Abstract
Protein structural biology came a long way since the determination of the first three-dimensional structure of myoglobin about six decades ago. Across this period, X-ray crystallography was the most important experimental method for gaining atomic-resolution insight into protein structures. However, as the role of dynamics gained importance in the function of proteins, the limitations of X-ray crystallography in not being able to capture dynamics came to the forefront. Computational methods proved to be immensely successful in understanding protein dynamics in solution, and they continue to improve in terms of both the scale and the types of systems that can be studied. In this review, we briefly discuss the limitations of X-ray crystallography in studying protein dynamics, and then provide an overview of different computational methods that are instrumental in understanding the dynamics of proteins and biomacromolecular complexes.
1. Introduction
Ever since the first protein structure was solved sixty years ago [1], X-ray crystallography has been the most important experimental method for understanding protein structure and relating it to its function. Out of over 140,000 structures in the Protein Data Bank (PDB), about 90% were determined using X-ray crystallography. These structures contributed to shedding light on many important biological phenomena by providing mechanistic insight into the functions of the proteins involved in biological processes [2]. The protein structures determined by X-ray crystallography were responsible for understanding the basic mechanisms involved in enzyme function, cellular signaling, cellular recognition, etc., as well as for the development of pharmaceutical interventions to modulate these mechanisms via structure-based drug discovery [2].
The structures determined by X-ray crystallography represent an averaged conformation. However, living systems are dynamic and so are their components including proteins. Consequently, the protein structure–function paradigm, entailing the intimate relationship between the three-dimensional structure and function of proteins was expanded to the protein structure–function–dynamics paradigm. As per recent studies [3,4], the role of protein structural dynamics seems not only limited to the function, but also to the evolution of function, making it crucial to take dynamics into consideration whilst trying to understand biological processes. Limitations of crystallography for providing insight into the conformational dynamics of biological macromolecules can be complemented by several experimental methods for studying solution dynamics. These include nuclear magnetic resonance (NMR) spectroscopy, small-angle X-ray scattering (SAXS), hydrogen exchange mass spectrometry (HX-MS), and recently, cryo-electron microscopy. However, these approaches have their own limitations, particularly with respect to the applicable systems, resolutions, etc. In this regard, computational methods proved to be crucial for studying the dynamics of biomacromolecules, particularly at shorter time scales on the order of nanoseconds to microseconds. With recent advances in supercomputer architecture, graphics processing unit (GPU)-based computing, and faster algorithms, microsecond- to millisecond-scale studies of large systems are becoming routine and, with the current pace, studying second-scale dynamics may be possible in the next few years [5]. In this review, we focus on how computational methods contributed to mitigating the limitations of biomolecular crystallography.
We begin with brief descriptions of some of the most pressing constraints that are encountered while studying biomacromolecules using X-ray crystallography. We then describe some of the recent advances in experimental methods, both in crystallography and otherwise, to remedy some of the limitations. Next, we expound the crucial contribution of computational methods in complementing, as well as supplementing, experimental methods in order to study protein structural dynamics. Finally, we conclude the review with some of the limitations of computational methods and the future prospects.
2. Constraints in Crystallography for Studying Protein Structure and Dynamics
X-ray crystallography has been the most important method for exploring the three-dimensional structure of biological macromolecules over time [1,2]. It is so far the most reliable and widely used method of obtaining atomic-resolution information about protein structure. Moreover, owing to well-established experimental protocols and data analysis methods, it remains a method of choice for the pharmaceutical industry to explore protein–drug interactions [2]. However, despite the various strengths of X-ray crystallography, it suffers from certain constraints (Figure 1), arising from a variety of different sources [6].
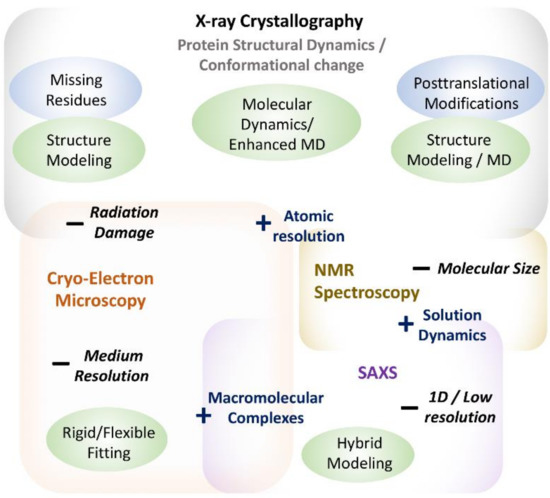
Figure 1.
Summative figure showing strengths and limitations of various experimental methods in understanding protein structure and dynamics. MD stands for Molecular Dynamics. Different boxes represent different experimental methods with “+” sign showing their strengths and “−” sign showing their limitations. Computational methods addressing the limitations are shown in green ellipses.
2.1. Challenges in Data Collection and Interpretation of Diffraction Data
One of the first challenges in studying protein structure using crystallography is obtaining good-quality protein crystals. Continued advances in the protocols and instrumentation allowed obtaining X-ray structures of larger systems at higher resolution; however, getting protein crystals that diffract to a high resolution remains a road block that protein crystallographers need to cross [7]. The protein crystallization process, being a multiparameter-dependent process, requires the tuning of several physical and biochemical factors for different proteins. The challenges include, but are not limited to, solubility of macromolecular complexes, membrane protein crystallization, disorder in the protein leading to unstable crystals, etc. The historical development, challenges, and future of protein crystallogenesis were previously reviewed in detail elsewhere [7].
Radiation damage to the protein crystals has long been a serious problem in biomacromolecular crystallography. Irradiation of crystals by X-rays causes the generation of free radicals that affect the overall diffraction intensity, and hence, the resolution and quality of structural model [8]. Apart from this overall reduction in quality, the damage caused by free radicals can cause specific changes in biomolecules, such as photoreduction, breaking of bonds, etc., particularly in the solvent-exposed regions of the proteins including active sites and cofactors bound to proteins, which can lead to incorrect conclusions [9]. Serial femtosecond crystallography, described in detail later, is a new approach for circumventing this issue.
Furthermore, once the diffraction data are collected, the models created using the data might sometimes be suboptimal, particularly for medium- to low-resolution structures. Such uncertainty was explored extensively in protein–ligand systems, where it was found that the model for ligands is often overinterpreted [10,11].
Since the model of the structure generated using X-ray crystallography is determined as an averaged model from the diffraction data obtained from multiple copies of the protein in crystal, the structural heterogeneity and anisotropy in the atomic motions is usually lost, except for very-high-resolution structures [12]. Considering that the function of proteins may be dependent on the sub-angstrom-scale movement of atoms [13,14,15], ambiguity or inaccuracy in the crystal structure model might confound the understanding of the structure–function relationship.
2.2. Challenges in Retrieving Biochemical Information from Crystal Structures
2.2.1. Crystal Environment Artefacts
Crystals comprise unit cells which are in turn composed of several asymmetric units. As a result of crystallization, the protein molecules in these asymmetric units or neighboring symmetry-related molecules interact, giving rise to spurious interactions that are absent in functional molecules in cells and can be referred to as crystal packing artefacts [16]. Despite using similar forces, crystal contacts are not considered to be strong interactions when compared to biologically relevant interactions [17,18,19]. However, these interactions might still lead to the stabilization of certain higher-energy conformations, as explored in λ Cro dimer structures using extensive molecular dynamics and replica-exchange molecular dynamics simulations [20,21]. It is sometimes observed that the same protein in different crystal forms takes different conformations, which suggests the role of crystal contacts in obtaining the given conformation [22,23].
Apart from crystal packing, the crystal chemical environment (buffer salt concentration, pH, etc.) can also affect the conformation observed in the crystal structure. In human protein kinase CK2, a high concentration of lyotropic salts in the crystallization buffer is known to support the closed conformation of the kinase, which was rarely observed in solution dynamics [24,25].
2.2.2. Cryo-Cooling Effects
Cooling the crystals to a very low temperature (typically 100 K) for diffraction data collection—referred to as cryo-cooling—has been an indispensable technique in X-ray crystallography for about three decades now [26]; currently, most X-ray crystallographic structures are obtained using this technique. Cryo-cooling not only decreases the incidence of radiation damage due to high-energy X-ray radiation, but also decreases the overall atomic motion, thus improving the diffraction resolution [26]. However, the protein assumes distinct dynamics between the cryogenic temperature and the physiological temperature. The dynamics of proteins in crystals are described by B factors despite their limitation [27], and typically, the B factor increases at higher temperatures, with inflection near 180 K, which is associated with glass transition [28]. Heterogeneity in cooling across crystals also poses challenges in determining the effects of cryo-cooling on protein structure, as discussed in Section 3.1.3. [29].
2.2.3. Missing Residues, High-Flexibility Regions
Crystal structures often contain multiple residues whose coordinates are missing from the model. As per a recent study, about 69% of PDB files were found to contain missing residues [30]. In particular, low-resolution structures (>3 Å) were found to be missing about 10% of the residues [30]. This is usually the result of high flexibility in the solvent-exposed loop regions of the proteins, which leads to highly ambiguous or no electron density for that region. These missing residues can make it difficult to understand the biological processes when they are at or near a functionally important region of the protein.
Intrinsically disordered proteins (IDPs) or regions of structured proteins with intrinsically disordered regions form some of the toughest targets for structural and dynamics analysis [31]. The high flexibility of such proteins makes it almost impossible to get good-quality crystals, and hence, X-ray diffraction data. The structural elucidation of such IDPs also remains challenging using other experimental techniques.
2.2.4. Missing Water Molecules and Solvent Information
Most biomolecules are surrounded by water within cells. Apart from playing crucial roles in regulating the physico-chemical properties of these molecules, water molecules also participate directly in biochemical reactions [32,33,34]. Usually, crystal structures at higher resolution have some ordered water molecules that show electron density, and hence, can be modeled. However, in most cases involving lower-resolution structures, the water molecules cannot be observed. Even at high resolution, the role of dynamic water molecules in function cannot be studied using crystallography.
Notwithstanding the challenges mentioned above, crystal structures determined so far provide crucial mechanistic insights into the biological phenomena under observation, and can be used to design further experimental and computational studies to fully characterize dynamics and functional mechanisms. Having described the limitations in obtaining structural and dynamic information using X-ray crystallography, we proceed to review the recent advances in experimental methods that contributed in overcoming these limitations.
3. Recent Experimental Advances for Enhancing/Complementing Crystallography
3.1. Advancements of Crystallography Methods
Several advances were made recently in the field of crystallography to address some of the limitations discussed above. Most of these advances were heralded by path-breaking developments in instrumentation and in experimental protocols.
The advent of increasingly high-energy X-ray sources, such as second- and third-generation synchrotrons, dramatically improved the resolution for biomacromolecular crystallography [35]. In addition, high-energy X-rays were also shown to cause less radiation damage, although there are conflicting reports about the relationship between the energy of X-rays used and radiation damage [9,36,37,38].
3.1.1. Crystallographic Refinement Methods
As mentioned in Section 2.1, despite the presence of structural heterogeneity and dynamics in protein crystals, most crystal structures in the Protein Data Bank contain single conformational models [39]. Several computational methods were developed to model this heterogeneity present in the electron density maps [39,40,41,42,43]. Most of these methods involve sampling different conformations to improve the fit to electron density. One such method is phenix.rosetta_refine, which combines the crystallography refinement protocol of Phenix [44] and the conformational sampling by popular modeling software Rosetta to provide good models from low-resolution data [45]. Recently, a crystallographic refinement method was proposed by Pearce et al. [46] that makes use of multiple diffraction datasets to model regions with weak or ambiguous electron density [46]. Further development and the broad usage of such computational methods can lead to abstraction of more accurate models from crystallographic data.
3.1.2. Serial Femtosecond Crystallography and Single-Particle Experiments with X-ray Free-Electron Laser (XFEL) Sources
The two most challenging methodological problems associated with crystallography are growing the crystals and radiation damage due to long-term exposure to high-energy radiation. The advent of X-ray free-electron lasers (XFELs) opened an avenue for solving both these problems [47,48]. The femtosecond-long XFEL pulses generate diffraction patterns before the destruction of protein crystals [49]. The high-energy pulse allows measurements from nanocrystals, thereby circumventing the need for growing large crystals. However, since the crystal is completely destroyed in a single encounter, a large number of diffraction measurements need to be collected using a stream of crystals [48]. This method holds enormous promise for determining the structures of conventionally challenging membrane proteins [50]. Femtosecond crystallography is also used to study the dynamics of proteins in time-resolved experiments [51,52,53,54], as was done with a synchrotron beam [55].
In addition to crystallographic studies, attempts began extracting the structural information of biological systems in near-native state using XFEL through single-particle analysis of non-crystalline samples [56,57,58]. Although, the experiments are currently performed for sub-μm systems and the resolution is quite limited, experimental techniques and computational algorithms are actively being developed toward the goal of structure analysis [59,60,61,62,63,64].
3.1.3. Multi-Temperature/Room-Temperature Crystallography
The effects of cryo-cooling on protein crystal structures are not fully understood. Experimentally, Fraser and coworkers investigated these low-temperature effects extensively [65,66]. They suggested that cryo-cooling affects the side-chain conformational ensemble in a significant and biologically relevant manner [65,66]. In particular, a side-chain conformer, suggested to be catalytically important, was only detectable with room-temperature diffraction of cyclophilin A [65]. By comparing NMR experimental and crystal structures of dihydrofolate reductase, they also demonstrated that the room-temperature crystal structure is indispensable for investigating protein dynamics [67]. In addition, the cooling process may introduce further heterogeneity because of different timelines of cooling [29].
The recent development of XFEL also provides the opportunity for room-temperature crystallography. Sirerra et al. [68] studied the room-temperature structure of ribosome in comparison with cryo-cooled synchrotron data, showing a 3.6-Å shift of the h28 phosphate backbone. Such a large change can have a significant impact on the ligand binding affinity [68].
3.1.4. Crystal Contact Free Space
Structures solved by X-ray crystallography capture only a snapshot or an averaged representation of a dynamic protein. However, these dynamic motions are essential for studying the biological function. Crystal contacts play a role in blocking these internal motions in a protein unless the dynamic region of the protein is located where it cannot interact with its neighboring molecule in the crystal. A new fusion protein method was developed for studying large dynamic motions within protein crystals by minimizing crystal contacts [69]. In this method, a target protein is fused with a tag protein via a rigid linker causing the formation of crystal contact free space (CCFS) in the protein crystal. The dynamic region is localized in the CCFS, and thus, is free from any crystal contact. This method was applied to a mitochondrial outer membrane protein, Tom20, which binds to presequence to import proteins into the mitochondria, and an archaeal oligosaccharyltransferase protein (AgIB) [69]. Although flexible regions free from crystal contacts result in weaker electron densities, using proper data0processing techniques and computational methods, dynamical information can be obtained.
3.2. Other Biophysical Techniques
In this section, we review some of the biophysical methods that give important information about protein structure and dynamics, which recently gained importance owing to improved experimental and computational protocols. As detailed analyses of each of these methods is beyond the scope of this review, we mention the strengths of each of the methods when compared to crystallography and cite recent reviews that provide details of these methods.
NMR spectroscopy is a powerful method for studying not only the structure and dynamics of biomacromolecules in solution, but also ligand binding [70], protein–protein interaction [71], and protein–nucleic-acid interaction [72]. Historically, one of the biggest limitations of studying biomacromolecules using NMR spectroscopy was its inability to deal with large molecules (>20 kDa). However, owing to recent advances in labeling techniques and new experimental protocols, structures of several protein complexes with sizes in the range of hundreds of kDa were determined [73,74]. Apart from this, NMR spectroscopy is currently being successfully utilized to understand the role of low-population conformations of proteins in solutions, which were previously impossible to investigate [75]. Further advancements, such as solid-state NMR spectroscopy, in combination with computational methods and scattering studies, enable the exploration of biological systems such as membrane proteins, which are challenging to study using crystallography [76,77].
Hydrogen exchange mass spectrometry (HX-MS) is a comparatively old technique for studying conformational changes in proteins that historically eluded scrutiny through crystallography or NMR spectroscopy. HX-MS quantifies the exchange of amide hydrogen with deuterium in solution and provides information about the tertiary structure and dynamics of the protein [78]. HX-MS is helpful in elucidating conformational changes in proteins upon ligand binding [79] and during allosteric regulation [80]. Recently, HX-MS was used to understand the conformational dynamics of DNA binding and Cas3 recruitment by the cascade complex [81].
Cross-linking mass spectrometry (XL-MS) is an important experimental technique for gaining insight into protein–protein interactions and structures of large macromolecular complexes. Recent years saw considerable development both in the cross-linking methodology and in data processing and visualization (recently reviewed by Leitner et al. [82]). XL-MS usually involves creating covalent bonds between spatially close regions of protein complexes, followed by proteolysis and subsequent mass spectrometry to determine the regions enriched in cross-linked peptides. The data obtained from XL-MS can be used to generate distance restraints and were recently used extensively to model large macromolecular complexes (See Section 4.2).
Transmission electron microscopy has been used to observe biological systems on the scale of micrometers to tens of nanometers for a long time. However, owing to recent advances in instrumentation, including better electron guns, electron detectors, automated mounting systems, and image-processing software and algorithms [83], there was a resolution revolution in the field of cryo-electron microscopy (cryo-EM) [84] with volume maps reaching resolutions of up to 2 Å. This made several difficult crystallographic target protein complexes accessible to structural and dynamical analysis [85,86,87,88].
Small-angle X-ray scattering (SAXS) is a powerful and well-established method for studying proteins in solution. It can provide several important parameters about the protein in its native state, including molecular weight, overall shape, and radius of gyration [89]. With recent advancements in synchrotron radiation sources, there was a resurgence in the use of SAXS for studying biomolecular structure and dynamics [90,91]. It is particularly helpful in determining low-resolution shape information about the oligomeric states of proteins [92], as well as the structure and dynamics of intrinsically disordered proteins [93,94] and integral membrane proteins difficult to study using other biophysical techniques [95].
As described in the previous section, there was immense advancement in experimental methods for studying protein structure and dynamics. However, each method has its own limitations, and computational methods became essential in mitigating these limitations, integrating results from multiple experimental sources and providing a better understanding of protein structure and dynamics. In the following section, we review the computational methods that are crucial in the exploration of protein structure and dynamics.
4. Computational Methods for Complementing/Supplementing Experiments
4.1. Protein Structural Modeling
The inability to obtain a protein of interest in considerable quantities, either via expression or purification, still remains one of the first challenges in studying the protein–structure–function relationship. For all such cases where protein structure determination proves challenging, structure prediction remains one of the most sought-after computational methods. With an ever increasing number of known protein structures and the development of better algorithms, there has been a considerable improvement in the quality of model predictions [96].
The most accurate and popular methods of protein structure prediction are still dependent on the availability of the structure for a homologous protein. Homology- or template-based modeling methods involve using the structural information from a sequentially similar region of a homologous protein for which the structure is already known. Template-based methods improved considerably over the years, primarily due to an increase in the number of proteins for which structures are known and due to improvements in sequence-alignment methods [97].
Despite the impressive increase in the number of experimentally determined protein structures, there is still a large gap between sequence space and structure space. Consequently, for a large number of proteins, templates are not available to assist in structure prediction. To understand the structure for such proteins, several ab initio or template-free methods were developed. Although such prediction methods are severely limited with regards to the length of the protein sequence, certain recent advances in reliable residue–residue contact predictions considerably improved their accuracy [96,98].
Apart from playing a crucial role in understanding the structure and function of proteins difficult to study using experiments, protein structural modeling is routinely used to study regions that are missing from the crystal structures. Modeling the regions of missing coordinates in protein crystal structures forms one of the first steps in protein structural and dynamics studies using varying computational methods.
4.2. Hybrid Methods for Studying Protein Complexes
As discussed in Section 3, there was immense advancement in experimental methods that provide low-resolution or complementary information about the overall structure and dynamics of proteins and their complexes in solution. Simultaneously, computational methods were developed to integrate such information coming from multiple sources, with existing high-resolution crystal and NMR structural data for gaining insight into solution structure and dynamics. Hybrid methods, utilizing information from multiple experimental sources and integrating them using computational algorithms, are particularly successful in understanding the structure and dynamics of large macromolecular complexes [99,100,101,102,103,104].
Volume maps obtained even from state-of-the-art cryo-EM experiments typically range around 3–4 Å resolution, which is insufficient to obtain atomic level information. Crystallography data, although high resolution, might not represent a functional or solution state. Computational methods integrating information obtained from these two complementary sources can, thus, supplement the overall information and provide novel insight into both large-scale, as well as atomic-level, structure and dynamics. Several methods were developed for fitting high-resolution crystal structures to EM density maps (for a recent review, see Kim et al. [105]). These methods rely on different approaches to deform coordinates of known high-resolution crystals or NMR structures to fit low-resolution EM maps, such as normal-mode-based flexible fitting [106,107], coarse-grained model-based flexible fitting [108,109], all-atom molecular-dynamics-based fitting [110], deformable elastic-network-based fitting [111], Monte Carlo method-based fitting [112], or a combination of these methods [113].
SAXS provides information about the overall shape of large macromolecule in solution. This is very helpful in understanding the large conformational changes associated with the function of macromolecules. The low-resolution information obtained from SAXS can be used to reconstruct high-resolution structural models using existing crystal structures [114,115,116,117]. Cross-linking mass spectrometry data provide spatial restraints between different components of large macromolecular complexes. These restraints are utilized by integrative modeling software to elucidate the structure and dynamics of these complexes [118,119,120,121,122]. Hybrid methodologies were also developed recently to utilize X-ray free-electron laser diffraction data to understand protein structure and dynamics [123].
4.3. Molecular Mechanics Methods
Biomacromolecules (Proteins, DNA, and RNA) of interest can be described at multiple scales of detail. At the small scale, the electron distribution of each atom in the molecule is described, and the dynamics of these atoms are studied using quantum mechanics. Although this approach is extremely useful and accurate in studying small molecules and mapping reactions in localized active-site pockets of enzymes [124], it becomes computationally infeasible for large systems involving thousands of atoms. At a lesser detailed level, the atoms of the biomacromolecules themselves are modeled as particles, and the forces between these particles are described and calculated based on semi-empirically-derived force-field potentials based on classical mechanics (Figure 2). The methods using such descriptions, referred to as molecular mechanics methods, form a quintessential part of computational simulations of biomacromolecules [125]. Molecular dynamics (MD) simulations help study the time evolution of dynamics of a molecular system by integrating the forces acting on each atom of the system at successive time points. Since the first MD simulations of colliding hard spheres [126], the method was developed immensely and is used extensively to study biological processes of varying length and time scales [125]. The emergence of faster processors, particularly graphics processing units, and the development of faster algorithms to perform calculations on these processors, led to a revolution in large-scale studies using MD simulations. This led to the structural and dynamic exploration of large macromolecular machines at atomistic resolution in unprecedented detail.
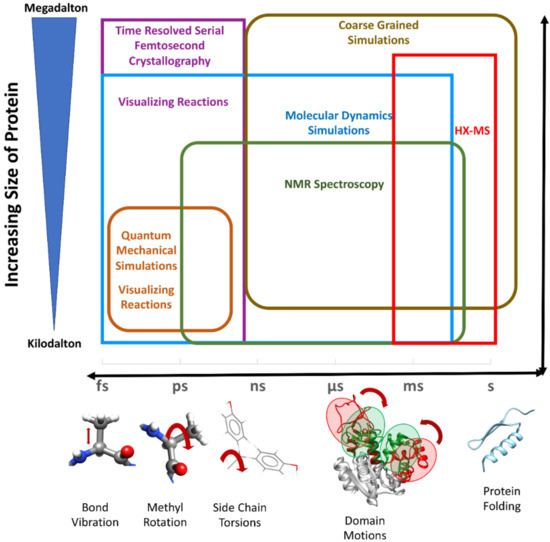
Figure 2.
Protein dynamics can be modeled at several length and time scales. Various experimental and computational methods (shown in same colored text and boxes) are used to understand the multiscale dynamics of proteins and their complexes. Red arrows show different motions in the molecules happening at femtosecond (fs), picosecond (ps), nanosecond (ns), microsecond (μs) and millisecond (ms) time scale. Protein folding can have a time scale of seconds (s) to minutes or even hours.
4.3.1. Coarse-Grained Modeling
Despite breakthroughs in hardware and software capabilities of modern computer systems, it is still a challenging task to study protein dynamics in the time range relevant for biological functions, especially for important macromolecules. In order to address this, simplified coarse-grained (CG) models were developed and are used successfully for the last four decades. CG models are simplified representations of biological macromolecules, often treating amino-acid residues, secondary structural elements, or whole domains as single entities. This leads to a dramatic reduction in the degrees of freedom, consequently making these methods computationally much less expensive than molecular dynamics simulations. Structure-based models like Go models that assume a smooth funnel-shaped energy landscape are particularly useful in studying the folding of proteins [127,128]. Inspired by the seminal work of Monique Tirion [129], elastic network models form another class of coarse-grained model that contributed to understanding protein dynamics in diverse biological systems [130,131,132,133]. Network-based coarse-graining of protein structures is also able to provide insight into protein folding [134] and allosteric communication [134,135,136].
4.3.2. Enhanced MD Simulations
The rugged conformational landscape of proteins makes conventional MD simulations vulnerable to be trapped in one of the local, non-functional free-energy minima [137]. In order to overcome this problem, several enhanced MD methods were developed [137]. As the range is broad, we can only review a limited number of methods. For more detail, readers are referred to extensive reviews by Bernardi et al. [137], Mitsutake et al. [138], and Valsson et al. [139].
Methods such as replica exchange [140], umbrella sampling [141], and multicanonical algorithms [142,143] utilize equilibrium sampling of non-Boltzmann weights, which can enhance sampling efficiency. The information of canonical ensembles can be obtained via proper post-processes. The replica-exchange method is especially suitable for parallel computing, and its post-process is straightforward. Hence, the MD version of the replica-exchange method, known as replica-exchange molecular dynamics (REMD) [140] is widely used in biomolecular systems. REMD and its related methods have interesting applications, including studying pH dependence of biomolecules [144], drug design [145], cold denaturation [146], and biological systems with strong phase transitions [147]. Furthermore, recent theoretical progress enabled the extraction of kinetic rates using these methods [148].
Some other methods use non/quasi-equilibrium sampling. In steered MD [149], a molecule or a part of it is dragged with an artificial external force so that desired conformational changes can be studied. Even with such an external force, it is possible to extract free energy via Jarzynski equality [149]. The Wang–Landau method [150] and metadynamics [139,151] employ dynamically updated potentials or weights of sampling so that the system can sample a wider range of energy or reaction coordinates which are specified a priori.
4.4. Crystal MD Simulations
Crystal structures typically portray a single averaged model of protein structure, which loses information regarding the dynamics of the protein not only in solution, but also in the crystal itself. In terms of exploring structural heterogeneity within the protein crystal, there were a few recent attempts to include the ensemble view of the structure in crystal model building. (See Section 4.2). However, molecular dynamics simulations have been used to understand the structural dynamics in the crystal environment for a long time [152,153].
In these simulations, the entire unit cell is simulated in an environment very similar to the experiment. This makes the comparison between X-ray data and simulations rather informative; however, these are computationally expensive simulations as compared to conventional MD simulations, owing to larger starting structures and delayed convergence [154]. Nevertheless, crystal MD simulations provided novel insights into the dynamics of protein and solvent in crystal [155], the energetics of the packing interface [21], crystal packing effects [156,157], protein–protein and protein–detergent interactions in crystals of membrane proteins [158], solute diffusion in protein crystals [159], and the behavior of water and ions in protein crystals [160].
5. Novel Insights from Computational Methods
The crucial role of computational methods in understanding protein structural dynamics cannot be overstated. In the following sections, we review novel insights gained using computational methods, either independently or in concert with experiments.
5.1. Exploring the Structure and Dynamics of Large Macromolecular Complexes
Biological systems are complex and, often, a multidisciplinary approach is required to understand these complex systems. This is particularly true when it comes to understanding the structure–function–dynamics of large macromolecular machines within cells like ribosomes, proteasomes, spliceosomes, transcription initiation complexes, etc. Until recently, the only experimental method that provided high-resolution information of these macromolecular assemblies was X-ray crystallography. Despite great achievements in determining crystal structures of some of these complexes, the information for most of macromolecular complexes is missing. Recent phenomenal advances in the cryo-EM led to enormous breakthroughs in the understanding of macromolecular structures and dynamics [84].
The ribosome is a macromolecular machine responsible for the synthesis of all proteins inside cells. Owing to its crucial role in all living systems, structure–function studies of the ribosome have gone on for decades, and the determination of an atomic-resolution crystal structure of the ribosome enhanced the understanding of one of the most fundamental biological process [161]. However, the ribosome is a highly dynamic macromolecular complex that undergoes several conformational changes during protein translation [162]. Static structures obtained from crystallography and cryo-EM often fail to capture the transient dynamics between different states of this complex. Whitford et al. applied a hybrid approach of using a flexible fitting program, MDfit, along with experimental information from multiple sources, including X-ray crystallography, cryo-EM, and biochemical experiments, to shed light on the transient conformations involving transfer RNA (tRNA) translocation into the ribosome during the process of translation [163]. Additionally, despite the large size of the ribosome, considerable progress was made toward the molecular simulations of whole ribosomes. This was recently reviewed by Bock et al. [164].
The C-terminal domain (CTD) of RNA polymerase II comprises several repeats and is structurally disordered, leaving it inaccessible to most structure determination methods, including X-ray crystallography and cryo-EM. Consequently, despite its important role involving multiple protein–protein interactions during transcription, there was limited structural information detailing the CTD in complex with its cognate binding partners. Using integrative structural modeling, Jasnovidova et al. [102] determined the molecular architecture of the C-terminal domain of RNA polymerase with termination factor Rtt103. This involved assimilating existing and novel structural and other experimental data into a computational algorithm to determine the overall molecular architecture [102].
The conformational dynamics of the spliceosome, a megadalton-sized machine that processes precursor messenger RNAs to construct coding regions (exons), was studied at atomic resolution to delineate functional motions that were missing from the static structures [165]. Computational modeling and simulation also enabled the study of previously unexplored areas, such as describing the crowding effects inside the cytoplasm [166].
Going a step further on the length scale, hybrid methods involving cryo-EM, X-ray crystallography, molecular modeling, and molecular dynamics simulations were used to determine the structure and dynamics of a whole viral capsid comprising millions of atoms [167,168]. These studies provided crucial insights into the maturation and assembly process of the viral capsid.
5.2. Structural and Dynamical Effects of Post-Translational Modifications
There are hundreds of types of post-translational modifications (PTMs) of proteins that were found to be biologically important. Post-translational modifications like phosphorylation, methylation, acetylation, glycosylation, etc. were shown to play major roles in almost all biological processes [169]. These modifications cause changes in the structure, as well as the dynamics, of the proteins, modulating the functions of these proteins and their interacting partners [170].
In view of the importance of PTMs in the biological processes, the experimentally derived structural information for such modifications is considerably low [171]. Molecular modeling and simulations contributed significantly in understanding the role of PTMs in the structure and dynamics of proteins. Molecular dynamics simulations were used to study the effect of phosphorylation on the polar properties of protein surfaces, which could be crucial in understanding the interaction of proteins with other molecules [172]. In another molecular dynamics study, the role of S-glutathionylation of a particular cysteine residue in Arabidopsis thaliana BRASSINOSTEROID INSENSITIVE 1-associated receptor kinase (BAK1) was explored, and the effect of this PTM on the conformational dynamics reconciled with previous experimental observations [173].
5.3. Structure-Based Drug Design
Computational methods have contributed to almost every stage of drug discovery for a long time now [174]. Structure-based drug design conventionally focuses on high-throughput virtual screening using crystal structures of the target proteins; however, the role of dynamics in protein–drug interactions is being increasingly realized [175]. At the stage of initial virtual screening, instead of a static crystal structure, using an ensemble of structures for finding a lead molecule takes into consideration the inherent solution dynamics of the target binding site [176,177].
Virtual screening entails searching through millions of compounds for their ability to bind to the target protein. Pharmacophore-based screening simplifies this task by extracting a number of electronic and chemical features from a known ligand that can be used to screen for compounds with similar features. An intuitive extension of this approach is three-dimensional pharmacophore-based screening, where not only the chemical features but also the steric features are considered based on available protein–drug structures [178]. Conventionally, static single crystal structures were used to extract such pharmacophores; however, recently, molecular dynamics simulations of protein–ligand complexes were performed and used to generate dynamic pharmacophore models [179,180]. Using multiple crystal structures or conformations obtained from MD simulations was shown to reveal certain features that were missed when considering only single structures [181,182].
5.4. Understanding the Role of Water in Protein Structure and Function
Computational methods, particularly molecular dynamics simulations and quantum mechanics simulations, play crucial roles in understanding the role of water in the structure and function of biomacromolecules. Molecular dynamics simulations were used to understand the role of water molecules in protein–DNA binding [183], enthalpy–entropy compensation during protein–ligand interactions [184], proton transfer reactions in channel rhodopsins [185], etc. Furthermore, MD simulations were used to complement experimental methods like terahertz absorption spectroscopy [186], neutron scattering [187], and time-resolved fluorescence spectroscopy [34] to understand the structure and dynamics of folded and intrinsically disordered proteins.
6. Limitations of Computational Methods
Computational methods for studying protein structural dynamics were instrumental in understanding numerous biological processes and successfully shed light on phenomena that were inaccessible to experimental methods. However, like any other method, they also have certain limitations. In the following sections, we discuss some of the common limitations of the previously discussed computational methods.
6.1. Force Fields
Results obtained from any computational method are only as reliable and accurate as the parameters used in the development of that method. Molecular dynamics simulations use a combination of functional forms defining the interactions between different particles in the system and a set of predetermined parameters that are used to calculate forces between the particles [188]. Such force fields are constantly evolving with improved parameters to reconcile experimental observations and simulation results [189]. However, there are some key limitations to what can be studied using these force fields.
The force fields used to study solution dynamics of proteins were developed to model the behavior of folded proteins. This led to certain insufficiencies in studying intrinsically disordered proteins (IDPs) using the same force fields. Although simple modifications in some parameters of the force field improve the agreement between simulations and experiments, a more thorough evaluation and parameterization of forcefields is required for IDPs [190,191].
Most of the current force fields use fixed partial charges on the atoms to calculate electrostatic forces. Although these force fields were conventionally successful in simulating the behavior of molecules in homogeneous environments, their performance is limited in conditions where there are variations in local electric fields, and continued efforts are being made to resolve these limitations [192].
6.2. Sampling
While the sampling issues were addressed by several methodological developments as described above, the time scale of MD simulations is ultimately limited by the computational speed. Apart from the MD-specific machine Anton, which can reach sub-millisecond time scales per day [193], commonly used MD software packages that are used with general-purpose personal computer (PC/PC) clusters reach only sub-microsecond time scales per day, while using hundreds of central processing units (CPUs) [194]. Thus, the microsecond time scale is a practical limit for a single trajectory in common research groups as of 2018, although it is possible to replicate a system in question to increase statistics. The Markov state model [195] is one promising approach for inferring longer-time dynamics from limited MD trajectories, and any new framework that can work within such restraints should be useful and will open new gates for dynamics studies of protein.
7. Summary and Future Directions
X-ray crystallography has long been the de facto method of choice to study protein structure and function. Despite several challenges and limitations, it continues to remain one of the most crucial sources of structural information. However, the realization of the importance of dynamics and the concomitant improvement in the accuracy and applicability of computational methods to be able to study the functional dynamics of proteins in native-like environments heralded the era of computation-driven structural biology. This was equally matched by the rise of novel experimental methods to elucidate the dynamics of proteins in solution at varying length and time scales. Considering the complexity of biological processes, it is now abundantly clear that a multipronged approach comprising inputs from several different experimental and computational methods is required to understand the important questions. Consequently, hybrid/integrative methods are starting to become the new norm in biomolecular structure and dynamics studies. This is evident from the fact that the Protein Data Bank now has a new field of classification named “hybrid methods”, consisting of structures determined using more than one kind of experimental method. Also, a wwPDB hybrid/integrative methods task force was established in order to formalize the representation, validation, archiving, and publication of structural and dynamics data generated using hybrid or integrative methods [196,197]. Computational methods form an essential part of such structure determination protocols as they enable combining and complementing data from multiple sources, and they will continue to play a crucial role in the field of structural biology with more advanced sampling methods, better supercomputers, and more experimental advances.
Author Contributions
Writing—original draft preparation, A.S. (Ashutosh Srivastava), T.N., and A.S. (Arpita Srivastava). Writing—review and editing, A.S. (Ashutosh Srivastava), T.N., A.S. (Arpita Srivastava), O.M., and F.T. Supervision, O.M. and F.T. Funding acquisition, O.M. and F.T.
Funding
This work was supported by JSPS KAKENHI Grant Numbers 26119006, 16K07286, and 17K07305, and FOCUS for Establishing Supercomputing Center of Excellence. Arpita Srivastava acknowledges financial support from the IGER program of Nagoya University and the JASSO scholarship.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.G.; Wyckoff, H.; Phillips, D.C. A three-dimensional model of the Myoglobin molecule obtained by X-ray analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. A Glimpse of Structural Biology through X-ray Crystallography. Cell 2014, 159, 995–1014. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.; Kaltenbach, M.; Correy, G.J.; Carr, P.D.; Porebski, B.T.; Livingstone, E.K.; Afriat-Jurnou, L.; Buckle, A.M.; Weik, M.; Hollfelder, F.; et al. The role of protein dynamics in the evolution of new enzyme function. Nat. Chem. Biol. 2016, 12, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Sailer, Z.R.; Harms, M.J. Molecular ensembles make evolution unpredictable. Proc. Natl. Acad. Sci. USA 2017, 114, 11938–11943. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rosell, G.; Giorgino, T.; Harvey, M.J.; De Fabritiis, G. Drug Discovery and Molecular Dynamics: Methods, Applications and Perspective beyond the Second Timescale. Curr. Top. Med. Chem. 2017, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chruszcz, M.; Wlodawer, A.; Minor, W. Determination of Protein Structures—A Series of Fortunate Events. Biophys. J. 2008, 95, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Giegé, R. A historical perspective on protein crystallization from 1840 to the present day. FEBS J. 2013, 280, 6456–6497. [Google Scholar] [CrossRef] [PubMed]
- Taberman, H. Radiation Damage in Macromolecular Crystallography—An Experimentalist’s View. Crystals 2018, 8, 157. [Google Scholar] [CrossRef]
- Yano, J.; Kern, J.; Irrgang, K.-D.; Latimer, M.J.; Bergmann, U.; Glatzel, P.; Pushkar, Y.; Loll, B.; Sauer, K.; Messinger, J.; et al. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. USA 2005, 102, 12047–12052. [Google Scholar] [CrossRef] [PubMed]
- Pozharski, E.; Weichenberger, C.X.; Rupp, B. Techniques, tools and best practices for ligand electron-density analysis and results from their application to deposited crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 150–167. [Google Scholar] [CrossRef] [PubMed]
- Weichenberger, C.X.; Pozharski, E.; Rupp, B. Visualizing ligand molecules in twilight electron density. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Depristo, M.A.; De Bakker, P.I.W.; Blundell, T.L. Heterogeneity and Inaccuracy in Protein Structures Solved by X-ray Crystallography. Structure 2004, 12, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E., Jr. Conformational changes: How small is big enough? Nat. Med. 1998, 4, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, A.; Thornton, J. Conformational Changes Observed in Enzyme Crystal Structures upon Substrate Binding. J. Mol. Biol. 2005, 346, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; del Sol, A.; Nussinov, R. Allostery: Absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 2009, 378, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakamura, H.; Kinoshita, K. Discrimination between biological interfaces and crystal-packing contacts. Adv. Appl. Bioinform. Chem. 2008, 1, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Rodier, F. Protein–Protein interaction at Crystal Contacts. Proteins Struct. Funct. Genet. 1995, 23, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, R.P.; Chakrabarti, P.; Rodier, F.; Janin, J. A Dissection of Specific and Non-specific Protein-Protein Interfaces. J. Mol. Biol. 2004, 336, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O.; Argos, P. Protein-protein crystal-packing contacts. Protein Sci. 1997, 6, 2261–2263. [Google Scholar] [CrossRef] [PubMed]
- Ahlstrom, L.S.; Miyashita, O. Molecular Simulation Uncovers the Conformational Space of the λ Cro Dimer in Solution. Biophys. J. 2011, 101, 2516–2524. [Google Scholar] [CrossRef] [PubMed]
- Ahlstrom, L.S.; Miyashita, O. Packing interface energetics in different crystal forms of the λ Cro dimer. Proteins Struct. Funct. Bioinform. 2014, 82, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Andrec, M.; Snyder, D.A.; Hou, Z.; Young, J.; Montelione, G.T.; Levy, R.M. A large data set comparison of protein structures determined by crystallography and NMR: Statistical test for structural differences and the effect of crystal packing Michael. Proteins Struct. Funct. Bioinform. 2007, 69, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Wozniak, J.A.; Matthews, B.W. Protein Flexibility and Adaptability Seen in 25 Crystal Forms of T4 Lysozyme. J. Mol. Biol. 1995, 250, 527–552. [Google Scholar] [CrossRef] [PubMed]
- Klopffleisch, K.; Issinger, O.G.; Niefind, K. Low-density crystal packing of human protein kinase CK2 catalytic subunit in complex with resorufin or other ligands: A tool to study the unique hinge-region plasticity of the enzyme without packing bias. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Hirota, T.; Irle, S.; Tama, F. Conformational dynamics of human protein kinase CK2α and its effect on function and inhibition. Proteins Struct. Funct. Bioinform. 2018, 86, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Garman, E.F.; Owen, R.L. Cryocooling and radiation damage in macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, D62, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Kuzmanic, A.; Pannu, N.S.; Zagrovic, B. X-ray refinement significantly underestimates the level of microscopic heterogeneity in biomolecular crystals. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Tilton, R.F.; Dewan, J.C.; Petsko, G.A. Effects of Temperature on Protein Structure and Dynamics: X-ray Crystallographic Studies of the Protein Ribonuclease-A at Nine Different Temperatures from 98 to 320. Biochemistry 1992, 31, 2469–2481. [Google Scholar] [CrossRef] [PubMed]
- Halle, B. Biomolecular cryocrystallography: Structural changes during flash-cooling. Proc. Natl. Acad. Sci. USA 2004, 101, 4793–4798. [Google Scholar] [CrossRef] [PubMed]
- Djinovic-Carugo, K.; Carugo, O. Missing strings of residues in protein crystal structures. Intrinsically Disord. Proteins 2015, 3, e1095697. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Linke, K.; Ho, F.M. Water in Photosystem II: Structural, functional and mechanistic considerations. BBA Bioenerg. 2014, 1837, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Ball, P. Water is an active matrix of life for cell and molecular biology. Proc. Natl. Acad. Sci. USA 2017, 114, 13327–13335. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.; Singh, A.K.; Bhasne, K.; Dogra, P.; Datta, A.; Das, P.; Mukhopadhyay, S. Femtosecond Hydration Map of Intrinsically Disordered α-Synuclein. Biophys. J. 2018, 114, 2540–2551. [Google Scholar] [CrossRef] [PubMed]
- Su, X.-D.; Zhang, H.; Terwilliger, T.C.; Liljas, A.; Xiao, J.; Dong, Y. Protein Crystallography from the Perspective of Technology Developments. Crystallogr. Rev. 2015, 21, 122–153. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Hirata, K.; Hasegawa, K.; Ueno, G.; Yamamoto, M. Synchrotron radiation dose dependence of radiation damage for protein crystals studied at various X-ray energies. J. Synchrotron Radiat. 2007, 14, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Rosenbaum, G.; Dauter, M.; Dauter, Z. Radiation decay of thaumatin crystals at three X-ray energies. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Holton, J.M. A beginner’s guide to radiation damage. J. Synchrotron Radiat. 2009, 16, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.T.; Ng, H.-L.; Fraser, J.S.; Corn, J.E.; Echols, N.; Sales, M.; Holton, J.M.; Alber, T. Automated electron-density sampling reveals widespread conformational polymorphism in proteins. Protein Sci. 2010, 19, 1420–1431. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.J.; Kondrashov, D.A.; Wesenberg, G.E.; Phillips, G.N. Ensemble Refinement of Protein Crystal Structures: Validation and Application. Structure 2007, 15, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Adams, P.D.; Moriarty, N.W.; Zwart, P.; Read, R.J.; Turk, D.; Hung, L.-W. Interpretation of ensembles created by multiple iterative rebuilding of macromolecular models. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bedem, H.; Dhanik, A.; Latombe, J.-C.; Deacon, A.M. Modeling discrete heterogeneity in X-ray diffraction data by fitting multi-conformers. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Tom Burnley, B.; Afonine, P.V.; Adams, P.D.; Gros, P. Modelling dynamics in protein crystal structures by ensemble refinement. Elife 2012, 1, 311. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, F.; Echols, N.; Headd, J.J.; Terwilliger, T.C.; Adams, P.D.; Baker, D. Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat. Methods 2013, 10, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Pearce, N.M.; Krojer, T.; Bradley, A.R.; Collins, P.; Nowak, R.P.; Talon, R.; Marsden, B.D.; Kelm, S.; Shi, J.; Deane, C.M.; et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Johansson, L.C.; Stauch, B.; Ishchenko, A.; Cherezov, V. A Bright Future for Serial Femtosecond Crystallography with XFELs. Trends Biochem. Sci. 2017, 42, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Martin-Garcia, J.M.; Conrad, C.E.; Coe, J.; Roy-Chowdhury, S.; Fromme, P. Serial femtosecond crystallography: A revolution in structural biology. Arch. Biochem. Biophys. 2016, 602, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Neutze, R.; Wouts, R.; van der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wacker, D.; Gati, C.; Han, G.W.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; Katritch, V.; Barty, A.; et al. Serial Femtosecond Crystallography of G Protein—Coupled Receptors. Science 2013, 342, 1521–1525. [Google Scholar] [CrossRef] [PubMed]
- Tosha, T.; Nomura, T.; Nishida, T.; Saeki, N.; Okubayashi, K.; Yamagiwa, R.; Sugahara, M.; Nakane, T.; Yamashita, K.; Hirata, K.; et al. Capturing an initial intermediate during the P450nor enzymatic reaction using time-resolved XFEL crystallography and caged-substrate. Nat. Commun. 2017, 8, 1585. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Kubo, M.; Baba, S.; Yamashita, K.; Hirata, K.; Ueno, G.; Nomura, T.; Kimura, T.; Shinzawa-Itoh, K.; Baba, J.; et al. A nanosecond time-resolved XFEL analysis of structural changes associated with CO release from cytochrome C oxidase. Sci. Adv. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Nogly, P.; Weinert, T.; James, D.; Carbajo, S.; Ozerov, D.; Furrer, A.; Gashi, D.; Borin, V.; Skopintsev, P.; Jaeger, K.; et al. Retinal isomerization in bacteriorhodopsin captured by a femtosecond X-ray laser. Science 2018, 361, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.; Tanaka, R.; et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 2016, 354, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.J.D. Femtosecond Crystallography with Ultrabright Electrons and X-rays: Capturing Chemistry in Action. Science 2014, 343, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, O.; Joti, Y. X-ray free electron laser single-particle analysis for biological systems. Curr. Opin. Struct. Biol. 2017, 43, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fan, J.; Li, H.; Jiang, H. Current Status of Single Particle Imaging with X-ray Lasers. Appl. Sci. 2018, 8, 132. [Google Scholar] [CrossRef]
- Aquila, A.; Barty, A.; Bostedt, C.; Boutet, S.; Carini, G.; Deponte, D.; Drell, P.; Doniach, S.; Downing, K.H.; Earnest, T.; et al. The linac coherent light source single particle imaging road map. Struct. Dyn. 2015, 2, 41701. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Miyashita, O.; Jonic, S.; Tokuhisa, A.; Tama, F. Single-particle XFEL 3D reconstruction of ribosome-size particles based on Fourier slice matching: Requirements to reach subnanometer resolution. J. Synchrotron Radiat. 2018, 25, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Hantke, M.F.; Ekeberg, T.; Maia, F.R.N.C. Condor: A simulation tool for flash X-ray imaging. J. Appl. Cryst. 2016, 49, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Joti, Y.; Shibuya, A.; Song, C.; Kim, S.; Tono, K.; Yabashi, M.; Tamakoshi, M.; Moriya, T.; Oshima, T.; et al. Imaging live cell in micro-liquid enclosure by X-ray laser diffraction. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Loh, N.-T.D.; Elser, V. Reconstruction algorithm for single-particle diffraction imaging experiments. Phys. Rev. E 2009, 80, 026705. [Google Scholar] [CrossRef] [PubMed]
- Ekeberg, T.; Svenda, M.; Abergel, C.; Maia, F.R.N.C.; Seltzer, V.; Claverie, J.-M.; Hantke, M.; Jönsson, O.; Nettelblad, C.; Van Der Schot, G.; et al. Three-Dimensional Reconstruction of the Giant Mimivirus Particle with an X-ray Free-Electron Laser. Phys. Rev. Lett. 2015, 114, 098102. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Mochizuki, Y.; Joti, Y.; Tama, F.; Miyashita, O. Gaussian mixture model for coarse-grained modeling from XFEL. Opt. Express 2018, 26, 26734. [Google Scholar] [CrossRef]
- Fraser, J.S.; Clarkson, M.W.; Degnan, S.C.; Erion, R.; Kern, D.; Alber, T. Hidden alternative structures of proline isomerase essential for catalysis. Nature 2009, 462, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.S.; van den Bedem, H.; Samelson, A.J.; Lang, P.T.; Holton, J.M.; Echols, N.; Alber, T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc. Natl. Acad. Sci. USA 2011, 108, 16247–16252. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, R.B.; van den Bedem, H.; Fraser, J.S.; Wright, P.E. Integrated description of protein dynamics from room-temperature X-ray crystallography and NMR. Proc. Natl. Acad. Sci. USA 2014, 111, E445–E454. [Google Scholar] [CrossRef] [PubMed]
- Sierra, R.G.; Gati, C.; Laksmono, H.; Dao, E.H.; Gul, S.; Fuller, F.; Kern, J.; Chatterjee, R.; Ibrahim, M.; Brewster, A.S.; et al. Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nat. Methods 2015, 13, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, R.; Shimada, A.; Komuro, Y.; Sugita, Y.; Kohda, D. Rational design of crystal contact-free space in protein crystals for analyzing spatial distribution of motions within protein molecules. Protein Sci. 2016, 25, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Bhattiprolu, K.C.; Gubensäk, N.; Zangger, K. Investigating Protein–Ligand Interactions by Solution Nuclear Magnetic Resonance Spectroscopy. ChemPhysChem 2018, 19, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Teilum, K.; Kunze, M.B.A.; Erlendsson, S.; Kragelund, B.B. (S)Pinning down protein interactions by NMR. Protein Sci. 2017, 26, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Chiliveri, S.C.; Deshmukh, M.V. Structure of RDE-4 dsRBDs and mutational studies provide insights into dsRNA recognition in the Caenorhabditis elegans RNAi pathway. Biochem. J. 2014, 458, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya Chiliveri, S.; Deshmukh, M.V. Recent excitements in protein NMR: Large proteins and biologically relevant dynamics. J. Biosci. 2016, 41, 787–803. [Google Scholar] [CrossRef]
- Huang, C.; Kalodimos, C.G. Structures of Large Protein Complexes Determined by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Biophys. 2017, 46, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.E. New Views of Functionally Dynamic Proteins by Solution NMR Spectroscopy. J. Mol. Biol. 2016, 428, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Becker-Baldus, J.; Bamann, C.; Saxena, K.; Gustmann, H.; Brown, L.J.; Brown, R.C.D.; Reiter, C.; Bamberg, E.; Wachtveitl, J.; Schwalbe, H.; et al. Enlightening the photoactive site of channelrhodopsin-2 by DNP-enhanced solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 9896–9901. [Google Scholar] [CrossRef] [PubMed]
- Opella, S.J.; Marassi, F.M. Applications of NMR to membrane proteins. Arch. Biochem. Biophys. 2017, 628, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Jaswal, S.S. Biological insights from hydrogen exchange mass spectrometry. BBA Proteins Proteom. 2013, 1834, 1188–1201. [Google Scholar] [CrossRef] [PubMed]
- Lorenz Eisinger, M.; Dörrbaum, A.R.; Michel, H.; Padan, E.; Langer, J.D.; Silberman, A.; Borchers, C.H.; Kaback, R.; Rand, K.D. Ligand-induced conformational dynamics of the Escherichia coli Na+/H+ antiporter NhaA revealed by hydrogen/deuterium exchange mass spectrometry. Proc. Natl. Acad. Sci. USA 2017, 114, 11691–11696. [Google Scholar] [CrossRef] [PubMed]
- Offenbacher, A.R.; Iavarone, A.T.; Klinman, J.P. Hydrogen-deuterium exchange reveals long-range dynamical allostery in soybean lipoxygenase. J. Biol. Chem. 2018, 293, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, P.B.G.; Patterson, A.; Kant, R.; Berry, L.; Golden, S.M.; Forsman, B.L.; Carter, J.; Jackson, R.N.; Bothner, B.; Wiedenheft, B. Conformational Dynamics of DNA Binding and Cas3 Recruitment by the CRISPR RNA-Guided Cascade Complex. ACS Chem. Biol. 2017, 13, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Leitner, A.; Faini, M.; Stengel, F.; Aebersold, R. Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines. Trends Biochem. Sci. 2016, 41. [Google Scholar] [CrossRef] [PubMed]
- Elmlund, D.; Le, S.N.; Elmlund, H. High-resolution cryo-EM: The nuts and bolts. Curr. Opin. Struct. Biol. 2017, 46, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Egelman, E.H. The Current Revolution in Cryo-EM. Biophys. J. 2016, 110, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Haselbach, D.; Komarov, I.; Agafonov, D.E.; Kastner, B.; Lü Hrmann, R.; Stark, H.; Hartmuth, K.; Graf, B.; Dybkov, O.; Urlaub, H.; et al. Structure and Conformational Dynamics of the Human Spliceosomal B act Complex. Cell 2018, 172, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Baker, M.L.; Wang, Z.; Baker, M.R.; Sinyagovskiy, P.A.; Chiu, W.; Ludtke, S.J.; Serysheva, I.I. Gating machinery of InsP 3 R channels revealed by electron cryomicroscopy. Nature 2015, 527, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Merk, A.; Bartesaghi, A.; Banerjee, S.; Earl, L.A.; Milne, J.L.S.; Falconieri, V.; Rao, P.; Davis, M.I.; Pragani, R.; Boxer, M.B.; et al. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell 2016, 165, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Meisburger, S.P.; Thomas, W.C.; Watkins, M.B.; Ando, N. X-ray Scattering Studies of Protein Structural Dynamics. Chem. Rev. 2017, 117, 7615–7672. [Google Scholar] [CrossRef] [PubMed]
- Tuukkanen, A.T.; Spilotros, A.; Svergun, D.I. Progress in small-angle scattering from biological solutions at high-brilliance synchrotrons. IUCrJ 2017, 4, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Rambo, R.P.; Tainer, J.A. Super-Resolution in Solution X-ray Scattering and Its Applications to Structural Systems Biology. Annu. Rev. Biophys. 2013, 42, 415–441. [Google Scholar] [CrossRef] [PubMed]
- Korasick, D.A.; Tanner, J.J. Determination of protein oligomeric structure from small-angle X-ray scattering. Protein Sci. 2018, 27, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Boon, P.L.S.; Saw, W.G.; Lim, X.X.; Raghuvamsi, P.V.; Huber, R.G.; Marzinek, J.K.; Holdbrook, D.A.; Anand, G.S.; Grüber, G.; Bond, P.J. Partial Intrinsic Disorder Governs the Dengue Capsid Protein Conformational Ensemble. ACS Chem. Biol. 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, T.N.; Herranz-Trillo, F.; Urbanek, A.; Estaña, A.; Cortés, J.; Sibille, N.; Bernadó, P. Small-angle scattering studies of intrinsically disordered proteins and their complexes. Curr. Opin. Struct. Biol. 2017, 42, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Josts, I.; Nitsche, J.; Maric, S.; Mertens, H.D.; Moulin, M.; Haertlein, M.; Prevost, S.; Svergun, D.I.; Busch, S.; Forsyth, V.T.; et al. Conformational States of ABC Transporter MsbA in a Lipid Environment Investigated by Small-Angle Scattering Using Stealth Carrier Nanodiscs. Structure 2018, 26, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)—Round XII. Proteins Struct. Funct. Bioinform. 2018, 86, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.D.; Das, S.; Sillitoe, I.; Orengo, C. An overview of comparative modelling and resources dedicated to large-scale modelling of genome sequences. Acta Crystallogr. Sect. D Struct. Biol. 2017, D73, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Schaarschmidt, J.; Monastyrskyy, B.; Kryshtafovych, A.; Bonvin, A.M.J.J. Assessment of contact predictions in CASP12: Co-evolution and deep learning coming of age. Proteins Struct. Funct. Bioinform. 2018, 86, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Bausewein, T.; Mills, D.J.; Langer, J.D.; Nitschke, B.; Nussberger, S.; Kühlbrandt, W. Cryo-EM Structure of the TOM Core Complex from Neurospora crassa. Cell 2017, 170, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, A.; Aufderheide, A.; Rudack, T.; Beck, F.; Pfeifer, G.; Plitzko, J.M.; Sakata, E.; Schulten, K.; Forster, F.; Baumeister, W. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc. Natl. Acad. Sci. USA 2016, 113, 7816–7821. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.; Ma, J.; Li, Z.; You, L.; Wang, J.; Wang, M.; Zhang, X.; Wang, Y. The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell 2017, 170, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Jasnovidova, O.; Klumpler, T.; Kubicek, K.; Kalynych, S.; Plevka, P.; Stefl, R. Structure and dynamics of the RNAPII CTDsome with Rtt103. Proc. Natl. Acad. Sci. USA 2017, 114, 11133–11138. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Kim, S.J.; An, S.; Cho, S.; Leitner, A.; Jung, T.; Aebersold, R.; Hebert, H.; Cho, U.S.; Song, J.J. Integrative Structural Investigation on the Architecture of Human Importin4_Histone H3/H4_Asf1a Complex and Its Histone H3 Tail Binding. J. Mol. Biol. 2018, 430, 822–841. [Google Scholar] [CrossRef] [PubMed]
- Kosinski, J.; Mosalaganti, S.; Von Appen, A.; Teimer, R.; Diguilio, A.L.; Wan, W.; Bui, K.H.; Hagen, W.J.H.; Briggs, J.A.G.; Glavy, J.S.; et al. Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science 2016, 352, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Nam Kim, D.; Sanbonmatsu, K.Y. Tools for the cryo-EM gold rush: Going from the cryo-EM map to the atomistic model. Biosci. Rep. 2017, 37, 20170072. [Google Scholar] [CrossRef] [PubMed]
- Tama, F.; Miyashita, O.; Brooks, C.L., III. Normal mode based flexible fitting of high-resolution structure into low-resolution experimental data from cryo-EM. J. Struct. Biol. 2004, 147, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Tama, F.; Miyashita, O.; Brooks, C.L., III. Flexible Multi-scale Fitting of Atomic Structures into Low-resolution Electron Density Maps with Elastic Network Normal Mode Analysis. J. Mol. Biol. 2004, 337, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Grubisic, I.; Shokhirev, M.N.; Orzechowski, M.; Miyashita, O.; Tama, F. Biased coarse-grained molecular dynamics simulation approach for flexible fitting of X-ray structure into cryo electron microscopy maps. J. Struct. Biol. 2009, 169, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Whitford, P.C.; Noel, J.K.; Gosavi, S.; Schug, A.; Sanbonmatsu, K.Y.; Onuchic, J.N. An all-atom structure-based potential for proteins: Bridging minimal models with all-atom empirical forcefields. Proteins Struct. Funct. Bioinform. 2009, 75, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Trabuco, L.G.; Villa, E.; Mitra, K.; Frank, J.; Schulten, K. Flexible Fitting of Atomic Structures into Electron Microscopy Maps Using Molecular Dynamics. Structure 2008, 16, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Schröder, G.F.; Brunger, A.T.; Levitt, M. Combining Efficient Conformational Sampling with a Deformable Elastic Network Model Facilitates Structure Refinement at Low Resolution. Structure 2007, 15, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, F.; Song, Y.; Li, X.; Brunner, M.J.; Xu, C.; Conticello, V.; Egelman, E.; Marlovits, T.C.; Cheng, Y.; Baker, D. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 2015, 12, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Topf, M.; Lasker, K.; Webb, B.; Wolfson, H.; Chiu, W.; Sali, A. Protein Structure Fitting and Refinement Guided by Cryo-EM Density. Structure 2008, 16, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S. Interpreting solution X-ray scattering data using molecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Gorba, C.; Miyashita, O.; Tama, F. Normal-Mode Flexible Fitting of High-Resolution Structure of Biological Molecules toward One-Dimensional Low-Resolution Data. Biophys. J. 2008, 94, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Tria, G.; Mertens, H.D.T.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Russel, D.; Lasker, K.; Webb, B.; Velázquez-Muriel, J.; Tjioe, E.; Schneidman-Duhovny, D.; Peterson, B.; Sali, A. Putting the Pieces Together: Integrative Modeling Platform Software for Structure Determination of Macromolecular Assemblies. PLoS Biol. 2012, 10, e1001244. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, A.; Herzog, F.; Leitner, A.; Rosenberger, G.; Aebersold, R. Cross-Link Guided Molecular Modeling with ROSETTA. PLoS ONE 2013, 8, e73411. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Bullock, J.M.A.; Sen, N.; Thalassinos, K.; Topf, M. Modeling Protein Complexes Using Restraints from Crosslinking Mass Spectrometry. Structure 2018, 26, 1015–1024.e2. [Google Scholar] [CrossRef] [PubMed]
- Fritz, B.G.; Roberts, S.A.; Ahmed, A.; Breci, L.; Li, W.; Weichsel, A.; Brailey, J.L.; Wysocki, V.H.; Tama, F.; Montfort, W.R. Molecular model of a soluble guanylyl cyclase fragment determined by small-angle X-ray scattering and chemical cross-linking. Biochemistry 2013, 52, 1568–1582. [Google Scholar] [CrossRef] [PubMed]
- Tokuhisa, A.; Jonic, S.; Tama, F.; Miyashita, O. Hybrid approach for structural modeling of biological systems from X-ray free electron laser diffraction patterns. J. Struct. Biol. 2016, 194, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Borowski, T.; De Visser, S.P. Quantum Mechanics/Molecular Mechanics Modeling of Enzymatic Processes: Caveats and Breakthroughs. Chem. A Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Alder, B.J.; Wainwright, T.E. Phase Transition for a Hard Sphere System. J. Chem. Phys. 1957, 27, 1208. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Vicatos, S.; Dryga, A.; Warshel, A. Coarse-Grained (Multiscale) Simulations in Studies of Biophysical and Chemical Systems. Annu. Rev. Phys. Chem. 2011, 62, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Go, N. Theoretical studies of protein folding. Ann. Rev. Biophys. Bioeng. 1983, 12, 183–210. [Google Scholar] [CrossRef] [PubMed]
- Tirion, M.M. Large Amplitude Elastic Motions in Proteins from a Single-Parameter, Atomic Analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Fuglebakk, E.; Tiwari, S.P.; Reuter, N. Comparing the intrinsic dynamics of multiple protein structures using elastic network models. BBA Gen. Subj. 2015, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Lezon, T.R.; Yang, L.-W.; Eyal, E. Global Dynamics of Proteins: Bridging Between Structure and Function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Sinha, S. Uncoupling of an ammonia channel as a mechanism of allosteric inhibition in anthranilate synthase of Serratia marcescens: Dynamic and graph theoretical analysis. Mol. BioSyst. 2017, 13, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Tama, F.; Sanejouand, Y.-H. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001, 14, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bagler, G.; Sinha, S. Assortative mixing in Protein Contact Networks and protein folding kinetics. Bioinformatics 2007, 23, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Vishveshwara, S. A Study of Communication Pathways in Methionyl- tRNA Synthetase by Molecular Dynamics Simulations and Structure Network Analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 15711–15716. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, L.; De Ruvo, M.; Paci, P.; Santoni, D.; Giuliani, A. Protein Contact Networks: An emerging paradigm in chemistry. Chem. Rev. 2013, 113, 1598–1613. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.C.; Melo, M.C.R.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, A.; Sugita, Y.; Okamoto, Y. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers 2001, 60, 96–123. [Google Scholar] [CrossRef]
- Valsson, O.; Tiwary, P.; Parrinello, M. Enhancing Important Fluctuations: Rare Events and Metadynamics from a Conceptual Viewpoint. Annu. Rev. Phys. Chem. 2016, 67, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Berg, B.; Neuhaus, T. Multicanonical ensemble: A new approach to simulate first-order phase transitions. Phys. Rev. Lett. 1992, 68, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, U.H.E.; Okamoto, Y.; Eisenmenger, F. Molecular dynamics, Langevin and hydrid Monte Carlo simulations in a multicanonical ensemble. Chem. Phys. Lett. 1996, 259, 321–330. [Google Scholar] [CrossRef]
- McGee, T.D.; Edwards, J.; Roitberg, A.E. pH-REMD Simulations Indicate That the Catalytic Aspartates of HIV-1 Protease Exist Primarily in a Monoprotonated State. J. Phys. Chem. B 2014, 118, 12577–12585. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.J.; Tirado-Rives, J.; Jorgensen, W.L. Enhanced Monte Carlo Sampling through Replica Exchange with Solute Tempering. J. Chem. Theory Comput. 2014, 10, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Palmer, J.C.; Debenedetti, P.G. Computational investigation of cold denaturation in the Trp-cage miniprotein. Proc. Natl. Acad. Sci. USA 2016, 113, 8991–8996. [Google Scholar] [CrossRef] [PubMed]
- Stelter, D.; Keyes, T. Enhanced Sampling of Phase Transitions in Coarse-Grained Lipid Bilayers. J. Phys. Chem. B 2017, 121, 5770–5780. [Google Scholar] [CrossRef] [PubMed]
- Stelzl, L.S.; Hummer, G. Kinetics from Replica Exchange Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 3927–3935. [Google Scholar] [CrossRef] [PubMed]
- Isralewitz, B.; Gao, M.; Schulten, K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001, 11, 224–230. [Google Scholar] [CrossRef]
- Wang, F.; Landau, D.P. Determining the density of states for classical statistical models: A random walk algorithm to produce a flat histogram. Phys. Rev. E 2001, 64, 056101. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Kitson, D.H.; Hagler, A.T. Theoretical Studies of the Structure and Molecular Dynamics of a Peptide Crystal. Biochemistry 1988, 27, 5246–5257. [Google Scholar] [CrossRef] [PubMed]
- Avbelj, F.; Moult, J.; Kitson, D.H.; James, M.N.G.; Hagler, A.T. Molecular dynamics study of the structure and dynamics of a protein molecule in a crystalline ionic environment, Streptomyces griseus protease A. Biochemistry 1990, 29, 8658–8676. [Google Scholar] [CrossRef] [PubMed]
- Janowski, P.A.; Liu, C.; Deckman, J.; Case, D.A. Molecular dynamics simulation of triclinic lysozyme in a crystal lattice. Protein Sci. 2016, 25, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Janowski, P.A.; Cerutti, D.S.; Holton, J.; Case, D.A. Peptide crystal simulations reveal hidden dynamics. J. Am. Chem. Soc. 2013, 135, 7938–7948. [Google Scholar] [CrossRef] [PubMed]
- Ahlstrom, L.S.; Vorontsov, I.I.; Shi, J.; Miyashita, O. Effect of the Crystal Environment on Side-Chain Conformational Dynamics in Cyanovirin-N Investigated through Crystal and Solution Molecular Dynamics Simulations. PLoS ONE 2017, 12, e0170337. [Google Scholar] [CrossRef] [PubMed]
- Vorontsov, I.I.; Miyashita, O. Solution and Crystal Molecular Dynamics Simulation Study of m4-Cyanovirin-N Mutants Complexed with Di-Mannose. Biophys. J. 2009, 97, 2532–2540. [Google Scholar] [CrossRef] [PubMed]
- Bond, P.J.; Faraldo-Gomez, J.D.; Deol, S.S.; Sansom, M.S.P. Membrane protein dynamics and detergent interactions within a crystal: A simulation study of OmpA. Proc. Natl. Acad. Sci. USA 2006, 103, 9518–9523. [Google Scholar] [CrossRef] [PubMed]
- Malek, K.; Coppens, M.-O. Molecular Simulations of Solute Transport in Xylose Isomerase Crystals. J. Phys. Chem. B 2008, 112, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Jiang, J. Molecular Dynamics Simulations for Water and Ions in Protein Crystals. Langmuir 2008, 24, 4215–4223. [Google Scholar] [CrossRef] [PubMed]
- Schmeing, T.M.; Ramakrishnan, V. What recent ribosome structures have revealed about the mechanism of translation. Nature 2009, 461, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A. A structural understanding of the dynamic ribosome machine. Nat. Rev. Mol. Cell Biol. 2008, 9, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Whitford, P.C.; Ahmed, A.; Yu, Y.; Hennelly, S.P.; Tama, F.; Spahn, C.M.T.; Onuchic, J.N.; Sanbonmatsu, K.Y. Excited states of ribosome translocation revealed through integrative molecular modeling. Proc. Natl. Acad. Sci. USA 2011, 108, 18943–18948. [Google Scholar] [CrossRef] [PubMed]
- Bock, L.V.; Kolář, M.H.; Grubmüller, H. Molecular simulations of the ribosome and associated translation factors. Curr. Opin. Struct. Biol. 2018, 49, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Palermo, G.; Spinello, A.; Rothlisberger, U.; Magistrato, A. All-atom simulations disentangle the functional dynamics underlying gene maturation in the intron lariat spliceosome. Proc. Natl. Acad. Sci. USA 2018, 115, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.; Mori, T.; Ando, T.; Harada, R.; Jung, J.; Sugita, Y.; Feig, M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. Elife 2016, 5, e19274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, F.; Liu, J.; Gao, B.; Liu, Y.; Zhai, Y.; Ma, J.; Zhang, K.; Baker, T.S.; Schulten, K.; et al. Atomic Model of Rabbit Hemorrhagic Disease Virus by Cryo-Electron Microscopy and Crystallography. PLoS Pathog. 2013, 9, e1003132. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.-J.; Xin, F.; Radivojac, P. Allosteric post-translational modification codes. Trends Biochem. Sci. 2012, 37, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Craveur, P.; Rebehmed, J.; De Brevern, A.G. PTM-SD: A database of structurally resolved and annotated posttranslational modifications in proteins. Database 2014, 2014, 41. [Google Scholar] [CrossRef] [PubMed]
- Polyansky, A.A.; Zagrovic, B.; Shemyakin, M.M.; Ovchinnikov, Y.A. Protein Electrostatic Properties Predefining the Level of Surface Hydrophobicity Change upon Phosphorylation. J. Phys. Chem. Lett. 2012, 3, 52. [Google Scholar] [CrossRef] [PubMed]
- Moffett, A.S.; Bender, K.W.; Huber, S.C.; Shukla, D. Allosteric Control of a Plant Receptor Kinase through S-Glutathionylation. Biophys. J. 2017, 113, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. The Many Roles of Computation in Drug Discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Feixas, F.; Lindert, S.; Sinko, W.; Mccammon, J.A. Exploring the role of receptor flexibility in structure-based drug discovery. Biophys. Chem. 2014, 186, 31–45. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Antolin, A.A.; Carotti, A.; Nuti, R.; Hakkaya, A.; Camaioni, E.; Mestres, J.; Pellicciari, R.; Macchiarulo, A. Exploring the effect of PARP-1 flexibility in docking studies. J. Mol. Graph. Model. 2013, 45, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Seidel, T.; khan Ibis, G.; Bendix, F.; Wolber, G. Strategies for 3D pharmacophore-based virtual screening. Drug Discov. Today Technol. 2010, 7, e221–e228. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, C.; Priyakumar, U.D.; Sastry, G.N. Dynamics Based Pharmacophore Models for Screening Potential Inhibitors of Mycobacterial Cyclopropane Synthase. J. Chem. Inf. Model. 2015, 55, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Wieder, M.; Garon, A.; Perricone, U.; Boresch, S.; Seidel, T.; Almerico, A.M.; Langer, T. Common Hits Approach: Combining Pharmacophore Modeling and Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Sinko, W.; de Oliveira, C.; Williams, S.; Van Wynsberghe, A.; Durrant, J.D.; Cao, R.; Oldfield, E.; Mccammon, J.A. Applying molecular dynamics simulations to identify rarely sampled ligand-bound conformational states of undecaprenyl pyrophosphate synthase, an antibacterial target. Chem. Biol. Drug Des. 2011, 77, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Perricone, U.; Wieder, M.; Seidel, T.; Langer, T.; Padova, A.; Almerico, A.M.; Tutone, M. A Molecular Dynamics–Shared Pharmacophore Approach to Boost Early-Enrichment Virtual Screening: A Case Study on Peroxisome Proliferator-Activated Receptor α. ChemMedChem 2017, 12, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Bradley, P. Probing the role of interfacial waters in protein-DNA recognition using a hybrid implicit/explicit solvation model. Proteins Struct. Funct. Bioinform. 2013, 81, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Breiten, B.; Lockett, M.R.; Sherman, W.; Fujita, S.; Al-Sayah, M.; Lange, H.; Bowers, C.M.; Heroux, A.; Krilov, G.; Whitesides, G.M. Water Networks Contribute to Enthalpy/Entropy Compensation in Protein−Ligand Binding. J. Am. Chem. Soc. 2013, 135, 15579–15584. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.; Bondar, A.-N. Mechanism by which water and protein electrostatic interactions control proton transfer at the active site of channelrhodopsin. PLoS ONE 2018, 13, e0201298. [Google Scholar] [CrossRef] [PubMed]
- Conti Nibali, V.; Havenith, M.; Nibali, V.C.; Havenith, M. New insights into the role of water in biological function: Studying solvated biomolecules using terahertz absorption spectroscopy in conjunction with molecular dynamics simulations. J. Am. Chem. Soc. 2014, 136, 12800–12807. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Fichou, Y.; Gallat, F.-X.; Wood, K.; Gabel, F.; Moulin, M.; Härtlein, M.; Heyden, M.; Colletier, J.-P.; Orecchini, A.; et al. Translational diffusion of hydration water correlates with functional motions in folded and intrinsically disordered proteins. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D. Empirical Force Fields for Biological Macromolecules: Overview and Issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O. Systematic Validation of Protein Force Fields against Experimental Data. PLoS ONE 2012, 7, e32131. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zheng, W.; Mittal, J. Balanced Protein−Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput 2014, 10, 5113–5124. [Google Scholar] [CrossRef] [PubMed]
- Henriques, J.; Cragnell, C.; Skepö, M. Molecular Dynamics Simulations of Intrinsically Disordered Proteins: Force Field Evaluation and Comparison with Experiment. J. Chem. Theory Comput. 2015, 11, 3420–3431. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A.; Huang, J.; Roux, B.; Mackerell, A.D. An Empirical Polarizable Force Field Based on the Classical Drude Oscillator Model: Development History and Recent Applications. Chem. Rev. 2016, 116, 4983–5013. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the SC14: IEEE International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páall, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Bowman, G.R.; Pande, V.S.; Noé, F. (Eds.) An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2014; Volume 797, ISBN 978-94-007-7605-0. [Google Scholar]
- Sali, A.; Berman, H.M.; Schwede, T.; Trewhella, J.; Kleywegt, G.; Burley, S.K.; Markley, J.; Nakamura, H.; Adams, P.; Bonvin, A.M.J.J.; et al. Meeting Review Outcome of the First wwPDB Hybrid/Integrative Methods Task Force Workshop. Struct. Des. 2015, 23, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Kurisu, G.; Markley, J.L.; Nakamura, H.; Velankar, S.; Berman, H.M.; Sali, A.; Schwede, T.; Trewhella, J. PDB-Dev: A Prototype System for Depositing Integrative/Hybrid Structural Models. Structure 2017, 25, 1317–1318. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).