Theoretical Studies on the Engagement of Interleukin 18 in the Immuno-Inflammatory Processes Underlying Atherosclerosis



Abstract
1. Introduction
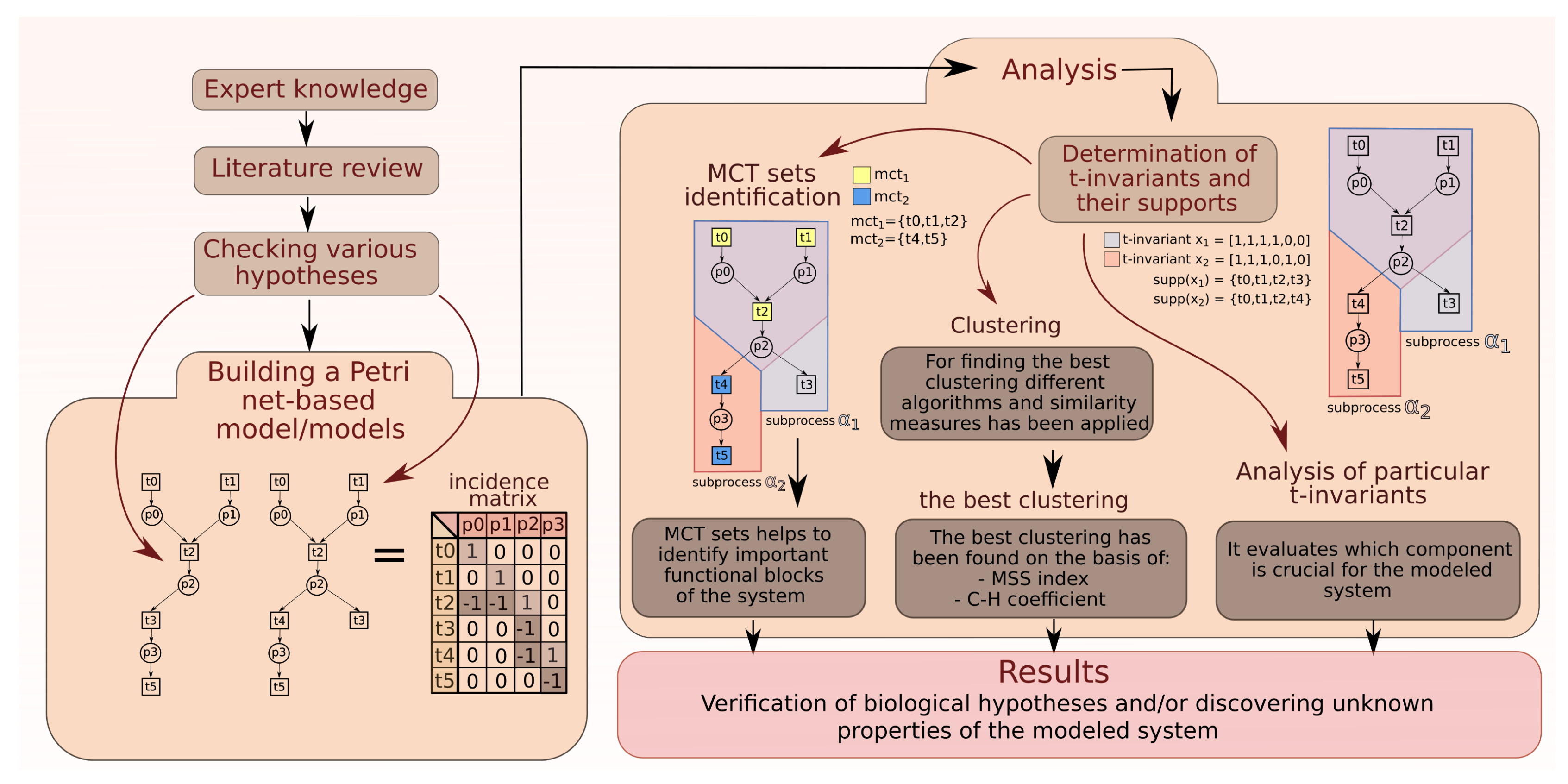
2. Analysis
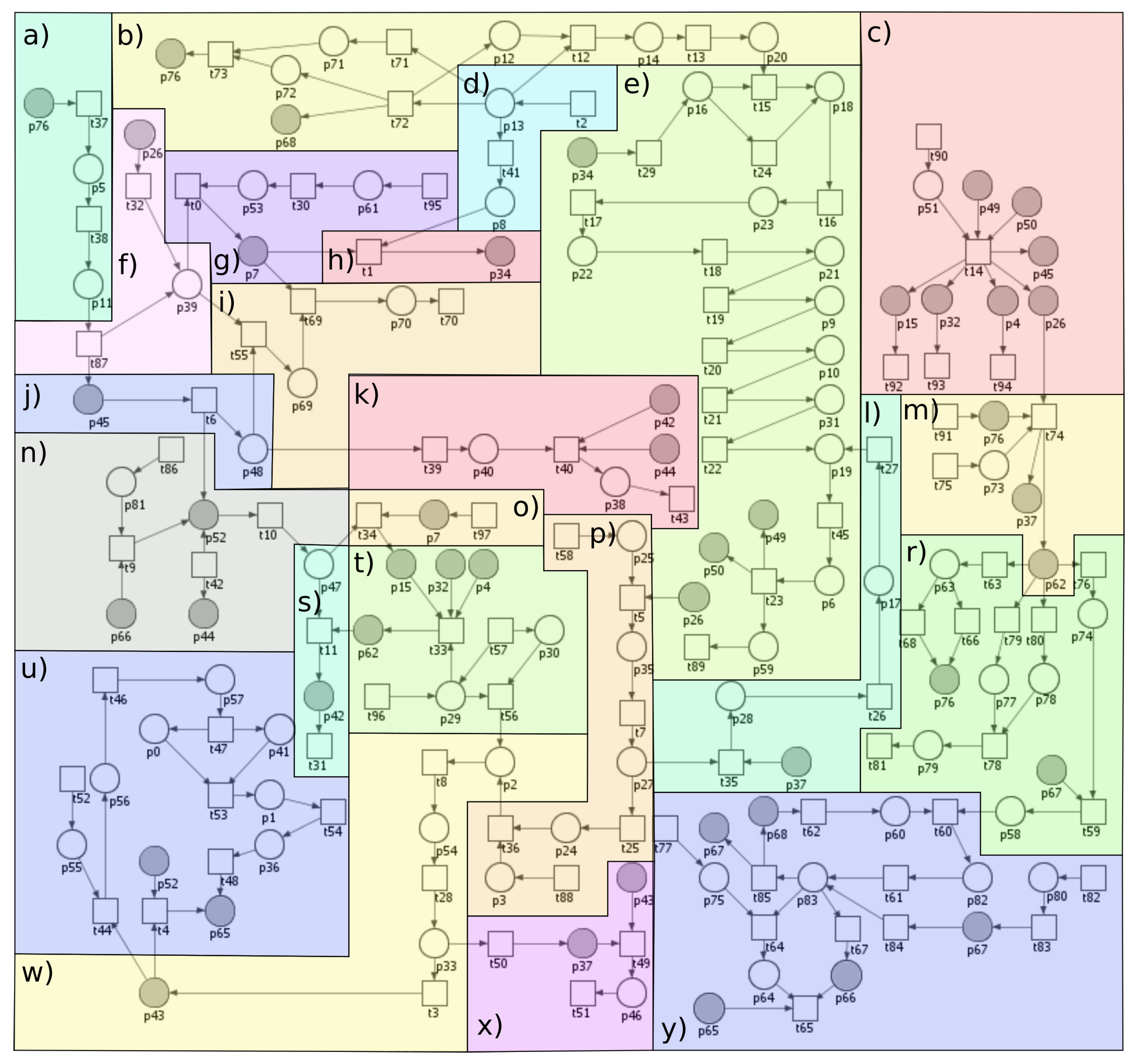
2.1. Analysis of MCT Sets
2.2. Analysis of t-Clusters
2.3. More Detailed Analysis of Particular t-Invariants
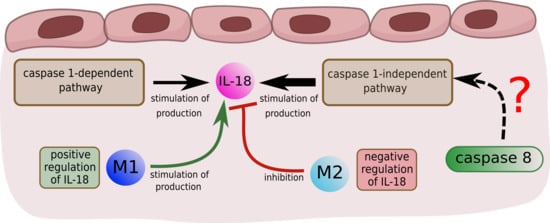
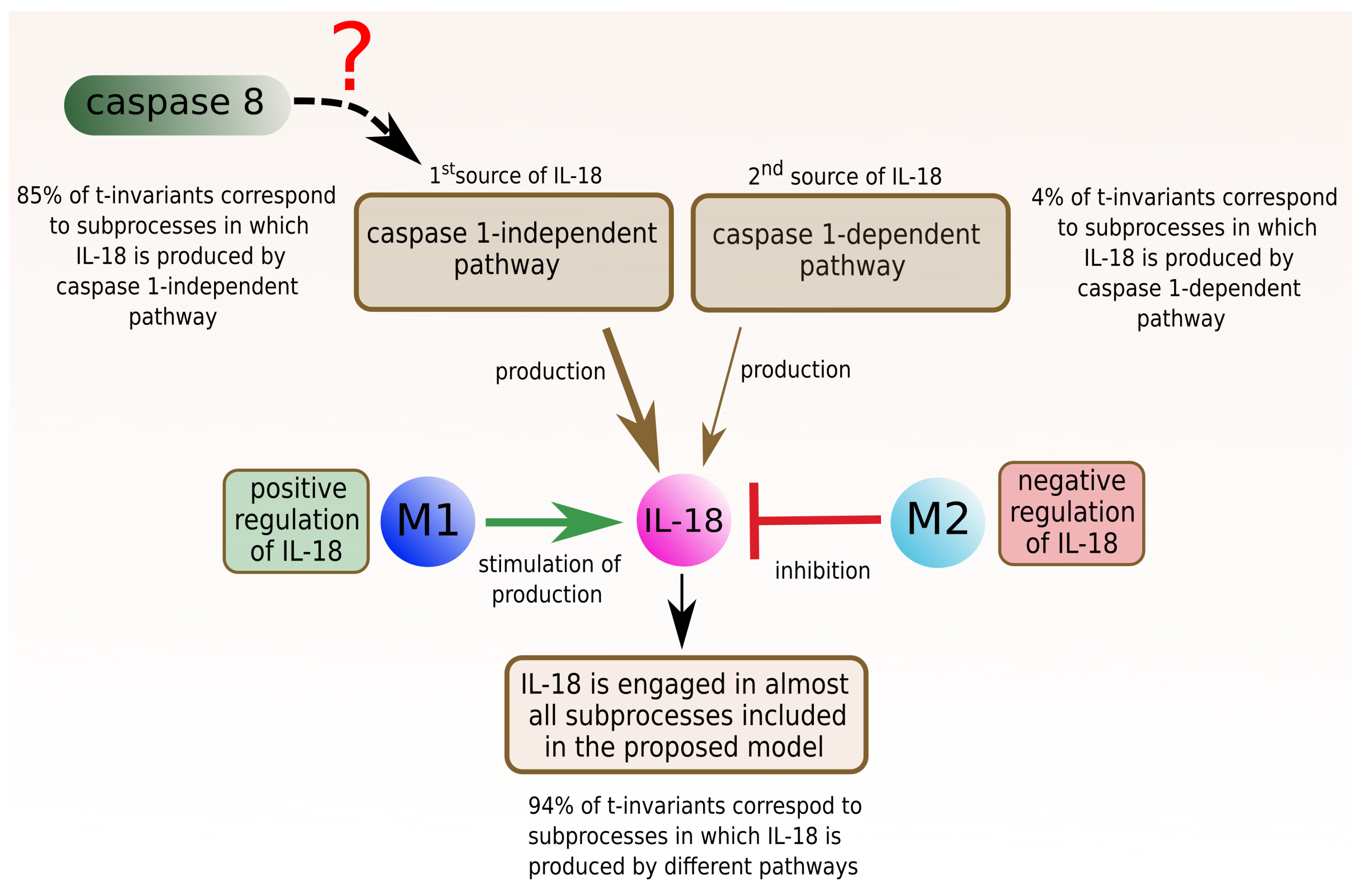
2.3.1. Interleukin 18
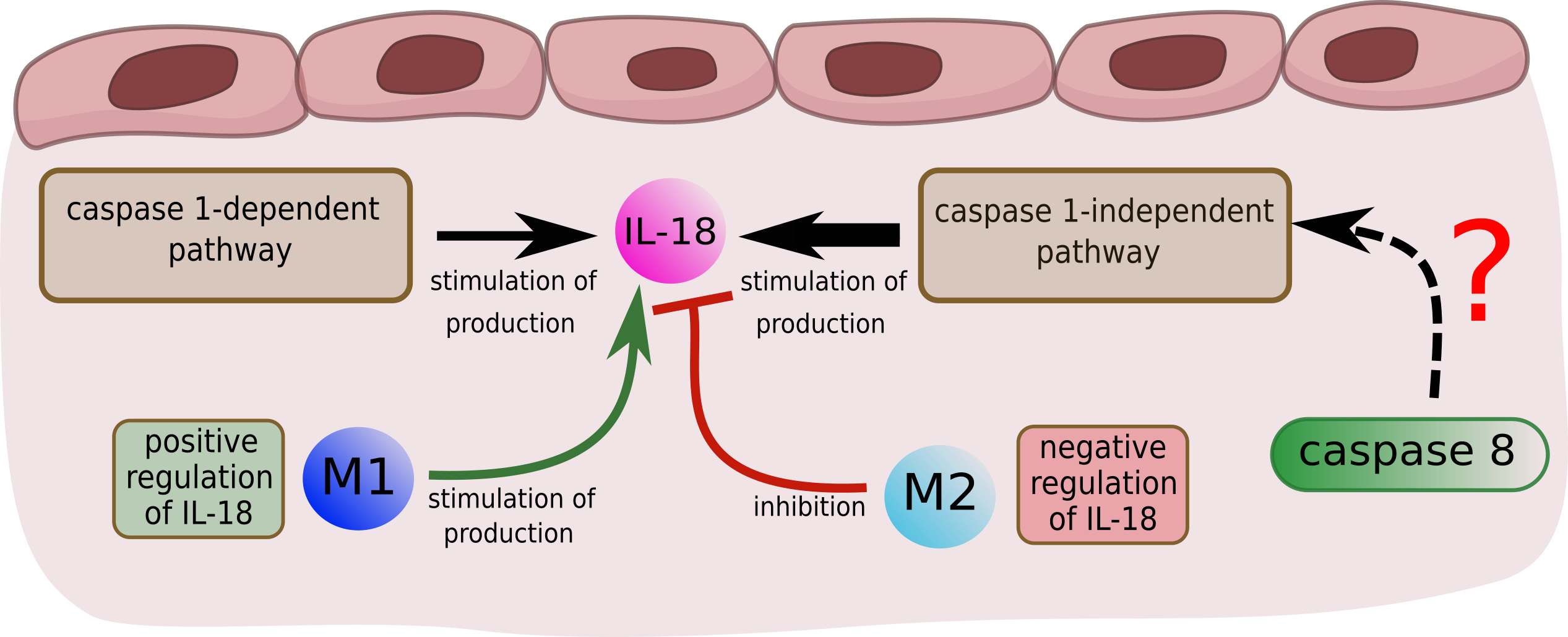
- 66 t-invariants (85%) correspond to subprocesses in which IL-18 produced by caspase 1-independent pathway is involved;
- 3 t-invariants (4%) correspond to subprocesses in which IL-18 produced by caspase 1-dependent pathway is involved;
- 4 t-invariants (5%) correspond to subprocesses in which IL-18 is produced by the two mentioned pathways (caspase 1-independent pathway and caspase 1-dependent pathway);
- 5 t-invariants (6%) correspond to subprocesses in which IL-18 is not involved.
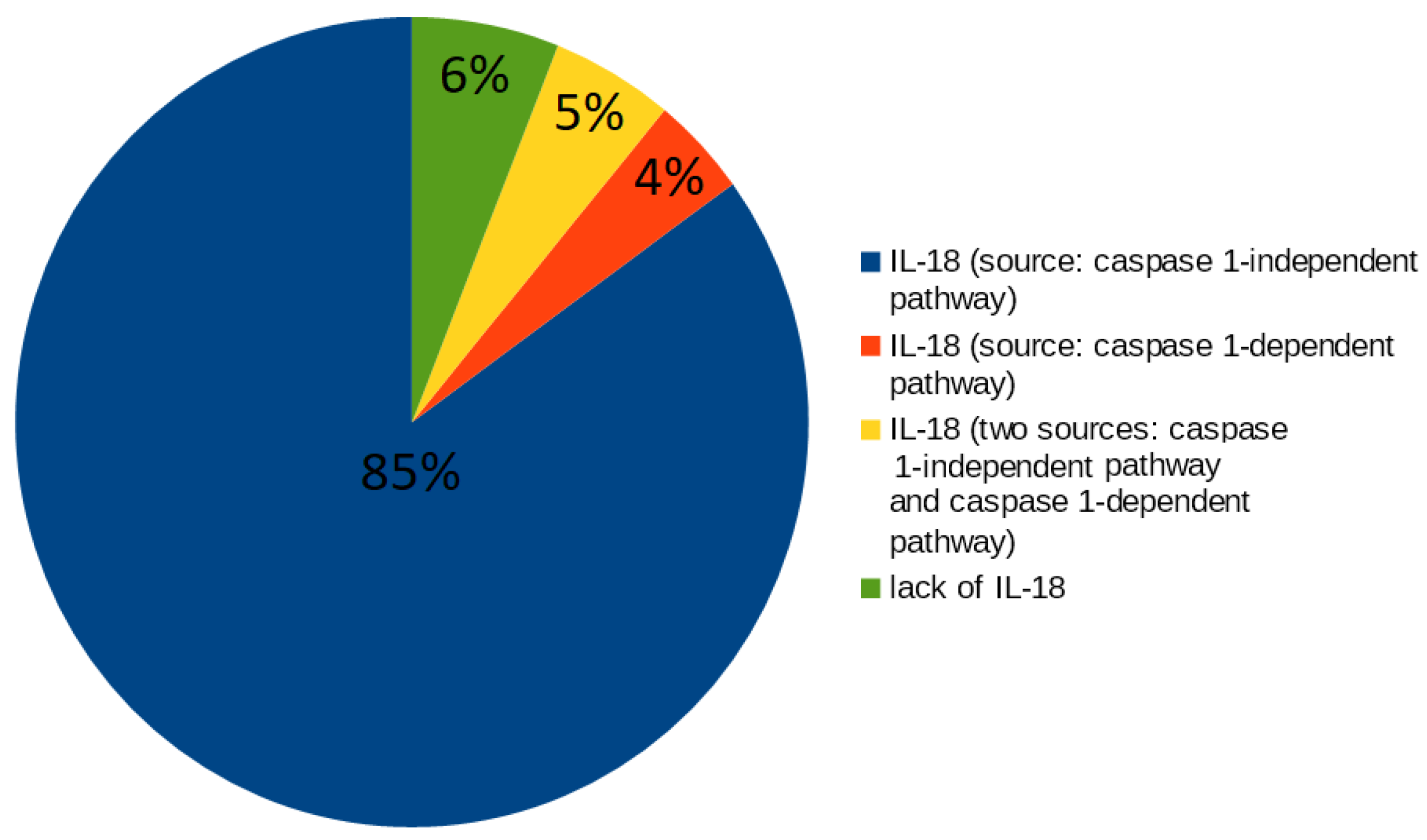
- 49 t-invariants (62.82%) correspond to subprocesses in which caspase 8 occurs along with IL-18 produced by caspase 1-independent pathway;
- 3 t-invariants (3.85%) correspond to subprocesses in which caspase 8 occurs along with IL-18 produced by caspase 1-dependent pathway;
- 2 t-invariants (2.56%) correspond to subprocesses in which caspase 8 occurs along with IL-18 produced by the two mentioned pathways (caspase 1-independent pathway and caspase 1-dependent pathway);
- 20 t-invariants (25.64%) correspond to subprocesses in which caspase 8 is not involved;
- 4 t-invariants (5.13%) correspond to subprocesses in which only caspase 8 is involved.
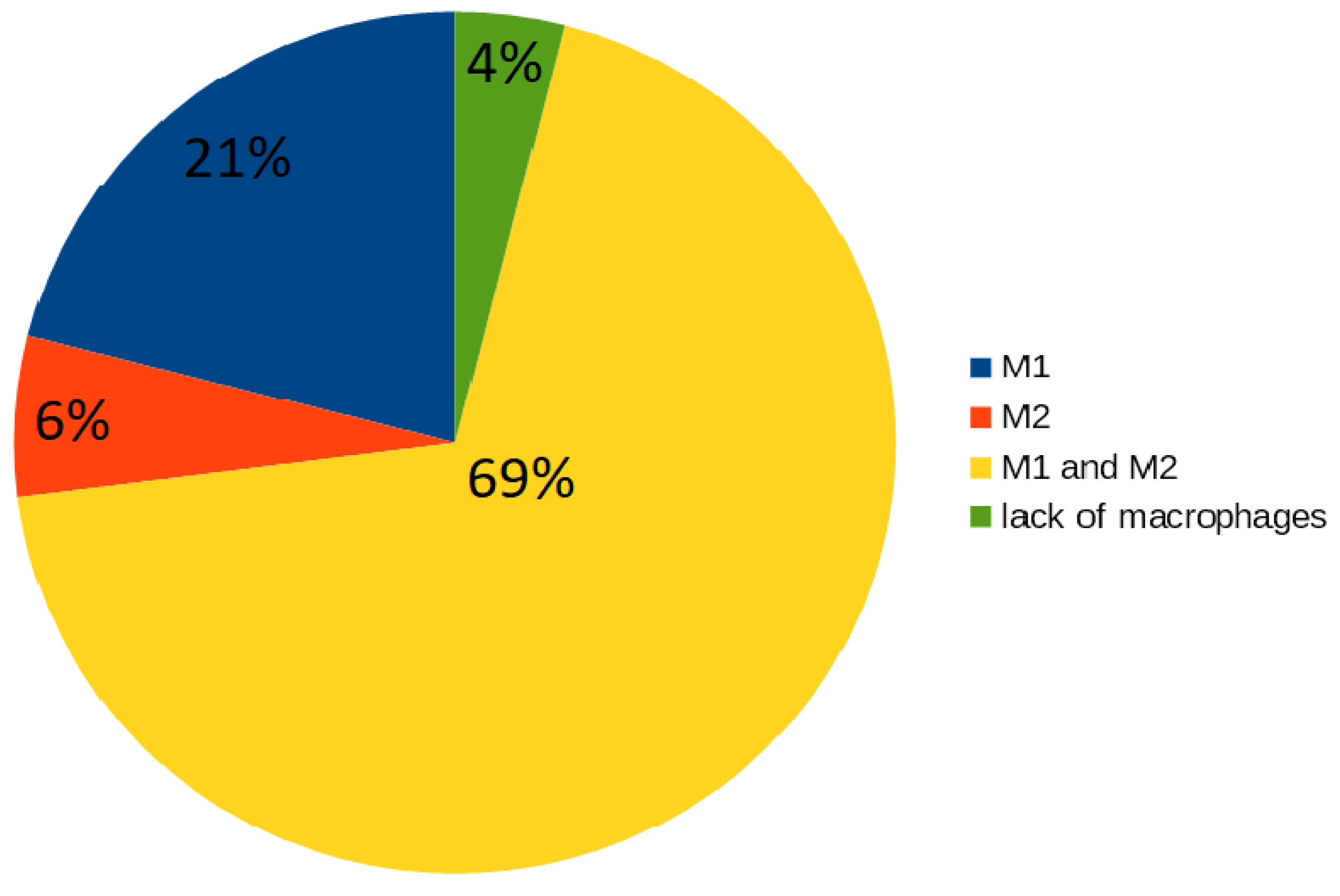
2.3.2. Macrophages M1 and M2
- 16 t-invariants (21%) correspond to subprocesses in which type M1 is involved;
- 5 t-invariants (6%) correspond to subprocesses in which type M2 is involved;
- 54 t-invariants (69%) correspond to subprocesses in which two types of macrophages are involved;
- 3 t-invariants (4%) correspond to subprocesses in which macrophages are not involved.
- macrophages M1:
- -
- inhibition of caspase 8,
- -
- positive regulation of IL-18 (NO synthesis inhibition),
- -
- pathway of IB phosphorylation by complex of receptors MyD88;
- macrophages M2:
- -
- activation of caspase 3, 6, 7 and 8,
- -
- high level of interleukin 10,
- -
- negative regulation of interleukin 18 (inhibition of IL-18),
- -
- apoptosis enhancement and anti-inflammation response.
2.3.3. Knockout Analysis
- 4 t-invariants (80%) correspond to subprocesses in which IL-18 is negatively regulated;
- 1 t-invariant (20%) corresponds to a subprocess in which IL-18 is positively regulated.
- 22 t-invariants (79%) correspond to subprocesses in which IL-18 is positively regulated;
- 6 t-invariants (21%) correspond to subprocesses in which IL-18 is negatively regulated.
2.3.4. Comparison of t-Clusters Analysis and the More Detailed Analysis of Particular t-Invariants
- 73 (94%) out of 78 t-invariants correspond to subprocesses in which IL-18 is produced;
- 75 (96%) out of 78 t-invariants correspond to subprocesses in which macrophages M1 and M2 are engaged.
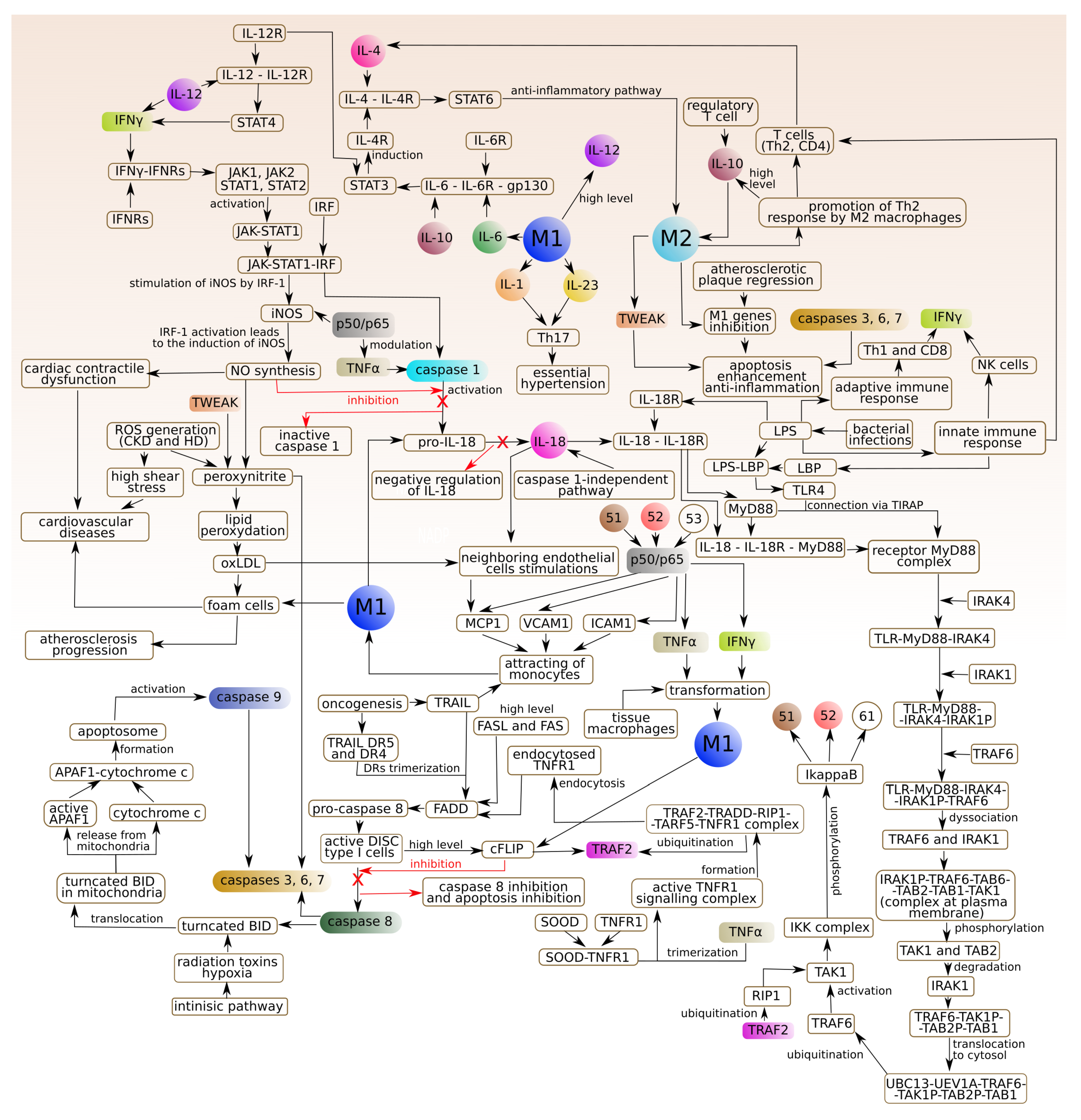
3. The Model of Phenomena That Underlie the Formation of Atherosclerotic Lesions in the Arterial Subendothelial Region
- M1 type (stimulated by IFN and TNF): the main features are the production of pro- inflammatory cytokines: IL-1, 6, 12 and 23. Activation of M1 type is associated with activation of caspase 1 and conversion of pro-IL-18 to active IL-18.
- M2 type: the main feature is the production of an anti-inflammatory cytokine: IL-10. IL-10 enhances the phenotype of M2 macrophages induced by IL-4. IL-4 acts via IL-4 receptor complex which activates STAT6, whereas IL-10 promotes M2 phenotype via activating STAT3 through IL-10 receptor [48].
4. Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A. List of Places

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Biological Meaning | No. | Biological Meaning |
|---|---|---|---|
| active apoptotic peptidase activating factor 1 (APAF1) [77] | foamy cells [49] | ||
| apoptotic protease activating factor 1 (APAF1) and cytochrome c complex [77] | high level of active caspase 8 active in type I cells [78] | ||
| FAS-associated protein with death domain (FADD) recruited [78] | high shear stress [49] | ||
| large amounts of cell-surface FAS receptor (FAS) and FAS ligand (FASL) [78] | inducible nitric oxide synthase (iNOS) [49] | ||
| intercellular adhesion molecule 1 (ICAM1) | low level of active caspase 8 in type I cells | ||
| interferon gamma (IFN) and interferon receptors (IFNRs) complex [79] | modified, oxidized LDL [49] | ||
| kinase complex (IKK complex made of two kinase IKK and IKK and regulatory subunit NF-B essential modulator (NEMO) also known as inhibitor of nuclear factor -B kinase subunit gamma (IKK) [80] | nitric oxide (NO) [49] | ||
| interleukin 18 (IL-18) [25,35,37] | p50/p65 proteasome inhibitors induce inhibitory B (IB) phosphorylated complex within macrophages [80] | ||
| interleukin 18 receptor (IL-18R) [38] | p50/p65 dimer nuclear factor kappa-light-chain-enhancer of activated B cells (NF-B) early phase in activated endothelial cell (EC) in atherosclerosis [80] | ||
IRAK1P-TRAF6-TAB2-TAB1-TAK1 complex at plasma membrane, where:
| p50-p65 dimer nuclear factor kappa-light-chain-enhancer of activated B cells (NF-B) early phase in activated smooth muscle cell (SMC) [81] | ||
| interleukin 1 receptor-associated kinase 1 (IRAK1) degraded | peroxynitrite [49] | ||
| Janus kinases and signal transducer and activator of transcription 1 (JAK/STAT1) pathway activated | pro-interleukin 18 (pro-IL18) [25,37] | ||
| lipopolysaccharide binding protein (LBP) [23,82] | pro-caspase 8 recruited | ||
| lipopolysaccharide (LPS) [23,25,26,39,82] | radiation and/or toxins and/or hypoxia | ||
| lipopolysaccharide (LPS) and lipopolysaccharide binding protein (LBP) complex [23,82] | truncated BH interacting-domain death agonist (BID) | ||
| monocyte chemotactic protein 1 (MCP1) | truncated BID in mitochondria | ||
| myeloid differentiation primary response gene 88 (MyD88) [23,25] | signal transducer and activator of transcription 3 (STAT3) activated proteins [25] | ||
| receptor-interacting protein 1 (RIP1) ubiquitinated | p50-p65 proteasome inhibitors induce inhibitory B (IB) phosphorylated complex in activated smooth muscle cell (SMC)[80] | ||
| receptors myeloid differentiation primary response gene 88 (MyD88) complex [23,25] | interleukin 4 (IL-4) [25] | ||
| TGF-beta activated kinase 1 (TAK1) activated | classically activated macrophages M1 [25] | ||
| toll-like receptor 4 (TLR4) activated [23,82] | a lot of classically activated macrophages M1 [25] | ||
TLR-MyD88-IRAK4-IRAK1P-TRAF6 complex, where:
| high level of interleukin 12 (IL-12) [25] | ||
| TLR-MyD88-IRAK4-IRAK1P complex | no inflammation | ||
| TLR-Myd88-IRAK4 complex | active caspases 3, 6 and 7 [77] | ||
| tumor necrosis factor receptor 1 (TNFR1) endocytosed | TNF-related weak inducer of apoptosis (TWEAK) changed concentration [83] | ||
| tumor necrosis factor receptor 1 (TNFR1) stable with silencer of death domains (SODD) [81] | high level of interleukin 10 (IL-10) [25] | ||
| tumor necrosis factor alpha (TNF) [35] | subpopulation of activated T cells, T helper 2 (Th2) cells and cluster of differentiation (CD4) [29] | ||
TRAF2-TRADD-RIP1-TRAF5-TNFR1 complex, where:
| inactive caspase 1 [84] | ||
| TNF receptor associated factors 2 (TRAF2) autoubiquitinated | lack of interleukin 18 (IL-18) [84] | ||
| TNF-related apoptosis-inducing ligand (TRAIL) | cluster of differentiation (CD) 4 T helper cells (Th1) and CD8 effector T cells (CD8 cytotoxic T cells) [29] | ||
| TNF-related apoptosis-inducing ligand (TRIAL) death receptors DR5 and DR4 | natural killer (NK) cells [27,28] | ||
UBC13-UEV1A-TRAF6-TAK1P-TAB2P-TAB1 complex, where:
| tissue macrophages | ||
| vascular cell adhesion molecule 1 (VCAM1) | interleukin 6 (IL-6) [25] | ||
| active death-inducing signaling complex (DISC) type I cells [78] | development of anti-inflammatory processes | ||
| active IL-18 and IL-18 receptors (IL-18R and IL-18R) complex [38,85] | interferon gamma (IFN) | ||
| active TNF receptor associated factors 1 (TNFR1) signaling complex | interleukin 1 (IL-1) [25,86,87] | ||
| apoptosome [77] | interleukin 23 (IL-23) [25,86,87] | ||
| cellular FLICE (FADD-like IL-1-converting enzyme)-inhibitory protein (cFLIP) [78] | cytokines secreted by Th17 cells (IL-17, IL-21, and IL-22) [87,88] | ||
| cardiovascular disease [49] | human regulatory T cells | ||
| interleukin 1 converting enzyme (ICE)/caspase 1 protease [37] | NADPH oxidase [89] | ||
| contractility failure [49] | interleukin 4 (IL-4) and interleukin 4 receptor (IL-4R) complex [25] | ||
| cytochrome c [77] | alternatively activated macrophages M2 with scavenger receptor A (SRA1) and cluster of differentiation 163 (CD163) [25] |
Appendix B. List of Transitions
| No. | Biological Meaning | No. | Biological Meaning |
|---|---|---|---|
| pro-interleukin 18 (pro-IL-18) activation by caspase 1 enzymatic cleavage [36,90] | inhibition of active caspase 8 in type I cells | ||
| IL-18 binding with IL-18R receptor [91] | increase quantity of cFLIP [92,93] | ||
| bacterial infection | apoptosis inhibition [91,93] | ||
| caspase 8 dimerization autocleavage and activation type I cells (cells in which caspase-8 activation is sufficient to induce apoptosis) [67,92] | intrinsic pathway of apoptosis [92,94] | ||
| cleavage and activation of caspases 3, 6 and 7 | cytochrome c binding to apoptotic protease activating factor 1 (APAF1) [94] | ||
| tumor necrosis factor receptor 1 (TNFR1) trimerization [81] | triggering the formation of apoptosome [94] | ||
| nitric oxide synthesis | caspase 1 inhibition [84] | ||
forming TRAF2-TRADD-RIP1-TRAF5-TNFR1 complex [81], where:
| trimerization of TNF-related apoptosis-inducing ligand (TRIAL) death receptors [93] | ||
| pro-caspase 8 recruitment influenced by FADD [93,95] | oncogenesis/inflammation [93] | ||
| NADPH oxidase activity enhancement by TWEAK [83,89] | silencer of death domains (SODD) binds to tumor necrosis factor receptor 1 (TNFR1) leading to its stabilization [81] | ||
| lipids peroxydation | activation of signal transducer and activator of transcription 3 (STAT3) proteins phosphorylation by JAK via interleukin 6, interleukin 6 receptor and glycoprotein 130 complex (IL-6-IL-6R-gp130) on target cells (classical signaling pathway) [96] | ||
| transformation into foamy cells | interleukin 4 receptor (type I IL-4R)) activation via binding with interleukin 4 (IL-4)and JAK1/JAK3 phosphorylation [97] | ||
| binding lipopolysaccharide (LPS) and lipopolysaccharide binding protein (LBP) [82,98] | signal transducer and activator of transcription 6 (STAT6) anti-inflammatory pathway [97] | ||
| binding lipopolysaccharide (LPS) presentation to toll-like receptor 4 (TLR4) and cluster of differentiation 14 (CD14) and myeloid differentiation primary response 2 (Myd2) [82,98] | IL-4 synthesis induced by M2 macrophages | ||
| p50-p65 translocation to the nucleus in macrophages M1, smooth muscle cell (SMC) and endothelial cell (EC) [80] | release of interleukin 12 (IL-12) by classically activated macrophages M1 | ||
| toll-like receptor 4 and myeloid differentiation primary response 88 (TLR4-MyD88) connection via toll-interleukin 1 receptor (TIR) domain containing adaptor protein (TIRAP) [98] | inhibition of M1 macrophages specific gene expression [47] | ||
| interleukin-1 receptor-associated kinase 4 (IRAK4) recruitment [91] | apoptosis enhancement and anti-inflammatory response [47] | ||
| connection with interleukin-1 receptor-associated kinase 1 (IRAK1) and its phosphorylation [91] | interferon gamma (INF) synthesis via IL-12 | ||
| TNF receptor associated factors 6 (TRAF6) recruitment and binding to interleukin 1 receptor- associated kinase 1 protein (IRAK1) [91] | TNF-like weak inducer of apoptosis (TWEAK) sequestration | ||
| dissociation of interleukin 1 receptor-associated kinase 1 protein (IRAK1P) and TNF receptor associated factors 6 (TRAF6) [91] | Interferon gamma (INF) via IL-12 and IL-12 receptors (IL-12Rs) complexes through signal transducer and activator of transcription 3 and 4 (STAT3 and STAT4) | ||
| phosphorylation of transforming growth factor (TGF) activated kinase 1 and 2 (TAK1 and TAK2) [91] | IL-18 negative regulation [84] | ||
TRAF6-TAK1P-TAB2P-TAB1 complex translocation to the cytosol [81,91]. Complex, where:
| no action of IL-18 [84] | ||
| TNF receptor-associated factor 6 (TRAF6) ubiquitination [91] | adaptive immune response [98] | ||
| phosphorylation of IKK- - activation of NF-B - the canonical pathway [80] | innate immune response [98] | ||
| connection of active IL-18, IL-18R and and also myeloid differentiation primary response 2 (IL-18-IL-18R-IL-18R-MyD88) | interferon gamma (IFN) synthesis | ||
| tumor necrosis factor receptor 1 (TNFR1) clathrin-dependent internalization [99] | transformation of tissue macrophages into M1 [25] | ||
| receptor-interacting protein 1 (RIP1) ubiquitination | normal state [25] | ||
| receptor-interacting protein 1 (RIP1) recruits transforming growth factor activated kinase 1 (TAK1) via transforming growth factor activated kinase 1 binding protein 2 (TAB2) | release of interleukin 6 (IL-6) by classically activated macrophages M1 [96] | ||
| connection through death domains (DDs) within FAS-associated protein with death domain (FADD) [93,95] | processes leading to atherosclerotic plaque regression [47] | ||
| recruitment of myeloid differentiation primary response gene 88 (MyD88) | T helper 17 (Th17) differentiation [86,88] | ||
| pro-interleukin 18 (pro-IL-18) synthesis citeDNK+13 | release of interleukin 1 (IL-1) by classically activated macrophages M1 [87,88] | ||
| atherosclerotic plaque progression | release of interleukin 23 (IL-23) by classically activated macrophages M1 [87,88] | ||
| modulation by TNF | autoimmune inflammation, essential hypertension [87,88] | ||
| attraction of monocytes | immune environment changes that stimulate human regulatory T cells (Treg) | ||
| neighboring endothelial cells stimulation | production of IL-10 by Treg | ||
| TNF receptor-associated factor 2 (TRAF2) ubiquitination | IL-10 enhances the M2 phenotype induced by IL-4 [2] | ||
| recruitment of FAS-associated protein with death domain (FADD) [99] | enhancement of M2 effector function | ||
| interferon gamma (IFN) and interferon receptor (IFNGR1 and IFNGR2) binding followed by interaction between IFNRs subunits and JAK 1 (IFNGR1) and JAK2 (IFNGR2) what activates latent cytoplasmic STAT1 [100] | NADPH oxidase activity source [89] | ||
| oligomerization of the IFN receptor and activation via trans-phosphorylation of the receptor-associated Janus kinases 1 and 2 (JAK1 and JAK2) followed by STAT 1 activation [79,100] | binding STAT1 with STAT binding element sequences of interferon regulatory factor 1 (IRF1) in IFN pathway [100] | ||
| cardiac contractile dysfunction [49] | the FAS-FASL interaction [99] | ||
| cardio-vascular disease symptoms [49] | the proteasome pathway in activated SMC [80] | ||
| modulation of T helper cells type 1 (Th1) differentiation during infections | smooth muscles cells (SMC) activation mechanisms [80] | ||
| reactive oxygen species (ROS) generation in chronic kidney disease (CKD) especially during hemodialysis (HD) [49] | IFN secretion | ||
| cardiovascular events [49] | regulation of migration and infiltration of monocytes/macrophages | ||
| BH interacting-domain death agonist (BID) cleavage influenced by high levels of active caspase 8 in type I cells [94] | mediation of the adhesion of lymphocytes, monocytes, eosinophils, and basophils to vascular endothelium [101] | ||
| kinase (IKK) complex activation [80] | mediation of the adhesion between integrins on leukocytes and endothelial or epithelial cells [101] | ||
| translocation of truncated BH interacting-domain death agonist (BID) to mitochondria [92,94] | an inflammatory environment [36] | ||
| release of cytochrome c from mitochondria to cytosol via mitochondrial perturbation [94] | TRAIL production by different cells | ||
| activation of caspase 9 [94] | IL-18 synthesis mediated by caspase 1-independent pathway |
Appendix C. List of t-Clusters
| t-Cluster | t-Invariants | MCT Set | Single Transitions |
|---|---|---|---|
| , , | , | ||
| , , , , | , | ||
| , , , , | , , | ||
| , | , , , , | ||
| , , , , , , , , | , , , , , , | ||
| , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , , , , | , , , , , , | ||
| , , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , , , | , , , , , , , , | ||
| , , , , , , , , , , | , , , , , , , , | ||
| , , , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , | , , , , , , | ||
| , , , , , , , , , , , , , | , , , , , , | ||
| , , , , , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , , | , , , , , | ||
| , , , , | , , , , , , , | ||
| , , , , , , , | , , , , , , , , | ||
| , , , , , , , | , , , , , , , | ||
| , , , , , , | , , , , , , , , | ||
| , , , , , , , , | , , , , , , , , , | ||
| , , , , , , | , , , , , , , , | ||
| , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , , , , | , , , , , , , , | ||
| , , , , , , , , , , , , | , , , , , , , , | ||
| , , , , , , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , , , , , | , , , , , , , | ||
| , , , , , , , | , , , , , , , | ||
| , , , , , , , , , | , , , , , , , , | ||
| , , , , , , , | , , , , , , , , | ||
| , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , | , , , , , , , , | ||
| , , , , , , , , , , | , , , , , , , , | ||
| , , , , , , | , , , , , , , , | ||
| , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , | , , , , , , , | ||
| , , , , , , , , , | , , , , , , , , | ||
| , , , , , , , , , , , , | , , , , , , | ||
| , , , , , , , , , , , | , , , , , , | ||
| , , , , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , , , | , , , , , , | ||
| , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , | , , , , , , , , | ||
| , , , | , , , , , , | ||
| , , , | , , , , , , | ||
| , , , , | , , , , , , | ||
| , , , , | , , , , , , , | ||
| , , , , | , , , , , , , , | ||
| , , , , | , , , , , , , , | ||
| , , , , , , , | , , , , , , , | ||
| , , , , , , , , | , , , , , , | ||
| , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , , | , , , , , | ||
| , , , , , , , , | , , , , , , , , | ||
| , , , , , , , , | , , , , , , , , | ||
| , , , , , , , | , , , , , , , , , | ||
| , , , , , , , | , , , , , , , , , | ||
| , , , , , , , | , , , , , , , | ||
| , , , , , , , | , , , , , , , , | ||
| , , , , , , , , | , , , , , , , , | ||
| , , , , , , , , | , , , , , , , , , | ||
| , , , , , | , , , , , | ||
| , | , , , , , | ||
| , , , , , , , , , , , | , , , , , , | ||
| , , , , , , , , | , , , , , | ||
| , , , , , , , , , , | , , , , , , , | ||
| , , , , , , , , , , , , | , , , , , , , , , | ||
| , , , , , , , , , , , , , | , , , , , , , , |
References
- Gupta, S.; Pablo, A.M.; Jiang, X.C.; Wang, N.; Tall, A.R.; Schindler, C. IFN-γ potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Investig. 1997, 99, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Hizukuri, Y.; Yamashiro, K.; Murakawa, M.; Hayashi, Y. IL-10 enhances the phenotype of M2 macrophages induced by IL-4 and confers the ability to increase eosinophil migration. Int. Immunol. 2015, 27, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Dinarello, C.A. IL-18 in autoimmunity: Review. Eur. Cytokine Netw. 2006, 17, 224–252. [Google Scholar] [PubMed]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An effector CD4 T cell lineage with regulatory T cell ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev. 2001, 12, 53–72. [Google Scholar] [CrossRef]
- Formanowicz, D.; Wanic-Kossowska, M.; Pawliczak, E.; Radom, M.; Formanowicz, P. Usefulness of serum interleukin-18 in predicting cardiovascular mortality in patients with chronic kidney disease—Systems and clinical approach. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, L.S.; Mogensen, C.K.; Rosendahl, A.; Cucak, H.; Nielsen, L.B.; Rasmussen, S.E.; Pedersen, T.X. Bone marrow-derived and peritoneal macrophages have different inflammatory response to oxLDL and M1/M2 marker expression—Implications for atherosclerosis research. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Rżosińska, K.; Formanowicz, D.; Formanowicz, P. The study of the influence of micro-environmental signals on macrophage differentiation using a quantitative Petri net based model. Arch. Control Sci. 2017, 2, 331–349. [Google Scholar] [CrossRef]
- Chen, L.; Wu, J. Systems biology for complex diseases. J. Mol. Cell Biol. 2012, 4, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Murata, T. Petri Nets: Properties, Analysis and Applications. Proc. IEEE 1989, 77, 541–580. [Google Scholar] [CrossRef]
- Petri, C.A. Communication with Automata; Schriften des Instituts fur Instrumentelle Mathematik: Bonn, Germany, 1962. (In German) [Google Scholar]
- Reising, W. Understanding Petri Nets. Modeling Techniques, Analysis Methods, Case Studies; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Formanowicz, D.; Kozak, A.; Głowacki, T.; Radom, M.; Formanowicz, P. Hemojuvelin–Hepcidin axis modeled and analyzed using Petri nets. J. Biomed. Inform. 2013, 46, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, L.; Rousseeuw, P.J. Finding Groups in Data: An Introduction to Cluster Analysis; John Wiley and Sons: New York, NY, USA, 1990; ISBN 0-471-87876-6. [Google Scholar] [CrossRef]
- Calinski, T.; Harabasz, J. A dendrite method for cluster analysis. Commun. Stat. 1974, 3, 1–27. [Google Scholar] [CrossRef]
- Radom, M.; Rybarczyk, A.; Szawulak, B.; Andrzejewski, H.; Chabelski, P.; Kozak, A.; Formanowicz, P. Holmes: A graphical tool for development, simulation and analysis of Petri net based models of complex biological systems. Bioinformatics 2017, 33, 3822–3823. [Google Scholar] [CrossRef] [PubMed]
- Formanowicz, D.; Radom, M.; Rybarczyk, A.; Formanowicz, P. The role of Fenton reaction in ROS-induced toxicity underlying atherosclerosis—Modeled and analyzed using a Petri net-based approach. BioSystems 2018, 165, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Formanowicz, D.; Radom, M.; Zawierucha, P.; Formanowicz, P. Petri net-based approach to modeling andanalysis of selected aspects of the molecular regulation of angiogenesis. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Heiner, M.; Herajy, M.; Liu, F.; Rohr, C.; Schwarick, M. Snoopy—A Unifying Petri Net Tool. In Application and Theory of Petri Nets; Haddad, S., Pomello, L., Eds.; PETRI NETS 2012, Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2012; Volume 7347, ISBN 978-3-642-31130-7. [Google Scholar] [CrossRef]
- Gambin, A.; Charzyńska, A.; Ellert-Miklaszewska, A.; Rybiński, M. Computational models of the JAK1/2-STAT1 signaling. JAK-STAT 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK–STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Kagan, J.C. A cross-disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Mol. Cell 2014, 54, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Valkov, E.; Stamp, A.; DiMaio, F.; Baker, D.; Verstak, B.; Roversi, P.; Kellie, S.; Sweet, J.; Mansell, A.; Gay, N.J.; et al. Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc. Natl. Acad. Sci. USA 2011, 108, 14879–14884. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed]
- Pastelin-Palacios, R.; Gil-Cruz, C.; Pérez-Shibayama, C.I.; Moreno-Eutimio, M.A.; Cervantes-Barragán, L.; Arriaga-Pizano, L.; Ludewig, B.; Cunningham, A.F.; García-Zepeda, E.A.; Becker, I.; et al. Subversion of innate and adaptive immune activation induced by structurally modified lipopolysaccharide from Salmonella typhimurium. Immunology 2011, 133, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Ramana, C.V.; Gil, M.P.; Schreiber, R.D.; Stark, G.R. Stark: Stat1-dependent and—Independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002, 23, 96–101. [Google Scholar] [CrossRef]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.; Reiser, H. IL-4: Role in disease and regulation of production. Clin. Exp. Immunol. 1998, 113, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Noursadeghi, M.; Tsang, J.; Haustein, T.; Miller, R.F.; Chain, B.M.; Katz, D.R. Quantitative imaging assay for NF-κB nuclear translocation in primary human macrophages. J. Immunol. Methods 2008, 329, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Strittmatter, G.E.; Garstkiewicz, M.; Sand, J.; Beer, H.D. Caspase-I: The inflammasome and beyond. Innate Immun. 2014, 20, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.L.; Shajahan, A.N.; Clarke, R. The Role of Interferon Regulatory Factor-1 (IRF1) in Overcoming Antiestrogen Resistance in the Treatment of Breast Cancer. Int. J. Breast Cancer 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Sudhakar, C.; Swarup, G. Tumor necrosis factor-alpha-induced caspase-1 gene expression. Role of p73. FEBS J. 2007, 274, 4396–4407. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Sekiyama, A.; Ueda, H.; Kashiwamura, S.; Sekiyama, R.; Takeda, M.; Rokutan, K.; Okamura, H. A stress-induced, superoxide-mediated caspase-1 activation pathway causes plasma IL-18 upregulation. Immunity 2005, 22, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Sakorafas, P.; Miller, R.; McCarthy, D.; Scesney, S.; Dixon, R.; Ghayur, T. IL-18 receptor beta-induced changes in the presentation of IL-18 binding sites affect ligand binding and signal transduction. J. Immunol. 2003, 170, 5571–5577. [Google Scholar] [CrossRef] [PubMed]
- Abu, E.M.; Lunenfeld, E.; Huleihel, M. LPS increases the expression levels of IL-18, ICE and IL-18 R in mouse testes. Am. J. Reprod. Immunol. 2008, 60, 361–371. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, D.; Maiuri, M.C.; Iovine, B.; Ialenti, A.; Bevilacqua, M.A.; Carnuccio, R. The role of NF-κB, IRF-1, and STAT-1alpha transcription factors in the iNOS gene induction by gliadin and IFN-gamma in RAW 264.7 macrophages. J. Mol. Med. 2006, 84, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Gunnett, C.A.; Lund, D.D.; McDowell, A.K.; Faraci, F.M.; Heistad, D.D. Mechanisms of inducible nitric oxide synthase–mediated vascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.M.; MacCarthy, P.A. Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol. Ther. 2000, 86, 49–86. [Google Scholar] [CrossRef]
- O’Donnell, M.A.; Legarda-Addison, D.; Skountzos, P.; Yeh, W.C.; Ting, A.T. Ubiquitination of RIP1 regulates an NF-κB-independent cell-death switch in TNF signaling. Curr. Biol. 2007, 17, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; He, M.; Wang, Y.; Liao, A.H. Modulators of the Balance between M1 and M2 Macrophages during Pregnancy. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.; Hung, M.E.; Cangelose, B.K.; Leonard, J.N. Regulation of the IL-10-driven macrophage phenotype under incoherent stimuli. Innate Immun. 2016, 22, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Naseem, K.M. The role of nitric oxide in cardiovascular diseases. Mol. Asp. Med. 2005, 26, 33–65. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Ley, S.C. RNF 11, a new piece in the A20 puzzle. EMBO J. 2009, 28, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Pobezinskaya, Y.L.; Kim, Y.S.; Choksi, S.; Morgan, M.J.; Li, T.; Liu, C.; Liu, Z. The function of TRADD in signaling via tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat. Immunol. 2008, 9, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yan, W.; Zheng, H.; Du, Q.; Zhang, L.; Ban, Y.; Li, N.; Wei, F. Regulation of IL-10 and IL-12 production and function in macrophages and dendritic cells. F1000Research 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, T.; Bot, I.; Kuiper, J. The IL-12 cytokine family in cardiovascular diseases. Cytokine 2017. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical Regulation of Early Th17 Cell Differentiation by Interleukin-1 Signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Madhur, M.S.; Lob, H.E.; McCann, L.A.; Iwakura, Y.; Blinder, Y.; Guzik, T.J.; Harrison, D.G. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 2010, 55. [Google Scholar] [CrossRef] [PubMed]
- Nordlohne, J.; von Vietinghoff, S. Interleukin 17A in atherosclerosis—Regulation and pathophysiologic effector function. Cytokine 2017. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, K.; Formanowicz, D.; Formanowicz, P. The effect of cigarette smoking on endothelial damage and atherosclerosis development—Modeled and analyzed using Petri nets. Arch. Control Sci. 2017, 27, 209–226. [Google Scholar] [CrossRef]
- Singh, R.B.; Mengi, S.A.; Xu, Y.J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar] [PubMed]
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel wall. Cardiovasc. Res. 2015, 107, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Zhong, J.; Sun, Q. The heterogenic properties of monocytes/macrophages and neutrophils in inflammatory response in diabetes. Life Sci. 2014, 116, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Baliga, B.; Kumar, S. Apaf-1/cytochrome c apoptosome: An essential initiatorof caspase activation or just a sideshow? Cell Death Differ. 2003, 10, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular Mechanisms Controlling Caspase Activation and Function. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Loreto, C.; La Rocca, G.; Anzalone, R.; Caltabiano, R.; Vespasiani, G.; Castorina, G.; Ralph, D.J.; Cellek, S.; Musumeci, G.; Giunta, S.; et al. The Role of Intrinsic Pathway in Apoptosis Activation and Progression in Peyronie’s Disease. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, M.A.; Wang, D.; Khan, S.M.; Vander Heiden, M.G.; Kelekar, A. Caspase-9 is activated in a cytochrome c-independent manner early during TNFα-induced apoptosis in murine cells. Cell Death Differ. 2003, 10, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schröter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kwon, M.J.; Kim, I.H.; Kim, Y.M.; Lee, M.K.; Nam, T.J. Pyropia yezoensis glycoprotein promotes the M1 to M2 macrophage phenotypic switch via the STAT3 and STAT6 transcription factors. Int. J. Mol. Med. 2016, 38, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Zhuo, X.; Ma, A. STAT6 Upregulation Promotes M2 Macrophage Polarization to Suppress Atherosclerosis. Med. Sci. Monit. Basic Res. 2017, 23, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Gutowska, K.; Formanowicz, D.; Formanowicz, P. Selected aspects of tobacco-induced prothrombotic state, inflammation and oxidative stress-modeled and analyzed using Petri nets. 2017; submitted. [Google Scholar]
- Sackmann, A.; Heiner, M.; Koch, I. Application of Petri net based analysis techniques to signal transduction pathways. BMC Bioinform. 2006, 7, 482. [Google Scholar] [CrossRef] [PubMed]
- Koch, I.; Reisig, W.; Schreiber, F. (Eds.) Modeling in Systems Biology; The Petri Net Approach; Springer: London, UK, 2011; ISBN 978-1-84996-474-6. [Google Scholar]
- Grafahrend-Belau, E.; Schreiber, F.; Heiner, M.; Sackmann, A.; Junker, B.H.; Grunwald, S.; Speer, A.; Winder, K.; Koch, I. Modularization of biochemical networks based on classification of Petri net t-invariants. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Heiner, M. Understanding Network Behavior by Structured Representations of Transition Invariants. In Algorithmic Bioprocesses; Condon, A., Harel, D., Kok, J., Salomaa, A., Winfree, E., Eds.; Natural Computing Series; Springer: Berlin/Heidelberg, Germany, 2009; ISBN 978-3-540-88869-7. [Google Scholar]
- Jiang, X.; Wang, X. Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J. Biol. Chem. 2000, 275, 31199–31203. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.R.; Lotocki, G.; Marcillo, A.E.; Dietrich, W.D.; Keane, R.W. FasL, Fas, and death-inducing signaling complex (DISC) proteins are recruited to membrane rafts after spinal cord injury. J. Neurotrauma 2007, 24, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.M.; Hultman, P.; Toomey, C.B.; Cauvi, D.M.; Hoffman, H.M.; Hamel, J.C.; Kono, D.H. Definition of IFN-γ-related pathways critical for chemically-induced systemic autoimmunity. J. Autoimmun. 2012, 39, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Israël, A. The IKK Complex, a Central Regulator of NF-κB Activation. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-κB and IκB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; de Vos, A.F.; Florquin, S.; Golenbock, D.T.; van der Poll, T. Lipopolysaccharide Binding Protein Is an Essential Component of the Innate Immune Response to Escherichia coli Peritonitis in Mice. Infect. Immun. 2003, 71, 6747–6753. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Billiar, T.R.; Talanian, R.V.; Kim, Y.M. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem. Biophys. Res. Commun. 1997, 240, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Azam, T.; Novick, D.; Bufler, P.; Yoon, D.Y.; Rubinstein, M.; Dinarello, C.A.; Kim, S.H. Identification of a critical Ig-like domain in IL-18 receptor alpha and characterization of a functional IL-18 receptor complex. J. Immunol. 2003, 171, 6574–6580. [Google Scholar] [CrossRef] [PubMed]
- Revu, S.; Wu, J.; Henkel, M.; Rittenhouse, N.; Menk, A.; Delgoffe, G.M.; Poholek, A.C.; McGeachy, M.J. IL-23 and IL-1β Drive Human Th17 Cell Differentiation and Metabolic Reprogramming in Absence of CD28 Costimulation. Cell Rep. 2018, 22, 2642–2653. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 Induce Innate IL-17 Production from γδ T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hsu, H.C.; Mountz, J.D. The Dynamic Duo–Inflammatory M1 macrophages and Th17 cells in Rheumatic Diseases. J. Orthop. Rheumatol. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Van Heerebeek, L.; Meischl, C.; Stooker, W.; Meijer, C.J.; Niessen, H.W.; Roos, D. NADPH oxidase(s): New source(s) of reactive oxygen species in the vascular system? J. Clin. Pathol. 2002, 55, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase Functions in Cell Death and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, Y. The IL-6/JAK/STAT3 pathway: Potential therapeutic strategies in treating colorectal cancer (Review). Int. J. Oncol. 2014, 44, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Ul-Haq, Z.; Naz, S.; Mesaik, M.A. Interleukin-4 receptor signaling and its binding mechanism: A therapeutic insight from inhibitors tool box. Cytokine Growth Fact. Rev. 2016, 32, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Brachert, W.; Heigl, U.; Ehrenschwender, M. Membrane Trafficking of Death Receptors: Implications on signaling. Int. J. Mol. Sci. 2013, 14, 14475–14503. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.R.; Gomes, E.A.; Russo, M.; Lepique, A.P. Interferon Regulatory Factor (IRF)-1 Is a Master Regulator of the Cross Talk between Macrophages and L929 Fibrosarcoma Cells for Nitric Oxide Dependent Tumoricidal Activity. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Melotti, P.; Nicolis, E.; Tamanini, A.; Rolfini, R.; Pavirani, A.; Cabrini, G. Activation of NF-κB mediates ICAM-1 induction in respiratory cells exposed to an adenovirus-derived vector. Gene Ther. 2001, 8, 1436–1442. [Google Scholar] [CrossRef] [PubMed]







| MCT Set | Subprocesses from | Contained Transitions | Biological Interpretation |
|---|---|---|---|
| d, e, h | , , , , , , , , , , | IL-18R-mediated Myd88 signaling pathway. | |
| u | , , , , , , | Intrinsic pathway of apoptosis. This pathway arises from signals that originate within the cell and is associated with BID cleavage via radiation, toxins or hypoxia. Truncated BID translocates to mitochondria and is involved in the release of active APAF1 and cytochrome c from mitochondria to the cytosol. Active APAF1 and cytochrome c create a complex, which leads to triggering the formation of apoptosome and activation of caspase 9. Active caspase (textcolorredinitiator caspase) initiates apoptosis by cleaving and thereby activating executioner caspases. | |
| b, e | , , , , , | Recognition of lipopolysaccharide pattern by TLR4 complexes. Activated TL4 is engaged on TLR and MyD88 connection via TIRAP. | |
| c, e | , , , , | Activation of NF-B - the canonical pathway via IB phosphorylation by IKK (MyD88-dependent signaling pathway). Translocation of p50-p65 to the nucleus in M1 macrophages, SMC and EC. | |
| y, r | , , , , | IL-4–induced M2 polarization. Firstly, STAT3 activated by IL-6-IL6-R-gp130complex, influenced by high level of IL-10, induces binding IL-4 with type I IL-4R, which results in STAT6 pathway activation. | |
| k, n | , , , | High shear stress (mediated by ROS generation) and cardiac contractile dysfunction (affected by nitric oxide) lead to CVD. | |
| r | , , , | A lot of classically activated macrophages M1 release IL-1 and IL-23, which influence Th17 formation. Th17 cells are engaged in essential hypertension. | |
| g | , , | Classically activated macrophages M1, under influence of an inflammatory microenvironment, induce IL-18 synthesis via cleavage of pro-IL-18 by caspase-1. | |
| p | , , | SODD binds to TNFR1 and prevents self-aggregation and spontaneous downstream signaling at ligand absent (TNFR1 stabilization). SODD dissociates from TNFR1 upon receptor ligation. Binding of trimeric TNF to TNFR1 induces receptor trimerization and recruitment of TNFR1-associated death domain protein (TRADD), which serves as a platform to recruit next additional mediators. Active TNFR1 signaling complex is engaged on the formation of TRAF2-TRADD-RIP1-TRAF5-TNFR1 complex. | |
| p | , , | Internalization of TNFR1 triggers pro-apoptotic signals (via the FAS-associated death domain adapter protein (FADD)). | |
| l | , , | TRAF2-TRADD-RIP1-TRAF5-TNFR1 complex and cFLIP lead to TRAF2 ubiquitination and next to RIP1 ubiquitination, which recruits TAK1 (via TAB2). | |
| a, f | , , | JAK-STAT pathway stimulated by IFN. Activation pathway of JAK1, JAK2 and STAT1, leads to binding with SBE sequences of IRF1. IRF-1 binds to the IFN regulatory factor element (IRF-E) present in the inducible NO synthase (iNOS) promoter, and together with NF-B and STAT1, activate its transcription. This process leads to NO synthesis. | |
| i | , , | Nitric oxide leads to caspase 1 inhibition by S-nitrosylation of cysteine residues at the active sites. Negative regulation of IL-18 synthesis. | |
| y | , , | Processes leading to atherosclerosis plaque regression have an influence on increasing anti-inflammatory pathways, which results in inhibition of M1 macrophage specific gene expression and alternatively in promotion of M2 macrophage specific gene expression. | |
| y | , , | TWEAK–CD163-expressing M2 macrophages interaction. | |
| w | , | TRAIL or FasL bind their cognate receptors which induces receptor trimerization and formation of the death inducing signaling complex (DISC), type I cells comprising FADD and caspase-8. | |
| n | , | Peroxynitrite formation catalyzed by NADPH oxidase. | |
| t | , | Attracting of monocytes (influenced by TRAIL). | |
| x | , | Increased quantity of cFLIP can inhibit DISC formation by competing with caspase-8/10 for binding to FADD. | |
| t | , | TRAIL and its cognate receptors: death receptors (DR5 and DR4) trimerization. | |
| m | , | Transformation of tissue macrophages to M1 (a lot of classically activated macrophages M1). | |
| c | , | The heterodimer p50-p60 translocation to the nucleus in M1 macrophages, SMC and EC where it binds to specific B sites and activates a variety of NF-B target genes, including VCAM1 and ICAM1. |
| t-Cluster | Biological Meaning |
|---|---|
| TRAIL-induced apoptosis signaling pathways. Increased quantity of cFLIP leads to inhibition of caspase 8. | |
| The mitochondria-involved intrinsic apoptotic pathway. The intrinsic pathway arises from signals that originate within the cell, as a consequence of cellular stress or DNA damage. High level of IL-10 has an influence on development of anti-inflammatory processes. Damage to mitochondria and subsequent apoptosome-mediated caspase 9 activation, which directly activate the effector caspase, caspase 3. | |
| The mitochondria-involved intrinsic apoptotic pathway, similar to , accompanied by cleavage and activation of caspases 3, 6 and 7. | |
| TWEAK leads to the reaction catalyzed by NADPH oxidase, which results in peroxynitrite production. Macrophages M2 cause TWEAK sequestration in case of high level of IL-10. Peroxynitrite is engaged in lipids peroxidation and results in the production of modified oxidized LDL. This modified oxidized LDL together with IL-18 lead to neighboring endothelial cell stimulation and secretion of MCP1 (IL-18 is produced in caspase 1-independent pathway). | |
This cluster contains almost all processes included in the model, however it is missing:
| |
| Attracting of monocytes caused by MCP1, VCAM1, ICAM1 (secreted via p50/p65 translocation to the nucleus in macrophages M1 and SMC and EC) and TRAIL (in oncogenesis) lead to a lot of classically activated macrophages M1. In oncogenesis TRAIL and death receptors trimerization leads to pro-caspase 8 recruitment by FADD. Connection through DDs within FADD and TNFR result in active DISC type I cells and increased quantity of cFLIP. cFLIP together with TRAF2-TRADD-RIP1-TRAF5-TNFR1 complex lead to TRAF2 ubiquitination and in consequence to RIP1 ubiquitination. RIP1 recruits TAK1 via TAB2, which is part of IKK complex engaged in phosphorylation of IB (which stimulate p50/p65 translocation to the nucleus in macrophages M1, SMC and EC). p50/p65 translocation to the nucleus in macrophages M1 and SMC and EC leads to secretion of iNOS, which is associated with NO synthesis. NO synthesis leads also to peroxynitrite production, which together with high level of caspase 8 result in activation of caspases 3, 6 and 7. The high level of caspase 8 is caused by TRAIL and TRAIL death receptors trimerization (in oncogenesis). Active caspases 3, 6, 7 and also TWEAK lead to apoptosis enhancement anti-inflammation. TWEAK sequestration is caused by macrophages M2 in case of high level of IL-10. Moreover, NO is engaged in cardiac contractile dysfunction, which leads to CVD symptoms and cardiovascular events. These symptoms are additionally stimulated by ROS generation and foamy cells, which are associated with NO synthesis. | |
This cluster contains almost all processes included in the model; however, it is missing:
| |
This cluster contains almost all processes included in the model, however it is missing:
| |
This cluster contains almost all processes included in the model, however it is missing:
| |
| Attracting of monocytes stimulated by TRAIL, MCP1 (by neighboring endothelial cells stimulation) and MCP1, VCAM1, ICAM1 (by p50/p65 translocation to the nucleus in macrophages M1 and SMC and EC) leads to high level of classically activated macrophages M1, which release IL-6, IL-12, IL-1, IL-23. Macrophages M1 are also engaged in the transformation of oxLDL into foamy cells via NO synthesis and lipid peroxidation, which results in progression of atherosclerotic plaque. IL-18 is produced in caspase 1-independent pathway and it can create complex with IL-18R (IL-18R is stimulated by bacterial infections). Active IL-18-IL-18R-IL18R complex recruits MyD88 which is engaged in a pathway of IB phosphorylation. IB phosphorylation stimulates attract of monocytes via p50/p65 translocation to the nucleus in macrophages M1 and SMC and EC, which stimulate MCP1, VCAM1, ICAM1, TNF alpha, iNOS. IL-1 and IL-23 induce Th17 differentiation. Th17 cells are engaged in essential hypertension. IL-6 and high level of IL-10 lead to activation of STAT3 protein, which is engaged in an induction of IL-4 receptor alpha and binding with IL-4. IL-4 and IL-4R complex stimulates anti-inflammatory pathway, which in consequence leads to high level of IL-10. This cluster includes also negative regulation of IL-18. To be precise, IFN gamma leads to interaction with IFNRs, which results in JAK1, 2 and STAT1, STAT2 activation. This activation leads to binding with SBE sequences of IRF1, which result in NO synthesis induced by iNOS. It is important that NO is engaged in caspase 1 inhibition and in consequence leads to negative regulation of IL-18. | |
| Attracting of monocytes stimulated by TRAIL, MCP1, VCAM1, ICAM1 (by p50/p65 translocation to the nucleus in macrophages M1 and SMC and EC) lead to high level of classically activated macrophages M1, which release IL-6, IL-12, IL-1, IL-23. IL18 is produced in caspase 1-independent pathway and it can create complex with IL-18R (IL-18R is stimulated by bacterial infections). Active IL-18-IL-18R-IL-18R complex recruits MyD88 which is engaged in a pathway of IB phosphorylation. Moreover, receptor MyD88 complex can be additionally stimulated by LPS-LBP complex (TLR4 and MyD88 connection via TIRAP). IB phosphorylation stimulates attracting of monocytes via p50/p65 translocation to the nucleus in macrophages M1 and SMC and EC, which stimulate MCP1, VCAM1, ICAM1, TNF alpha, iNOS. IL-1 and IL-23 lead to the formation of Th17. Th17 cells are engaged in essential hypertension. IL-6 and high level of IL-10 lead to activation of STAT3 protein, which is engaged in an induction of IL-4 receptor alpha and binding with IL-4. IL-4 and IL-4R complex stimulates anti-inflammatory pathway, which in consequence leads to high level of IL-10. This cluster includes also negative regulation of IL-18. To be precise, IFN gamma leads to interaction with IFNRs, which results in JAK1, 2 and STAT1, STAT2 activation. This activation leads to binding with SBE sequences of IRF1, which result in NO synthesis induced by iNOS. Important is fact, that NO is engaged in caspase 1 inhibition and in consequence leads to negative regulation of IL-18. Binding of TRAIL to death receptors (DR5 and DR4) leads to the recruitment of an adaptor protein FADD, which leads to pro caspase 8 recruitment. The connection through DDs within FADD and TNFR leads to an activation of DISC type I cells. High level of caspase 8 together with peroxynitrite production result in activation of caspases 3, 6 and 7. Active caspases 3, 6, 7 and also TWEAK lead to apoptosis enhancement anti-inflammation. | |
| Attract of monocytes stimulated by TRAIL, MCP1 (by neighboring endothelial cells stimulation) and MCP1, VCAM1, ICAM1 (by p50/p65 translocation to the nucleus in macrophages M1 and SMC and EC) leads to high level of classically activated macrophages M1, which release IL-6, IL-12, IL-1, IL-23. Macrophages M1 are also engaged in the transformation of oxLDL into foamy cells via NO synthesis and lipid peroxidation, which results in progression of atherosclerotic plaque. IL-18 is produced in caspase 1-independent pathway and it can create complex with IL-18R (IL-18R is stimulated by bacterial infections). Active IL-18-IL-18R-IL-18R complex recruits MyD88 which is engaged in a pathway of IB phosphorylation. IB phosphorylation stimulates attract of monocytes via p50/p65 translocation to the nucleus in macrophages M1 and SMC and EC, which stimulates MCP1, VCAM1, ICAM1, TNF alpha and iNOS. IL-1 and IL-23 lead to the formation of Th17. Th17 cells are engaged in essential hypertension. IL-6 and high level of IL-10 lead to an activation of STAT3 protein, which is engaged in an induction of IL-4 receptor alpha and binding with IL-4. IL-4 and IL-4R complex stimulates anti-inflammatory pathway, which in consequence, leads to high level of IL-10. Binding of TRAIL to death receptors (DR5 and DR4) leads to the recruitment of an adaptor protein FADD, which leads to pro caspase 8 recruitment. The connection through DDs within FADD and TNFR leads to an activation of DISC type I cells. Increased quantity of cFLIP leads to inhibition of caspase 8. | |
| IFN gamma leads to interaction with IFNRs, which results in JAK1, JAK2 and STAT1, STAT2 activation. Activation of pathway of JAK-STAT1 leads to binding with SBE sequences of IRF-1. This process leads to NO synthesis induced by iNOS. NO is engaged in caspase 1 inhibition and in consequence leads to negative regulation of IL-18. NO synthesis leads also to peroxynitrite production, which together with high level of caspase 8 results in an activation of caspases 3, 6 and 7. High level of caspase 8 is caused by TRAIL and TRAIL death receptors trimerization (in oncogenesis). Active caspases 3, 6, 7 and also TWEAK lead to apoptosis enhancement anti-inflammation. TWEAK sequestration is caused by macrophages M2 in case of high level of IL-10. | |
| IFN gamma leads to interaction with IFNRs, which results in JAK1, 2 and STAT1, STAT2 activation. Activation of pathway of JAK-STAT1 leads to binding with SBE sequences of IRF1. This process leads to NO synthesis induced by iNOS. NO is engaged in caspase 1 inhibition and in consequence leads to negative regulation of IL-18. NO synthesis leads also to peroxynitrite production, which is engaged in lipid peroxidation and results in production of modified oxLDL. This modified oxLDL together with IL-18 lead to neighboring endothelial cell stimulation and secretion of MCP1 (IL-18 is produced in caspase 1-independent pathway). | |
This cluster contains almost all processes included in the model, however it is missing:
|
| Analysis of t-Clusters | The More Detailed Analysis of Particular t-Invariants | |
|---|---|---|
| Subprocesses in Which IL-18 Is Engaged | ||
| Sources of IL-18: | Number of t-Clusters | Number of t-Invariants |
| Caspase 1-independent pathway | 10 (67%) | 66 (85%) |
| Caspase 1-dependent pathway | 0 (0%) | 3 (4%) |
| Caspase 1-independent pathway and caspase 1-dependent pathway | 2 (13%) | 4 (5%) |
| lack of IL-18 | 3 (20%) | 5 (6%) |
| Subprocesses in Which Macrophages Are Engaged | ||
| Type of Macrophages: | Number of t-Clusters | Number of t-Invariants |
| M1 | 1 (7%) | 16 (21%) |
| M2 | 4 (27%) | 5 (6%) |
| M1 and M2 | 8 (53%) | 54 (69%) |
| lack of macrophages | 2 (13%) | 3 (4%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Formanowicz, D.; Gutowska, K.; Formanowicz, P. Theoretical Studies on the Engagement of Interleukin 18 in the Immuno-Inflammatory Processes Underlying Atherosclerosis. Int. J. Mol. Sci. 2018, 19, 3476. https://doi.org/10.3390/ijms19113476
Formanowicz D, Gutowska K, Formanowicz P. Theoretical Studies on the Engagement of Interleukin 18 in the Immuno-Inflammatory Processes Underlying Atherosclerosis. International Journal of Molecular Sciences. 2018; 19(11):3476. https://doi.org/10.3390/ijms19113476
Chicago/Turabian StyleFormanowicz, Dorota, Kaja Gutowska, and Piotr Formanowicz. 2018. "Theoretical Studies on the Engagement of Interleukin 18 in the Immuno-Inflammatory Processes Underlying Atherosclerosis" International Journal of Molecular Sciences 19, no. 11: 3476. https://doi.org/10.3390/ijms19113476
APA StyleFormanowicz, D., Gutowska, K., & Formanowicz, P. (2018). Theoretical Studies on the Engagement of Interleukin 18 in the Immuno-Inflammatory Processes Underlying Atherosclerosis. International Journal of Molecular Sciences, 19(11), 3476. https://doi.org/10.3390/ijms19113476