. 2018. "Studies on Anticonvulsant Effects of Novel Histamine H3R Antagonists in Electrically and Chemically Induced Seizures in Rats" International Journal of Molecular Sciences 19, no. 11: 3386. https://doi.org/10.3390/ijms19113386
APA StyleAlachkar, A., Łażewska, D., Latacz, G., Frank, A., Siwek, A., Lubelska, A., Honkisz-Orzechowska, E., Handzlik, J., Stark, H., Kieć-Kononowicz, K., & Sadek, B. (2018). Studies on Anticonvulsant Effects of Novel Histamine H3R Antagonists in Electrically and Chemically Induced Seizures in Rats. International Journal of Molecular Sciences, 19(11), 3386. https://doi.org/10.3390/ijms19113386