Repression of Transcriptional Activity of Forkhead Box O1 by Histone Deacetylase Inhibitors Ameliorates Hyperglycemia in Type 2 Diabetic Rats


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
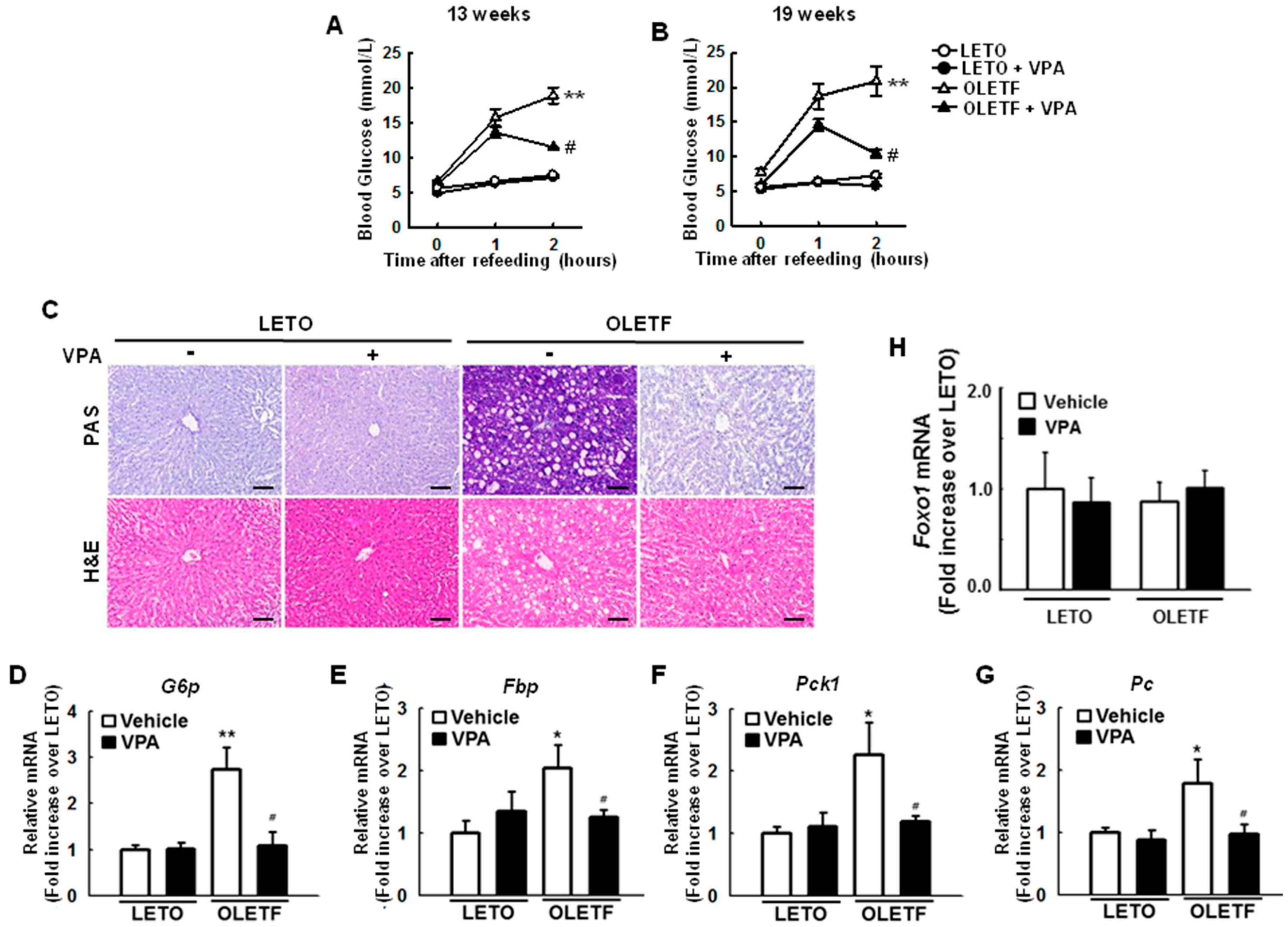
2.1. Administration of Sodium Valproate (VPA) Ameliorates Blood Glucose Levels in Otsuka Long-Evans Tokushima Fatty (OLETF) Rats
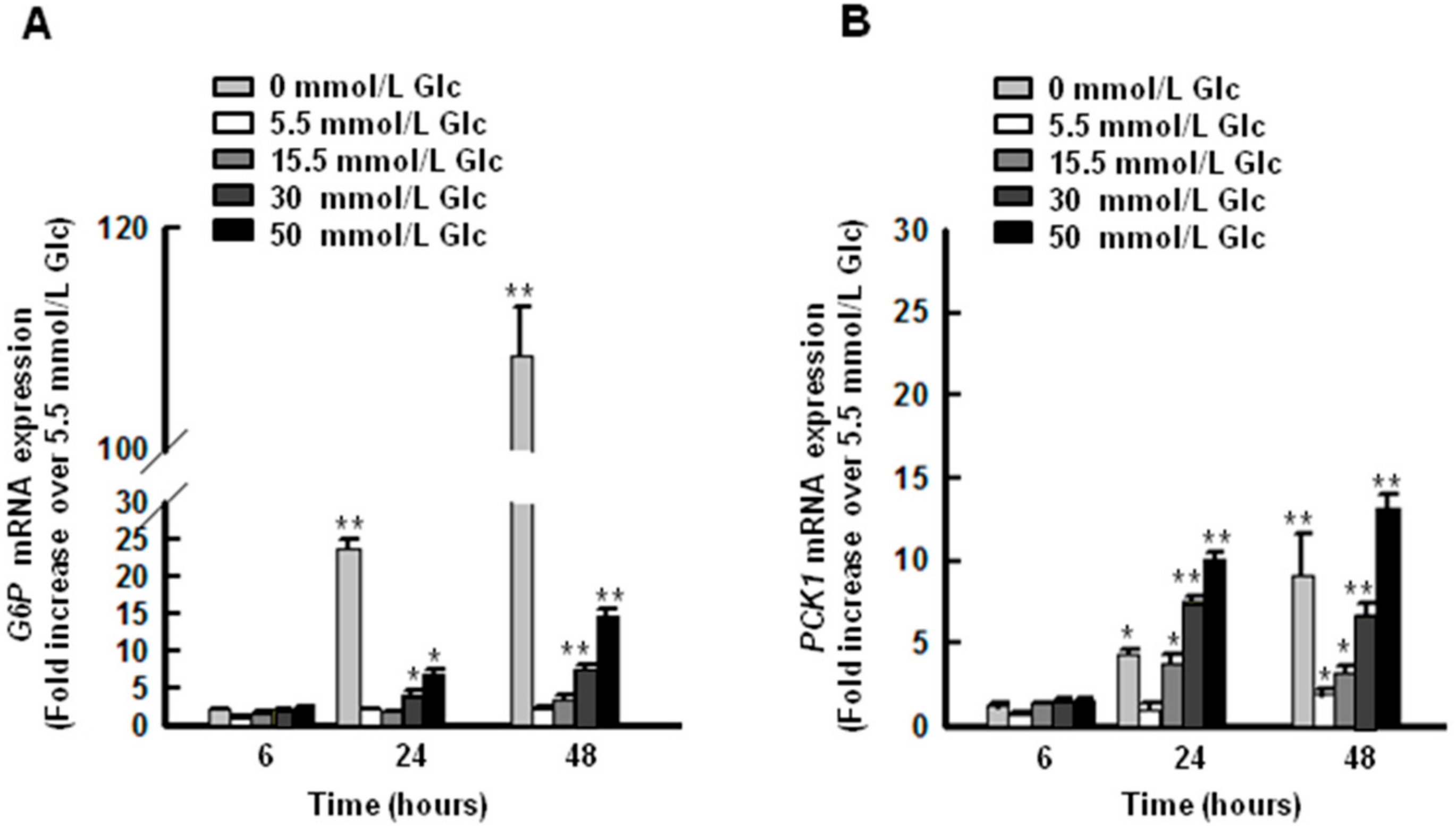
2.2. High Glucose Conditions Induce the Expression of G6P and PCK1 Genes in HepG2 Cells
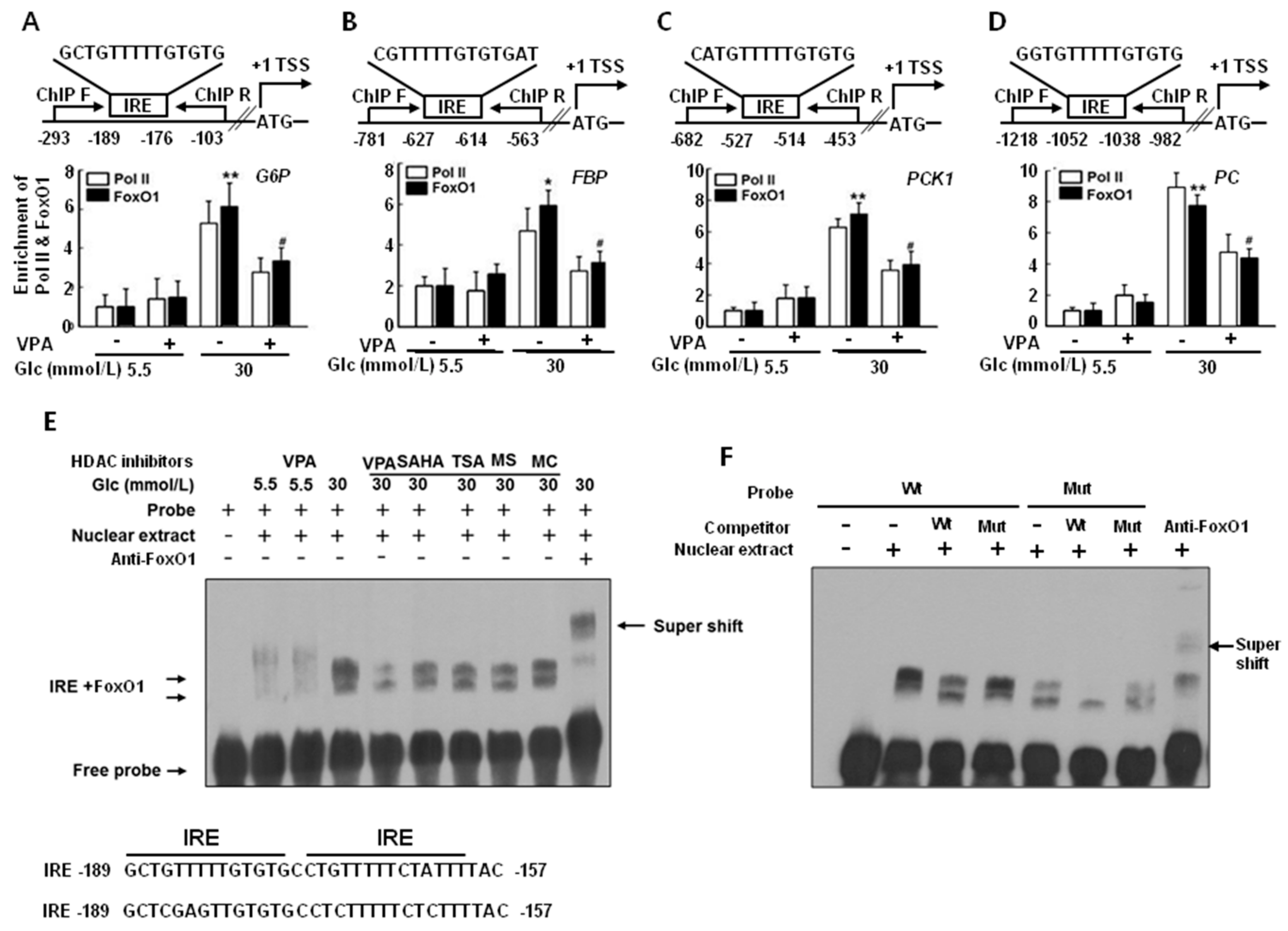
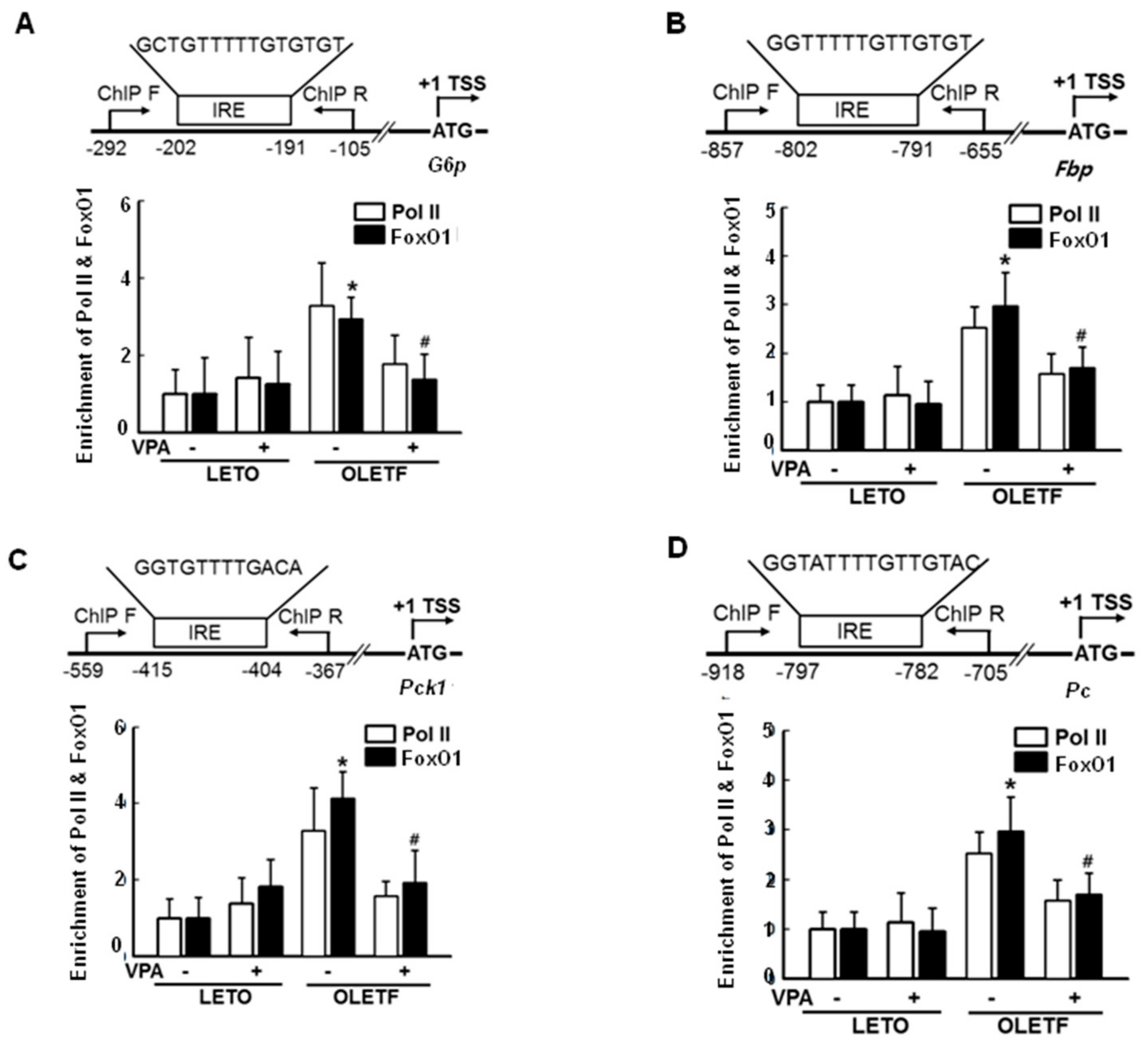
2.3. FoxO1 Enrichment at Target Genes Is Increased under Hyperglycemic Conditions and Is Disturbed by HDAC Inhibitors in HepG2 Cells
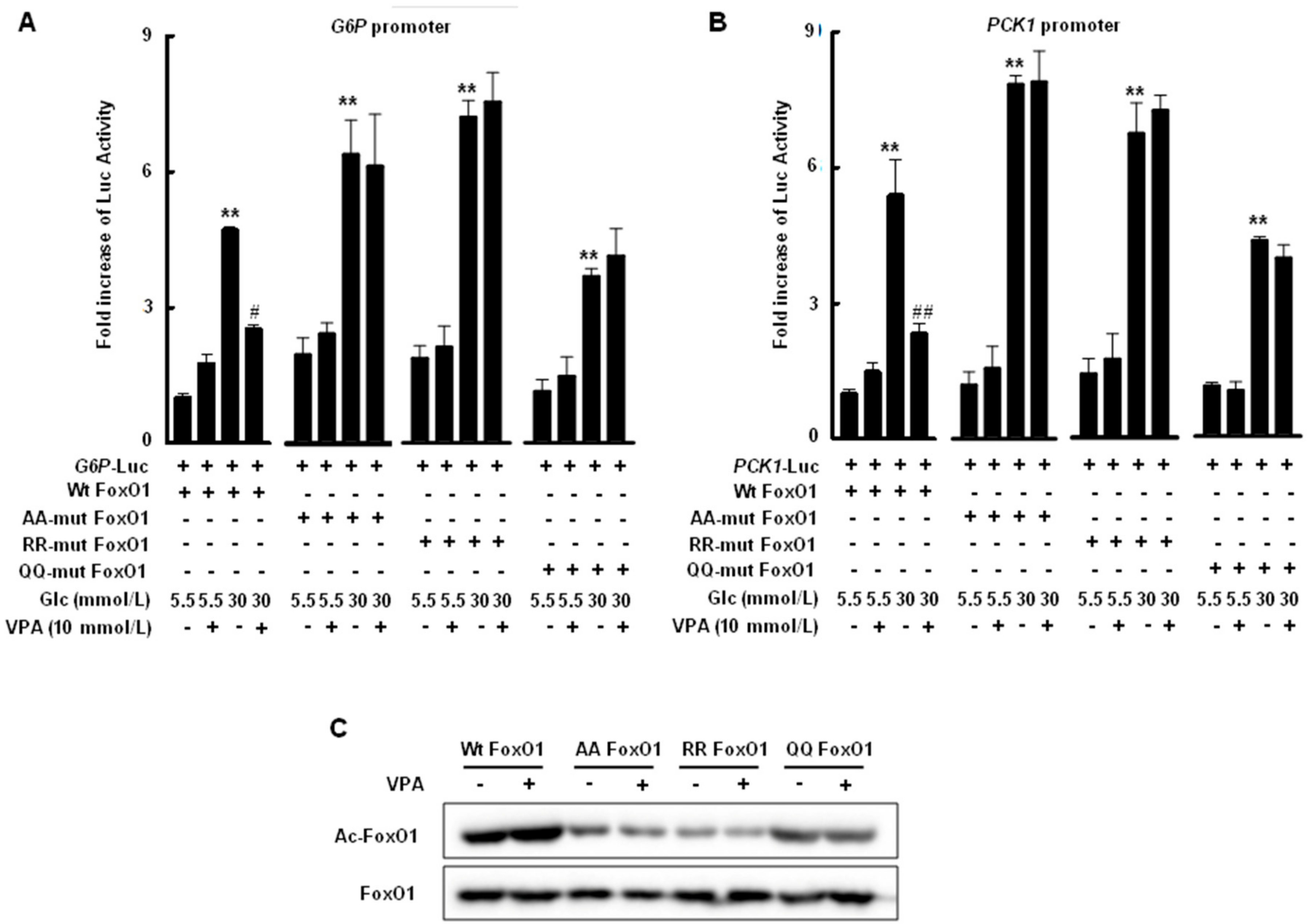
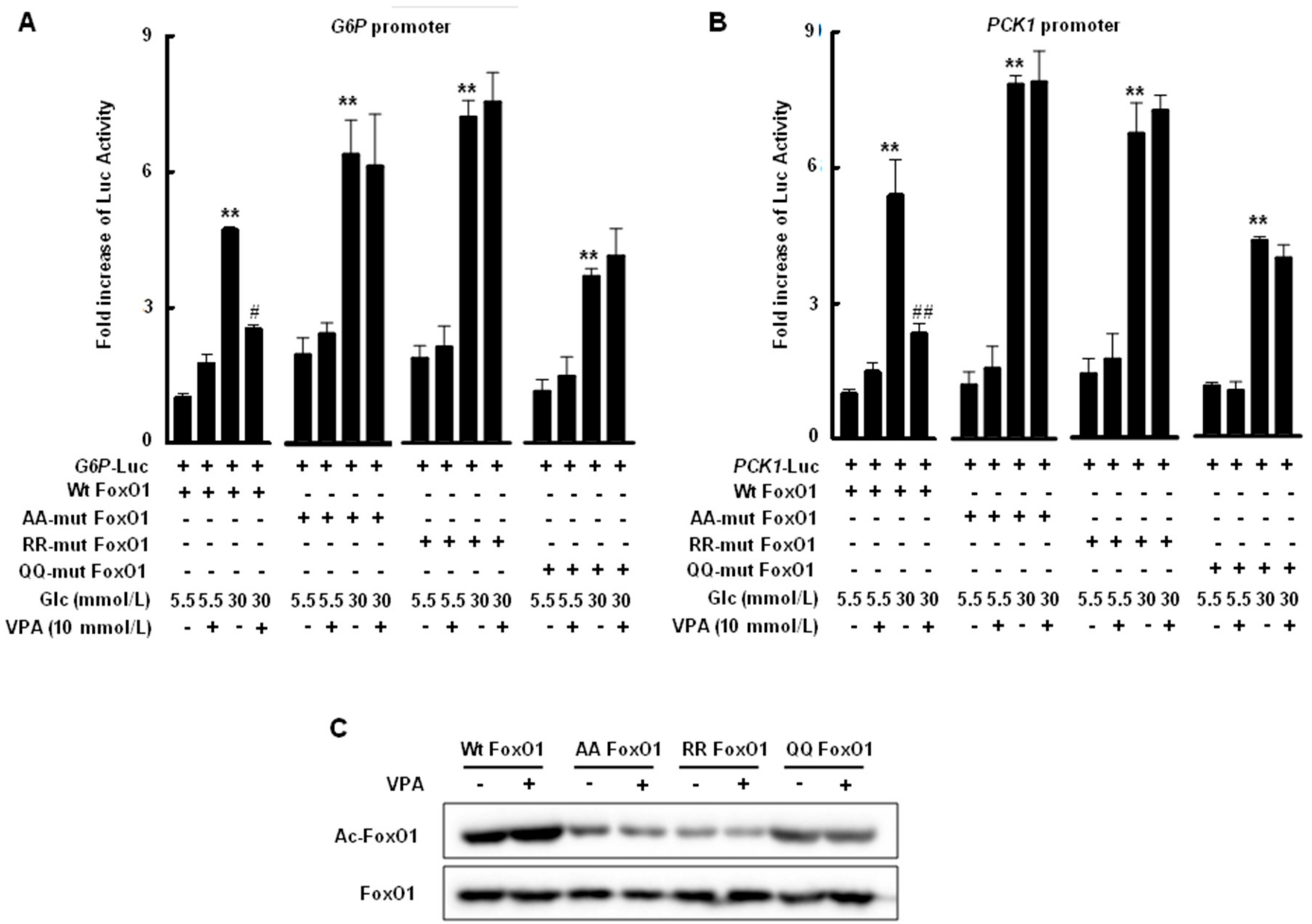
2.4. Transcriptional Activity of FoxO1 Is Attenuated by Acetylation
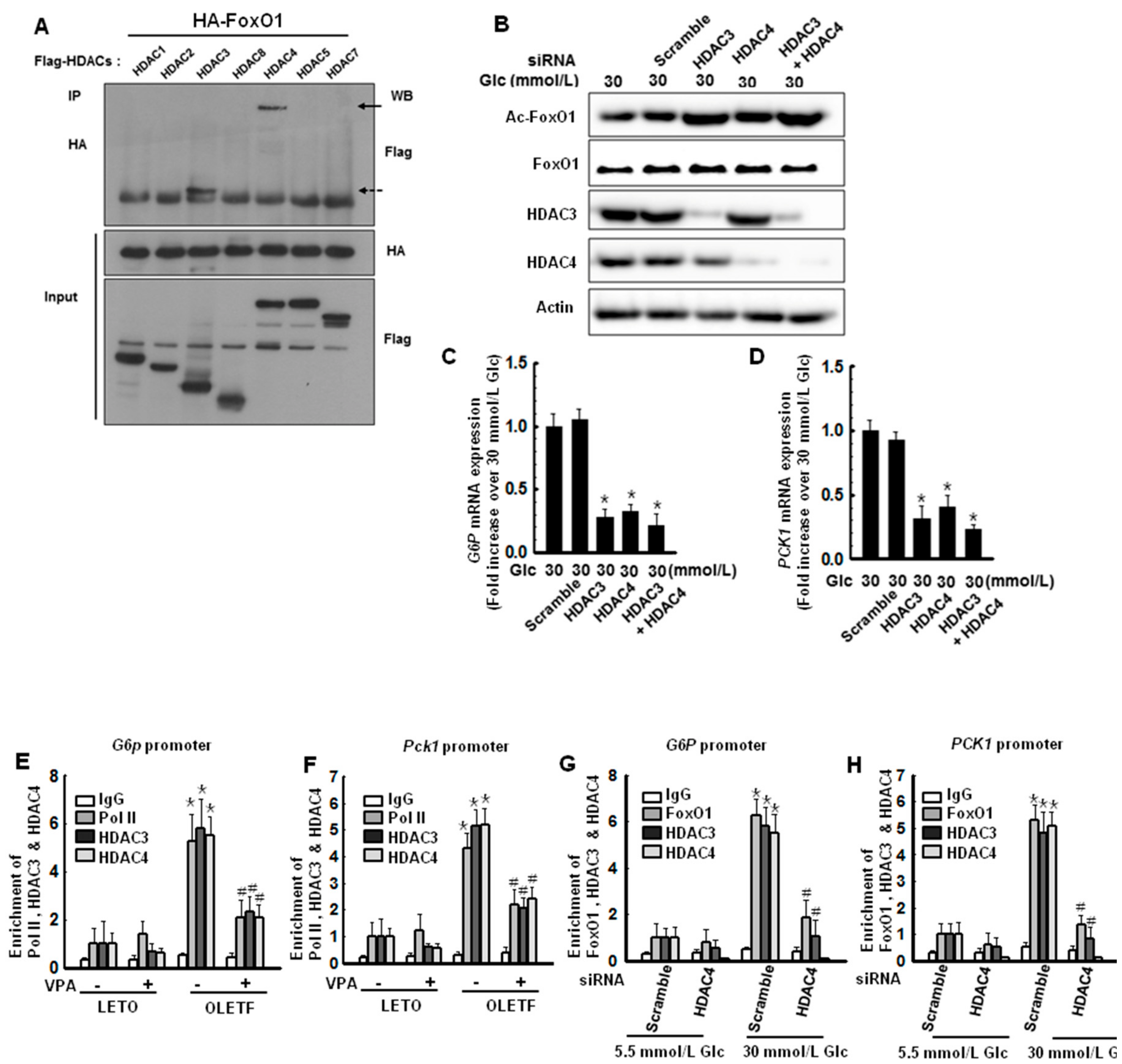
2.5. Hyperglycemic Conditions Induce Interactions between FoxO1 and HDAC3, as Well as FoxO1 and HDAC4, Which Regulate the Transcriptional Activity of FoxO1
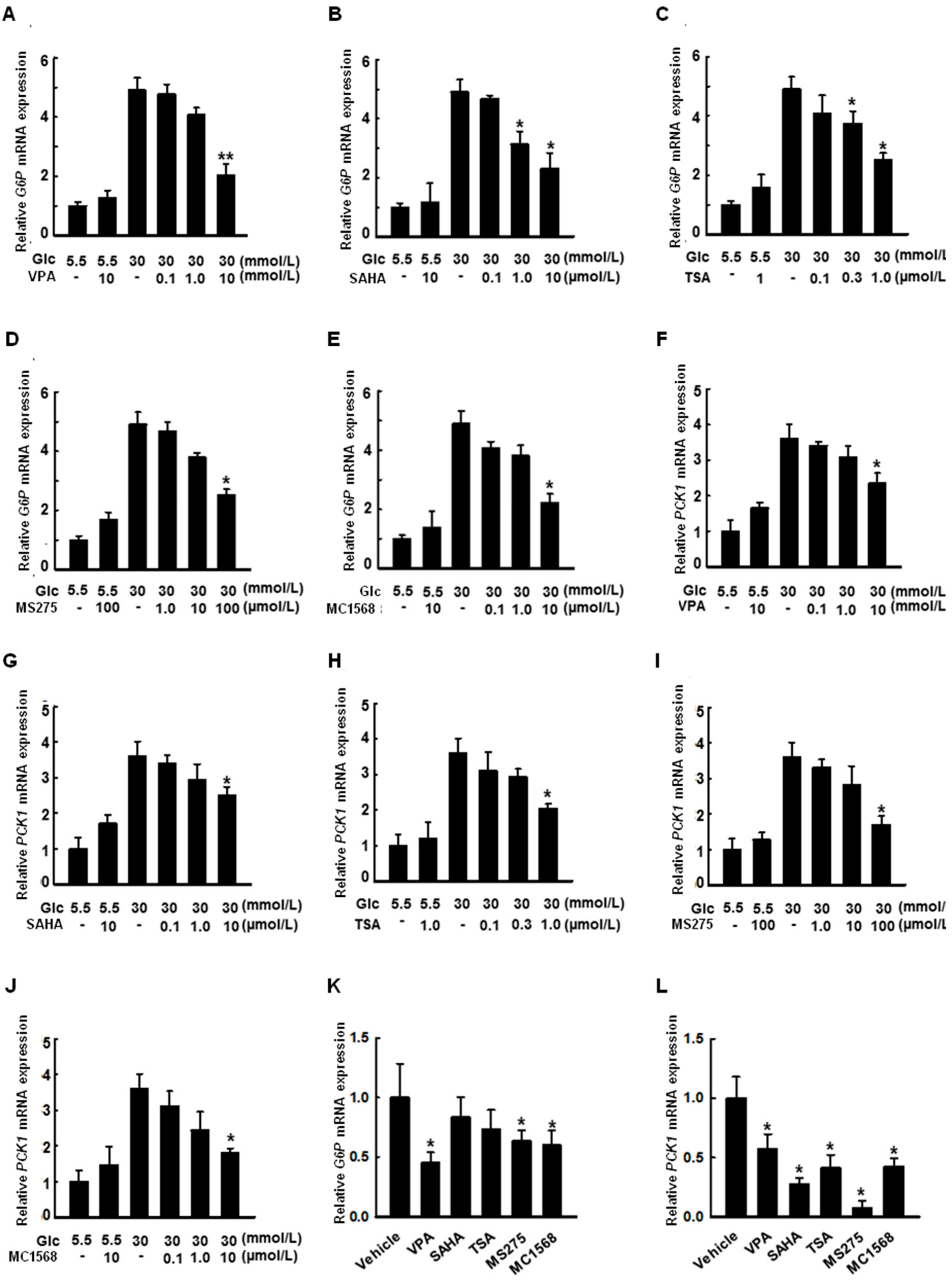
2.6. HDAC Inhibitors Decrease the Expression of G6P and PCK1 Genes under Hyperglycemic Conditions
2.7. VPA Administration Decreases FoxO1 Enrichment at the Target Genes in Type 2 Diabetic Rats
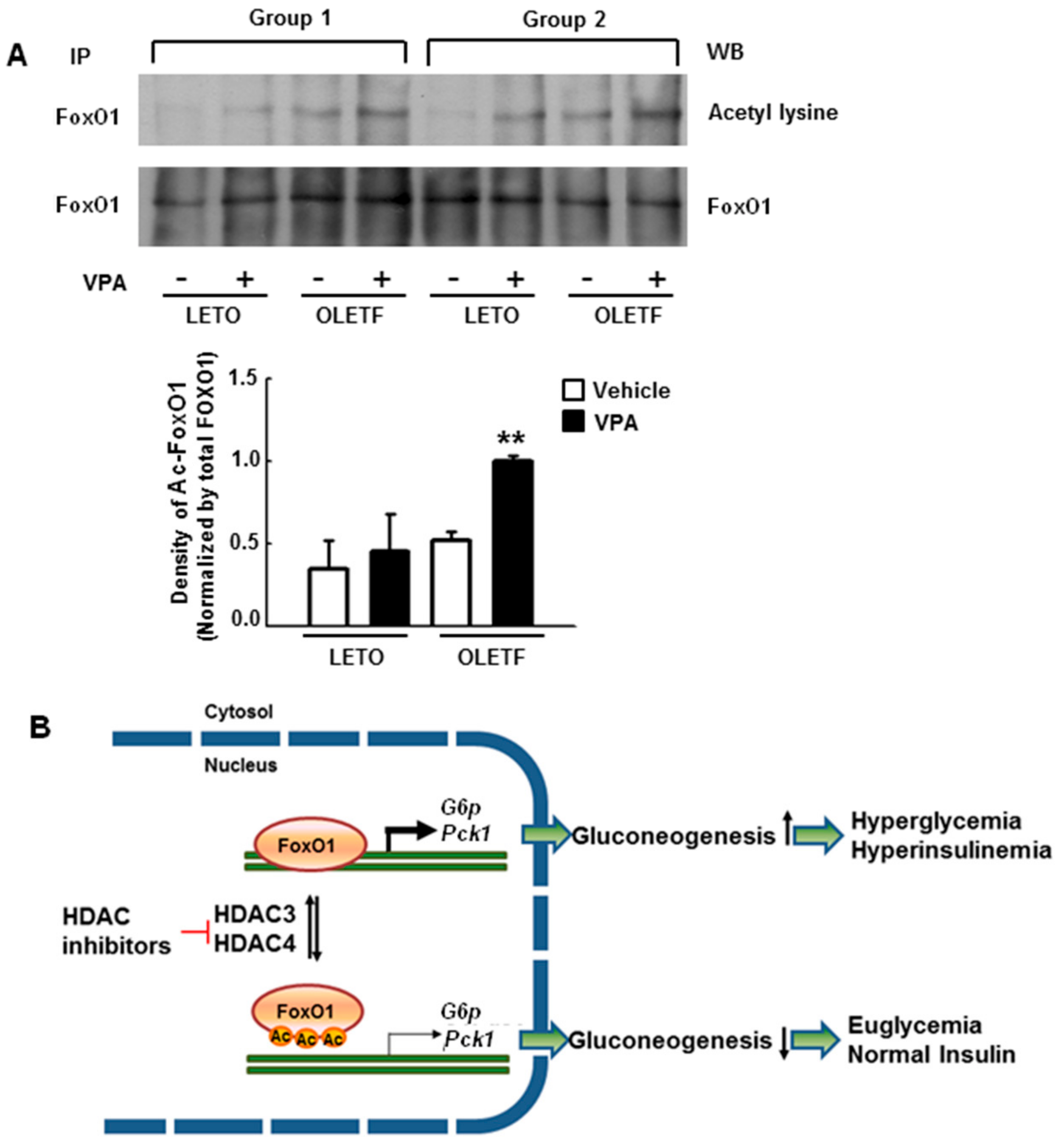
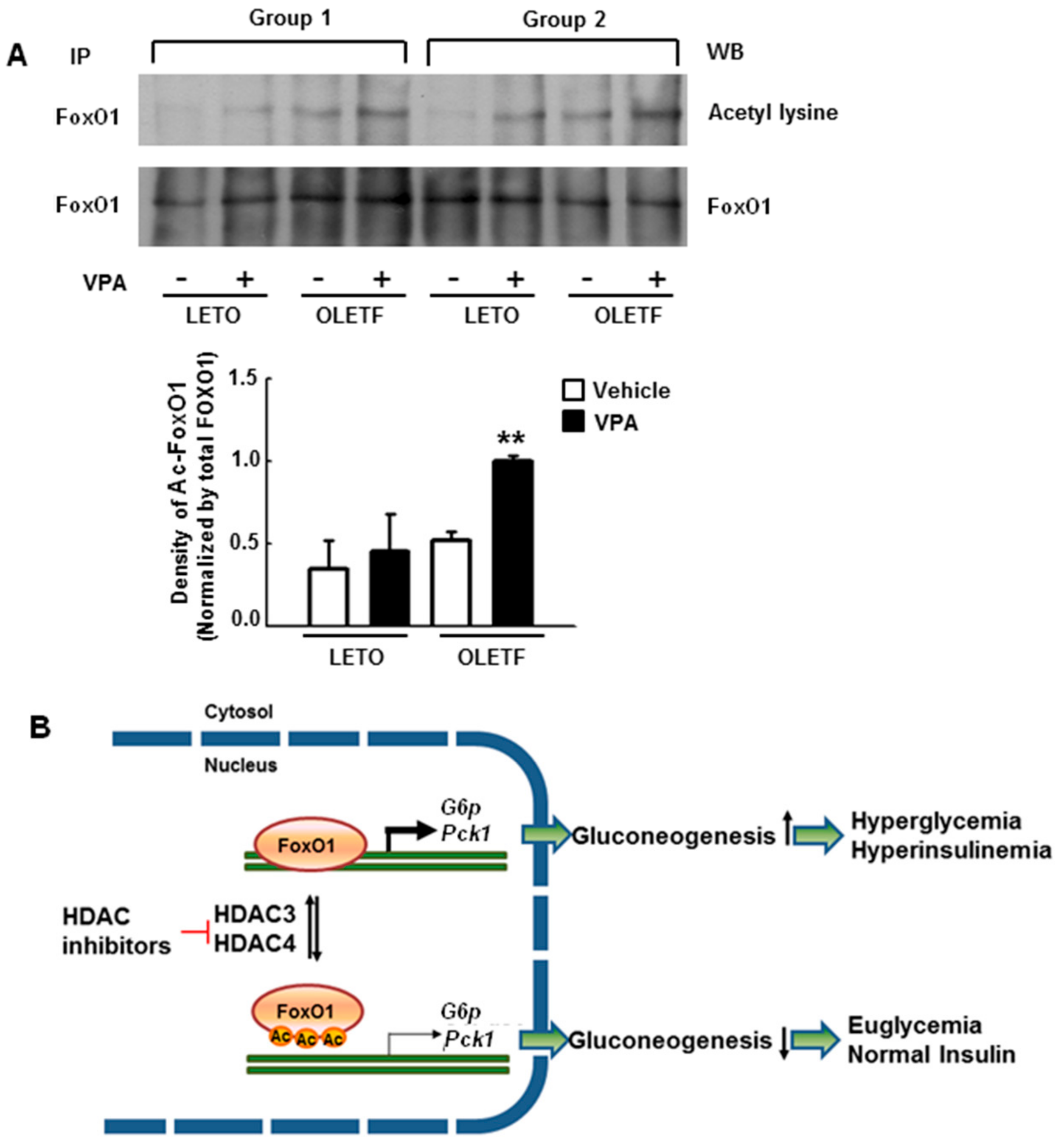
2.8. VPA Administration Increases FoxO1 Acetylation in Type 2 Diabetic Rats
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Periodic Acid–Schiff (PAS) Stain
4.3. Quantitative Real Time PCR (qPCR) and Western Blot Analysis
4.4. Immunoprecipitation and Western Blot
4.5. Cell Culture
4.6. Electrophoretic Mobility Shift Assay
4.7. RNA Interference
4.8. Chromatin Immunoprecipitation (ChIP) Assay
4.9. Luciferase Assay
4.10. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FoxO1 | forkhead box O1 |
| FBP | fructose-1,6-bisphosphatase |
| G6P | glucose 6-phosphatase |
| HDACs | histone deacetylases |
| HDACi | histone deacetylase inhibitors |
| IR | insulin receptor |
| IRE | insulin response element |
| LETO | male Long-Evans Tokushima Otsuka |
| NFκB | nuclear factor kappa B |
| OLETF | Otsuka Long-Evans Tokushima Fatty |
| PGC1α | peroxisome proliferator-activated receptor gamma coactivator 1 alpha |
| PI3K | phosphatidylinositol-3-kinase |
| PCK1 | phosphoenolpyruvate carboxykinase |
| PC | pyruvate carboxylase |
| VPA | sodium valproate |
| SAHA | suberoylanilide hydroxamic acid |
| TSA | trichostatin A |
| T2DM | Type 2 diabetes mellitus |
References
- Guariguata, L. By the Numbers: New Estimates from the Idf Diabetes Atlas Update for 2012. Diabetes Res. Clin. Pract. 2012, 98, 524–525. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M. Diabetes: Advances in Diagnosis and Treatment. JAMA 2015, 314, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2012, 35 (Suppl. 1), S64–S71. [Google Scholar] [CrossRef] [PubMed]
- Marinho, R.; Mekary, R.A.; Munoz, V.R.; Gomes, R.J.; Pauli, J.R.; de Moura, L.P. Regulation of Hepatic Trb3/Akt Interaction Induced by Physical Exercise and Its Effect on the Hepatic Glucose Production in an Insulin Resistance State. Diabetol. Metab. Syndr. 2015, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Liu, Y.; Koo, S.H.; Hedrick, S.; Vargas, T.; Heredia, J.; Yates, J., 3rd; Montminy, M. Insulin Modulates Gluconeogenesis by Inhibition of the Coactivator Torc2. Nature 2007, 449, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer Activities of Histone Deacetylase Inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-Wide Mapping of Hats and Hdacs Reveals Distinct Functions in Active and Inactive Genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.X.; Lee, J. Dietary Regulation of Histone Acetylases and Deacetylases for the Prevention of Metabolic Diseases. Nutrients 2012, 4, 1868–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peserico, A.; Simone, C. Physical and Functional Hat/Hdac Interplay Regulates Protein Acetylation Balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.A.; Lee, D.Y.; Cho, H.M.; Kim, S.Y.; Iwasaki, Y.; Kim, I.K. Histone Deacetylase Inhibition Attenuates Transcriptional Activity of Mineralocorticoid Receptor through Its Acetylation and Prevents Development of Hypertension. Circ. Res. 2013, 112, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Fenning, A.; Lim, J.; Le, G.T.; Reid, R.C.; Halili, M.A.; Fairlie, D.P.; Brown, L. Antifibrotic Activity of an Inhibitor of Histone Deacetylases in Doca-Salt Hypertensive Rats. Br. J. Pharmacol. 2010, 159, 1408–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinale, J.P.; Sriramula, S.; Pariaut, R.; Guggilam, A.; Mariappan, N.; Elks, C.M.; Francis, J. Hdac Inhibition Attenuates Inflammatory, Hypertrophic, and Hypertensive Responses in Spontaneously Hypertensive Rats. Hypertension 2010, 56, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Mosley, A.L.; Ozcan, S. Glucose Regulates Insulin Gene Transcription by Hyperacetylation of Histone H4. J. Biol. Chem. 2003, 278, 19660–19666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, C.; James, S.R. Acetylation of Insulin Receptor Substrate-1 Is Permissive for Tyrosine Phosphorylation. BMC Biol. 2004, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D.; et al. Inhibition of Class I Histone Deacetylases Unveils a Mitochondrial Signature and Enhances Oxidative Metabolism in Skeletal Muscle and Adipose Tissue. Diabetes 2013, 62, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.; Tonnesen, M.; Ronn, S.G.; Storling, J.; Jorgensen, S.; Mascagni, P.; Dinarello, C.A.; Billestrup, N.; Mandrup-Poulsen, T. Inhibition of Histone Deacetylases Prevents Cytokine-Induced Toxicity in Beta Cells. Diabetologia 2007, 50, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Kawano, K.; Hirashima, T.; Mori, S.; Saitoh, Y.; Kurosumi, M.; Natori, T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes 1992, 41, 1422–1428. [Google Scholar] [PubMed]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class Iia Histone Deacetylases Are Hormone-Activated Regulators of Foxo and Mammalian Glucose Homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The Foxo Code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Han, S.; Kitamura, T.; Accili, D. Dual Role of Transcription Factor Foxo1 in Controlling Hepatic Insulin Sensitivity and Lipid Metabolism. J. Clin. Investig. 2006, 116, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Brent, M.M.; Anand, R.; Marmorstein, R. Structural Basis for DNA Recognition by Foxo1 and Its Regulation by Posttranslational Modification. Structure 2008, 16, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Allis, C.D. Roles of Histone Acetyltransferases and Deacetylases in Gene Regulation. Bioessays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Lee, H.A.; Song, M.J.; Seok, Y.M.; Kang, S.H.; Kim, S.Y.; Kim, I. Histone Deacetylase 3 and 4 Complex Stimulates the Transcriptional Activity of the Mineralocorticoid Receptor. PLoS ONE 2015, 10, e0136801. [Google Scholar] [CrossRef] [PubMed]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Surdo, P.L.; Carfi, A.; et al. Unraveling the Hidden Catalytic Activity of Vertebrate Class Iia Histone Deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef] [PubMed]
- Cornell, S. Continual Evolution of Type 2 Diabetes: An Update on Pathophysiology and Emerging Treatment Options. Ther. Clin. Risk Manag. 2015, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, K.; Tavares, C.D.; Rines, A.K.; Puigserver, P. Molecular Pathophysiology of Hepatic Glucose Production. Mol. Asp. Med. 2015, 46, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, F. Tissue-Specific Insulin Signaling in the Regulation of Metabolism and Aging. IUBMB Life 2014, 66, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.M.; Schwartz, M.W. Control of Hepatic Glucose Metabolism by Islet and Brain. Diabetes Obes. Metab. 2014, 16 (Suppl. 1), 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Y.; Zhu, W.G. Applications of Post-Translational Modifications of Foxo Family Proteins in Biological Functions. J. Mol. Cell Biol. 2011, 3, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wan, M.; Leavens, K.F.; Chu, Q.; Monks, B.R.; Fernandez, S.; Ahima, R.S.; Ueki, K.; Kahn, C.R.; Birnbaum, M.J. Insulin Regulates Liver Metabolism in Vivo in the Absence of Hepatic Akt and Foxo1. Nat. Med. 2012, 18, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.; Zilberfarb, V.; Gangneux, N.; Christeff, N.; Issad, T. O-Glycosylation of Foxo1 Increases Its Transcriptional Activity Towards the Glucose 6-Phosphatase Gene. FEBS Lett. 2008, 582, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Tabata, M.; Rodgers, J.T.; Puigserver, P. Usp7 Attenuates Hepatic Gluconeogenesis through Modulation of Foxo1 Gene Promoter Occupancy. Mol. Endocrinol. 2014, 28, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Parhofer, K.G. Interaction between Glucose and Lipid Metabolism: More Than Diabetic Dyslipidemia. Diabetes Metab. J. 2015, 39, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.D.; Dong, H.H. Foxo1 and Hepatic Lipid Metabolism. Curr. Opin. Lipidol. 2009, 20, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Kamagate, A.; Qu, S.; Perdomo, G.; Su, D.; Kim, D.H.; Slusher, S.; Meseck, M.; Dong, H.H. Foxo1 Mediates Insulin-Dependent Regulation of Hepatic Vldl Production in Mice. J. Clin. Investig. 2008, 118, 2347–2364. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Hong, W.; Cai, P.; Chen, Y.; Xu, C.; Cao, D.; Yu, W.; Zhao, Z.; Huang, M.; Jin, J. Valproate Induced Hepatic Steatosis by Enhanced Fatty Acid Uptake and Triglyceride Synthesis. Toxicol. Appl. Pharmacol. 2017, 324, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Klover, P.J.; Mooney, R.A. Hepatocytes: Critical for Glucose Homeostasis. Int. J. Biochem. Cell Biol. 2004, 36, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Lin, J.K. Epigallocatechin Gallate (EGCG) Attenuates High Glucose-Induced Insulin Signaling Blockade in Human Hepg2 Hepatoma Cells. Mol. Nutr. Food Res. 2008, 52, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Herrera, I.; Martin, M.A.; Goya, L.; Ramos, S. Cocoa Flavonoids Attenuate High Glucose-Induced Insulin Signalling Blockade and Modulate Glucose Uptake and Production in Human Hepg2 Cells. Food Chem. Toxicol. 2014, 64, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Le, L.; Jiang, B.; Wan, W.; Zhai, W.; Xu, L.; Hu, K.; Xiao, P. Metabolomics Reveals the Protective of Dihydromyricetin on Glucose Homeostasis by Enhancing Insulin Sensitivity. Sci. Rep. 2016, 6, 36184. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, H.M.; Seok, Y.M.; Lee, H.A.; Song, M.; Kim, I. Repression of Transcriptional Activity of Forkhead Box O1 by Histone Deacetylase Inhibitors Ameliorates Hyperglycemia in Type 2 Diabetic Rats. Int. J. Mol. Sci. 2018, 19, 3539. https://doi.org/10.3390/ijms19113539
Cho HM, Seok YM, Lee HA, Song M, Kim I. Repression of Transcriptional Activity of Forkhead Box O1 by Histone Deacetylase Inhibitors Ameliorates Hyperglycemia in Type 2 Diabetic Rats. International Journal of Molecular Sciences. 2018; 19(11):3539. https://doi.org/10.3390/ijms19113539
Chicago/Turabian StyleCho, Hyun Min, Young Mi Seok, Hae Ahm Lee, Minji Song, and InKyeom Kim. 2018. "Repression of Transcriptional Activity of Forkhead Box O1 by Histone Deacetylase Inhibitors Ameliorates Hyperglycemia in Type 2 Diabetic Rats" International Journal of Molecular Sciences 19, no. 11: 3539. https://doi.org/10.3390/ijms19113539
APA StyleCho, H. M., Seok, Y. M., Lee, H. A., Song, M., & Kim, I. (2018). Repression of Transcriptional Activity of Forkhead Box O1 by Histone Deacetylase Inhibitors Ameliorates Hyperglycemia in Type 2 Diabetic Rats. International Journal of Molecular Sciences, 19(11), 3539. https://doi.org/10.3390/ijms19113539