1. Introduction
Natural products, in the form of different types of plant extracts or as pure active phytochemicals, have historically been known to offer important therapeutic support for both traditional and well-established medical approaches. It is unanimously accepted that plants and natural products have played a vital role in medicine and health since the beginning of mankind [
1,
2]. Being aware of the inherent therapeutic potential of phytochemicals, important lines of research have been headed towards this direction, thus, an impressive number of bioactive compounds have been isolated and deeply investigated over the past decades. Nowadays, we can outline that natural products, together with their chemical derivatives, count more than half of the FDA-approved medication and moreover continue to be important sources for the discovery of active drugs [
3].
Recent studies have described that the global incidence of cutaneous melanoma continues to grow year after year and, directly correlated to this, the mortality caused by unresectable or metastatic melanoma unfortunately represents a parameter in evolution [
4,
5]. Moreover, in the field of oncology, but not only, molecules derived from natural sources represent a huge contribution to drug discovery today. It is worth mentioning that a screening of the number of chemotherapeutic agents, in relation to their source, concludes that over 60% of approved drugs are derived from natural molecules. Vincristine, vinblastine, paclitaxel, docetaxel, topotecan, and irinotecan are only some examples of important plant-derived anticancer agents [
6,
7]. Among the phytochemicals with anticancer properties, flavonoids represent one of the most studied classes of natural compounds [
8,
9]. A flavonoid that arouses interest to an increased number of research groups, due to its plethora of biological activities, also including anticancer potential, is 4′,5,7-trihydroxyflavone, commonly known under the name of apigenin. Research in the field has assigned apigenin both in vitro and in vivo antiproliferative, pro-apoptotic, and anti-metastatic effects in experimental models of melanoma [
10,
11,
12].
Chamomile, parsley, and celery represent important sources of apigenin, both for daily human intake and for the industrial extraction of this flavone.
Chamomile, with its scientific name
Matricaria chamomilla L., also known as German chamomile, is an aromatic plant belonging to the Asteraceae family. The Asteraceae Bercht. & J.Presl family is also called Compositae, due to the composite character of flowers within this family. It is one of the largest families comprising more than 23,000 species included in over 1900 genera [
13]. It was asserted that the biological activity of different types of extracts is due to the phytochemicals included in the class of flavonoids (apigenin, luteolin, quercetin, patuletin) and essential oils (α-bisabolol and its oxides, azulenes) [
14]. The main biological activities include antioxidant, antimicrobial, anti-inflammatory, cytotoxic, antispasmodic, antiviral, and sedative potential [
15]. The antiproliferative potential of chamomile extract was described for various cell lines, including human prostate epithelial PZ-HPV-7 cells, human prostate cancer LNCaP, DU145, PC-3 cells, T-47D breast carcinoma, HeLa -cervical adenocarcinoma, HT1080- fibrosarcoma, and RKO-colon carcinoma cells [
16].
Parsley and celery are also aromatic plants belonging to the family Apiaceae. Apiaceae Lindl., also known as Umbelliferae Juss. This family represents the 16th-largest family of flowering plants, and comprises approximately 3000–3750 species included in 300–455 genera [
17]. Parsley and celery are two important constituents of this family, used both for their culinary and medical benefits. A comprehensive review that presents the ethnopharmacology, phytochemistry, and biological activities of parsley, also known under the scientific name of
Petroselinum crispum (Mill.) Nym. ex A. W. Hill, concludes that the seed extract has in vitro antioxidant, analgesic, spasmolytic, immunosuppressant, laxative, and diuretic properties [
18]. A recent study has shown that extracts obtained from the leaves and stem of English parsley indicate an antioxidant capacity, as well as a protective effect against DNA damage induced by H
2O
2. Moreover, the extract has been shown to inhibit the proliferation and migration of MCF7 breast cancer cell line [
19]. Celery seeds extracts have been described for their antioxidant, antimicrobial, antiarthritic, and antiulcer potential [
20,
21]. The group of Mansi et al. have also found that the extract can induce a hypolipidemic effect in rats [
22]. Anti-inflammatory, gastro-protective, anti-
Helicobacter pylori activity, and no toxicologically significant subchronic effects in experimental models using rats, were reported by the group of Powanda et al. [
23]. Wild celery oil was assigned with antiproliferative potential against HCT116 human colon carcinoma cells [
24].
The aim of this study is to assess the phytochemical composition, and antioxidant and anti-inflammatory potential of some major botanical sources of apigenin—chamomile, parsley, and celery methanolic extracts—as well as their biological activity against A375 human melanoma and human dendritic cells.
3. Discussion
All three investigated vegetal products are postulated in the literature as major botanical sources of apigenin but, according to their scientific classification, chamomile belongs to the Magnoliophyta clade, Asteraceae family, while parsley and celery belong to the Angiosperms clade, Apiaceae family. This is the reason why we have decided to discuss the phytochemical composition in these two directions, namely presentation of chamomile extract, and parsley and celery extracts, respectively. The main classes of phytochemicals with biological activity that can be found in the chamomile flowers include volatile oil (which, in turn, is composed by sesquiterpenes azulenogens and non-azulenogens, as well as monoterpenes), flavonoids, coumarins, terpenoids, and mucilages. In a comprehensive review about this medicinal aromatic plant, Srivastava et al. have estimated the presence of approximately 120 secondary metabolites, including 28 terpenoids and 36 flavonoids [
26]. From the class of flavonoids, apigenin represents one of the most promising compounds from the point of view of bioactivity. Within the vegetal product, the glycosidic form is predominant, while the aglycone can be found in small amounts [
27]. It is unanimously accepted that the most usual source of apigenin intake for the human body is represented by chamomile tea [
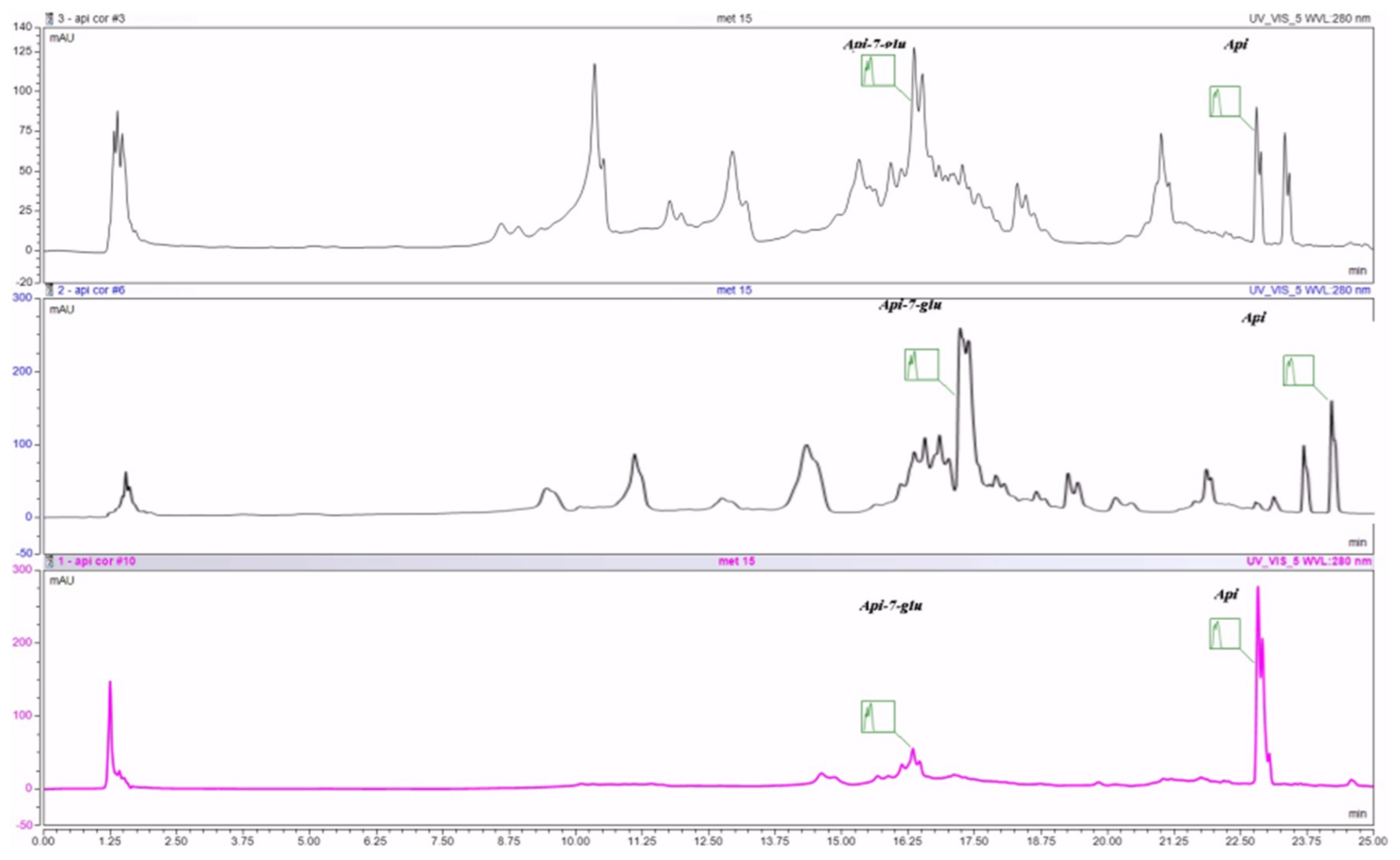
28].
The study analyzed a hydroalcoholic extract based on the idea postulated in the literature that optimum extracts obtained from this vegetal product contain about 50% alcohol [
26]. According to the European Pharmacopeia (EP), in order to have a biological effect, the minimum percentage of apigenin 7-glucoside in the flowers should be 0.25% [
29]. Our results confirm that the vegetal product complies with the pharmacopoeia provisions. In a similar approach, Fonseca et al. concluded that the percentage of free and glycosylated apigenin in the methanol extract is 106 and 903 μg/g whereas, in the ethanol extract, the amount is 11 and 247 μg/g [
30]. Analyzing the amount of pure and conjugated apigenin in different types of extracts, Haghi et al. have observed that the methanol extract leads to the highest amount of pure apigenin [
29]. In an aqueous extract, apigenin 7-
O-glucoside was found to be the major constituent [
31].
In a similar approach using different solvents for the extraction of active compounds from the aerial parts in the flowering stage of chamomile (e.g., water, methanol, ethanol), Haghi et al. have concluded that the amount of total phenolic compounds and total flavonoids range in the interval (1.77–50.75 g GAE/100 g in dry material) and (0.82–36.75 g quercetin equivalent (QE)/100 g in dry material), respectively [
29].
As revealed in the results section by different assays, it is obvious that the MC extract demonstrates free radical-scavenging potential. The practical approach is that intake of different food supplements or tea based on chamomile flower extract can lead to prevention of an increased number of pathologies related to oxidative stress and, also, prevent cell mutation. In a recent study, Cvetanovic et al. have assessed the antioxidant potential of different types of extracts obtained from chamomile flowers, namely Soxhlet, microwave-assisted, ultrasound-assisted, and subcritical water extraction. They concluded that the best free radical-scavenging ability was generated by subcritical water extraction [
32]. The stamp that confirms the antioxidant potential of water and alcohol extracts of this vegetal product was put down also by Al-Ismail et al., who used a linoleic acid and liposome model system, as well as the well-known DPPH free radical-scavenging assay [
33]. The phytochemicals that might be involved in this type of effect are the flavonoids, due to their polyphenolic structure; and the terpenoids, due to their double bond system. A recent study showed the protective role of chamomile decoction extract, using a rat model of alcohol-induced injury of gastric mucosa. This potential was assigned to its capacity to reverse the depletion of antioxidant enzymes activity, such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) induced by ethanol administration [
34]. Moreover, our results confirm that MC extract has a better activity against LOX than against free radicals, suggesting that different mechanisms are involved for such properties. The LOX-inhibiting activity can be correlated with an anti-inflammatory potential, which may decrease the membrane permeability.
In regard to parsley and celery extracts, the majority of research has involved different plant organs, usually leaves, stems, and culture cells [
35,
36]. Moreover, the essential and fatty oils were mostly investigated. In terms of our choice of vegetal product, the seeds are commonly believed to contain the genuine and simple compounds found in the future plant. The chemical analysis of parsley and celery seed alcoholic extracts revealed that the chosen vegetal material contains flavone aglycons and apigenin glucoside [
37]. Other studies indicated the presence of gallic acid, catechins, and its derivatives, in ethanolic leaf extract, which were also present in our samples in small quantities. The authors concluded, then, that polyphenols, and mainly kaempferol derivatives, were responsible for the antioxidant activity [
38].
At the same time, the calculated EC
50 value for all the antioxidant tests allowed us to observe differences in regard to their activity. Taken together, all investigated extracts showed a better scavenger activity against free radicals, a good inhibitory activity against lipoxygenase, but a lower sensitivity against iron. These confirm that the type of bioactive compound is highly important for the activity of the extract. Such claims have been previously discussed by Csepregi et al. [
39] in terms of structure–activity relationships, when the presence of certain free hydroxyl groups on the phenolic or flavonoidic skeleton can enhance the antioxidant/scavenger/chelation potential, depending on the assay.
However, the total phenolic quantification is not in direct correlation with the extract antioxidant capacity. There is still a correlation between the flavonoid content, the presence of aglycons, and the antioxidant potential for both parsley and celery samples. Every extract reacts differently depending on the assay, and the ratio and distribution of each component; for example, parsley and celery are more potent as enzyme inhibitors, whereas the chamomile extract acts more strongly as a radical scavenger. Moreover, all extracts show a weaker iron-binding activity. Our results sustain the observations made previously on the antioxidant capacity of parsley and celery extracts [
36,
40]. Some studies also suggest that the type of solvent used for extraction and the temperature used during processing has a great impact on the chemical composition and the biological implication of the final extract. The current study suggests that the chosen extraction method and solvent ensure a good chemical composition of the investigated extracts.
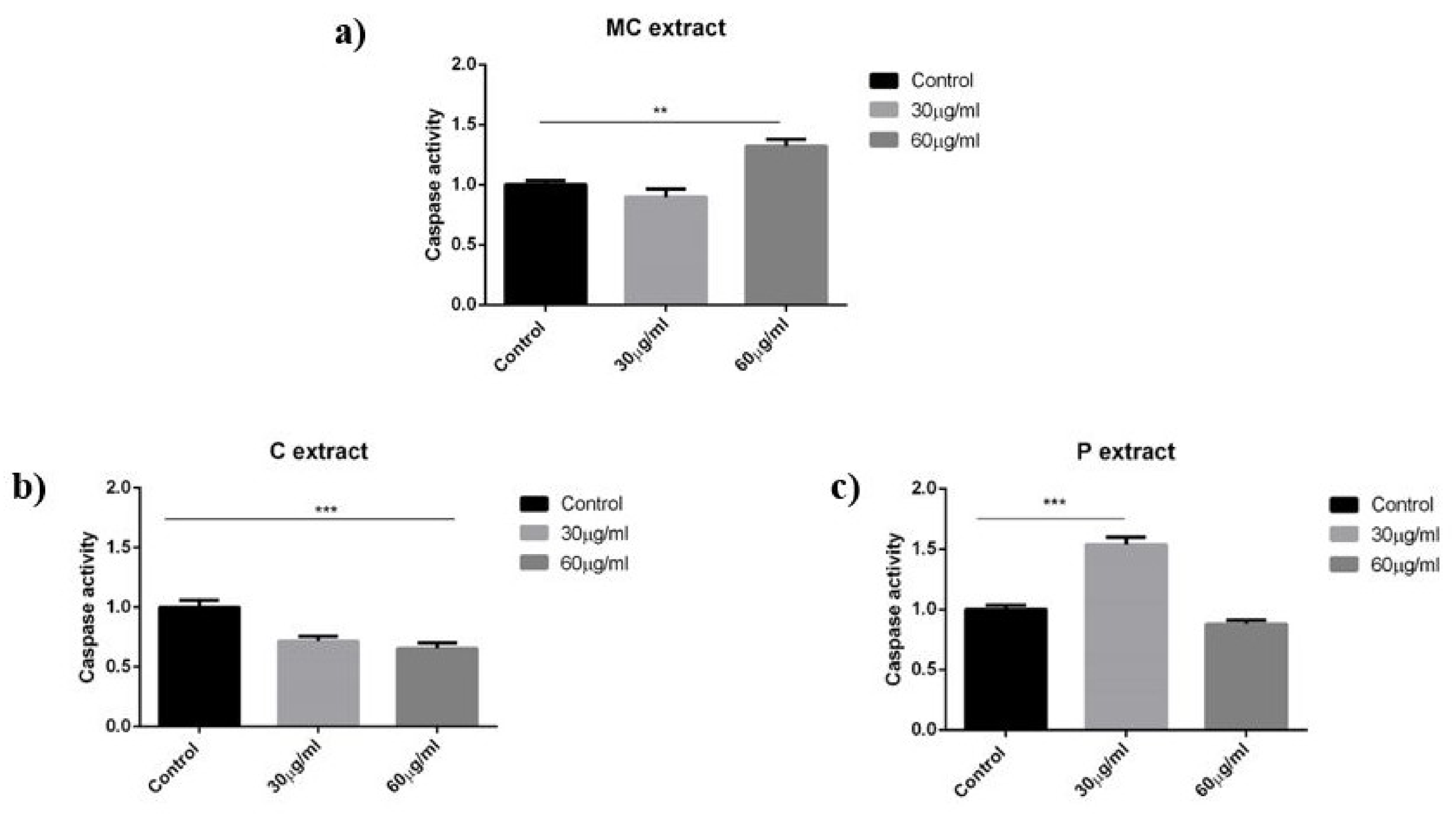
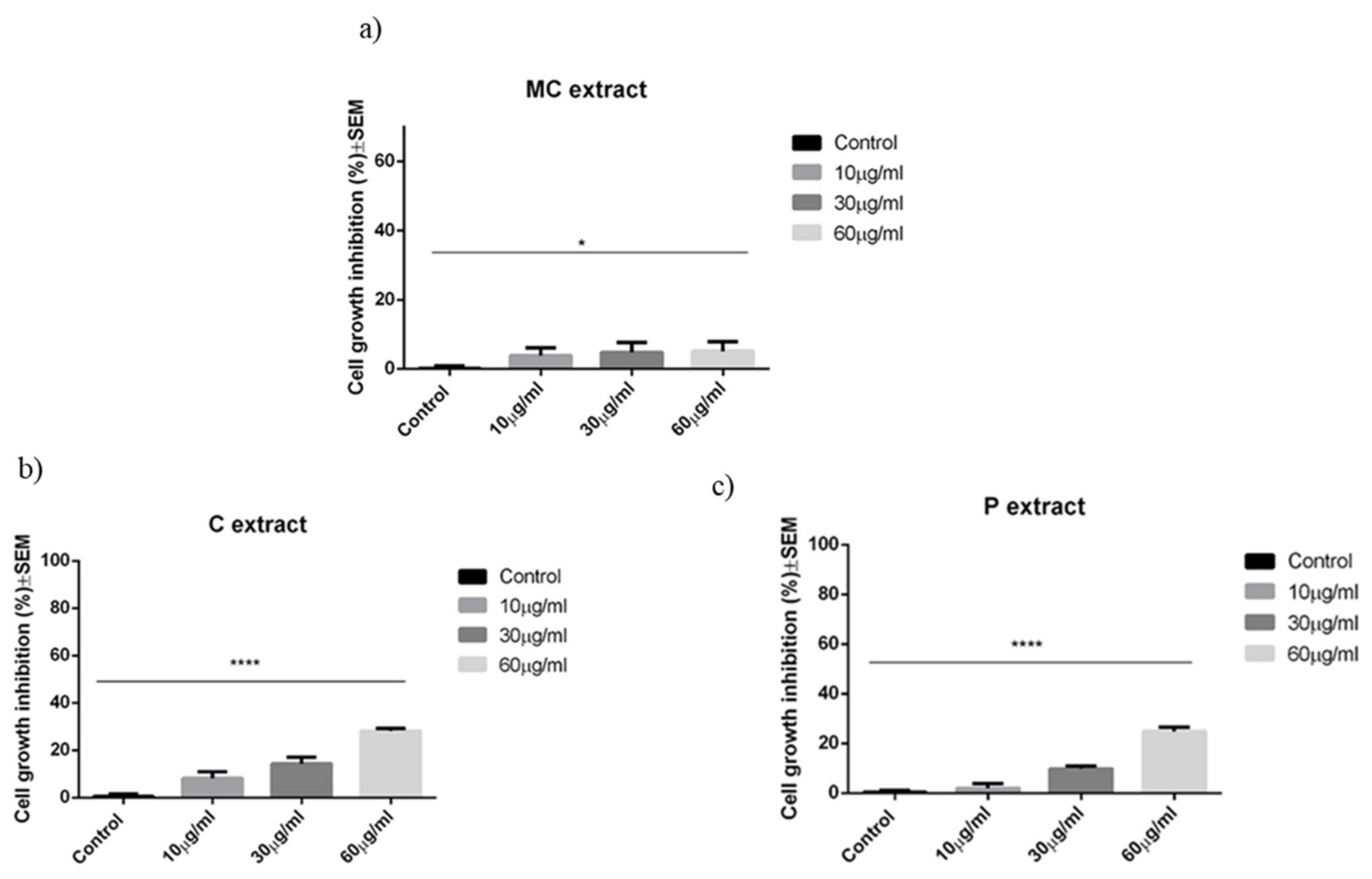
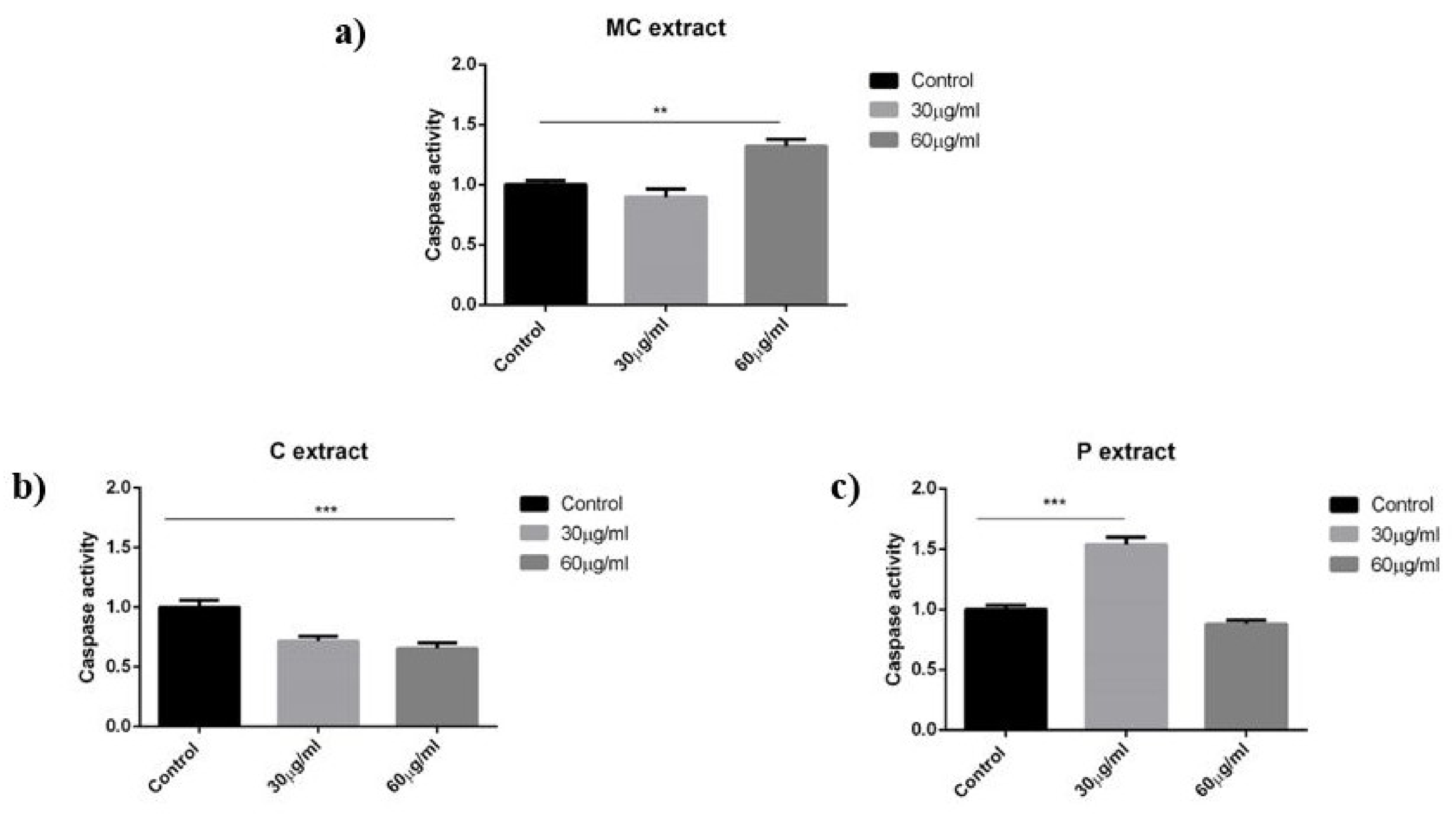
As discussed in the results section for the selected concentrations, the chamomile extract presents weak antiproliferative and pro-apoptotic potential against A375 human melanoma cell line. In a recent paper, Sak et al. have shown that methanolic extract obtained from German chamomile (
Matricaria recutita L.) presents a cytotoxic effect against SK-MEL-2 human melanoma cells with an IC
50 value of 40.7 μg/mL [
41]. In a comparative study that analyzed the anticancer activity of chamomile and marigold tea extract on different cancer cell lines, including a human melanoma cell line (Fem-x), the IC
50 value was >16.67 mg/mL for chamomile extract, while marigold tea extract generated a higher anti-melanoma effect, translated to an IC
50 value of 0.36 ± 0.12 [
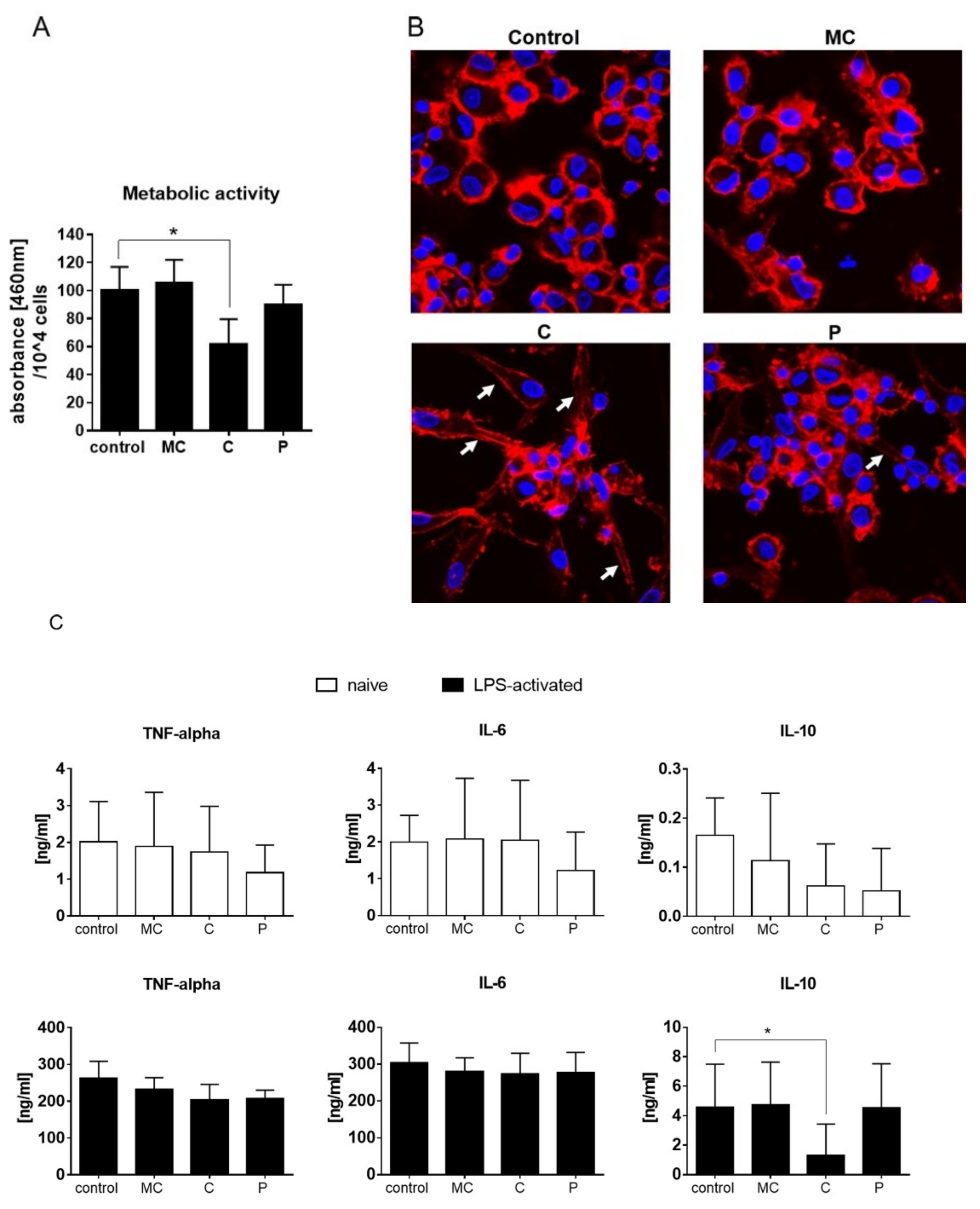
42]. On human dendritic cells, chamomile extract had a slight effect on LPS-mediated activation which however, did not change the metabolic activity or cytokine secretion of TNF-alpha, IL-6, or IL-10.
To the best of our knowledge, until this moment, there are no reports in the literature regarding the effect of celery extract on human melanoma cells. Our study indicates a weak antiproliferative potential against A375 human melanoma cells linear with the concentration. Celery extract was assigned with cytotoxic potential for other cancer cell lines, which gave us the idea to perform a screening for the selected melanoma cell line. For example, in a recent paper, Ahmand et al. have pointed toward the antiproliferative and pro-apoptotic activity of celery seeds extract against BGC-823 human stomach cancer cell line [
43]. Methanolic celery extracts have produced cytotoxic events and apoptosis in the case of two different human cell lines, namely, DLA, Dalton’s lymphoma ascites, and L929, mouse lung fibroblast [
44]. Polyacetylenes obtained from celery root have shown in vitro cytotoxic effect against five different cancer cell lines [
45]. Moreover, methanolic celery extracts have indicated a protective effect against an in vivo model of chemically induced hepatocarcinogenesis [
46]. Celery slightly reduced the ability of dendritic cells to proliferate during LPS activation, and led the cells acquire a spindle-like shape. Notably, this effect did not cause reduction in the pro-inflammatory cytokine levels of TNF-alpha and IL-6 but, rather, reduced secretion of the anti-inflammatory IL-10. Thus, celery extract might be a potential source to further enhance active immune responses.
The parsley extract was analyzed by the group of Dorman et al. They have tested the cytotoxic potential of parsley extract obtained from the leaves of
Petroselinum crispum (Mill.) Nym. ex A. W. Hill against non-cancerous CV1-P fibroblast and A375 human melanoma cells. The MTT assay has shown that cancerous cells were more sensitive to parsley extract treatment, in comparison to normal fibroblasts. The highest tested concentration, namely 2 mg/mL, led to a 50% decrease of the metabolic activity of cancerous cells. The leakage of lactate dehydrogenase, as a sign of disintegration of the cell membrane, increased by 210% for A375 cells and 179% for CV1-P cells. The expression of caspase 3 as a predictor of apoptosis was assessed, but the extract had no effect on the activation of this pro-apoptotic protein [
47]. The literature demonstrates that breast cancer cells can also be sensitive to parsley extract. For example, the root extract at a concentration of 500 μg/mL inhibited the DNA synthesis and metabolic activity of MCF712A and MCF7 estrogen receptor-positive benign and malignant mammary cells [
48]. Tang et al. have shown that dichloromethane extracts obtained from leaves and stems inhibit proliferation and migration of MCF7 cells [
19]. On dendritic cells, parsley extract had no anti- or pro-proliferative effect.
4. Materials and Methods
4.1. Extracts
Dry chamomile flowers (Matricaria chamomilla L.) were purchased from a Romanian cultivator (Iasi, Romania), the material was identified, and a voucher specimen (code Mc10/2016) was deposited at the Department of Pharmacognosy, “Grigore T. Popa” University of Medicine and Pharmacy Iasi, Romania. Flavonoids and polyphenolic acids were isolated from chamomile flowers by hydroalcoholic extraction (methanol 60%), in which the drug extract ratio (RDE) was 2.5 g of plant product per 100 mL of final extract. The fluid extract was concentrated to dry consistency with a Buchi rotary evaporator.
Parsley (Petroselinum crispum var. radicosum Miller) and celery (Apium graveolens var. radicosum L.) seeds were purchased from specialized providers, and their batch numbers were given to the voucher specimens (code Pc01/2016 for parsley and code Ag05/2016 for celery) which are currently kept along with the chamomile specimen.
After initial testing for certification of its identity, both parsley and celery were extracted with a methanolic solution for which the methanol/water ratio was 60:40. The extracts were obtained on a thermostatic water bath, starting with 2.5 g of powdered plant material. The final solution (100 mL) was leveled in a volumetric flask. After concentration in a rotary evaporator, the extracts were weighted and stored at 4 °C until further testing.
Apigenin ≥99% (HPLC) (CAS Number 520-36-5) was acquired from Sigma-Aldrich, Steinheim, Germany.
4.2. Sample Coding
For easier reference in the present paper, all extracts were coded as follows: MC for chamomile extract, P for parsley extract, and C for celery extract. The plant name has been checked against
http://www.theplantlist.org.
4.3. RP-UHPLC Quantification
UPLC was performed with a Thermo UltiMate3000 gradient chromatograph equipped with a quaternary pump controlled by Chromeleon interface, an autosampler, and multidiode array detector (DAD). Solvents were filtered using a Millipore system, and analysis was performed on an Accucore XL C18 column (150 × 4, 6 × 4). The mobile phase was acetonitrile (A) and water containing 0.1% acetic acid (B), and the gradient was 10–23% (A) in 5 min; 23% (A) isocratic for 10 min and then 23–35% (A) in 12 min; 35–70% (A) for 5 min. The injection volume was 20 µL, and scanning absorbance wavelengths from 240 to 520 nm, typical for phenols. The flow rate increased from 0.2 to 1 mL/min. HPLC grade solvents and double-distilled water were used in the chromatographic studies. All chromatographic experiments were performed at 25 °C.
All polyphenol standards of analytical grade were purchased from Sigma Chemical Co. (Steinheim, Germany) Triplicate injections were used for both samples, and standards and their calibration curves were obtained using the average value.
4.4. Spectrophotometric Quantification of Flavonoids
The quantification of total flavonoids from plant material employed its complexation properties with aluminum chloride (AlCl
3) according to known techniques [
49]. An aliquot of the dry extract was solubilized in 1.25 mL water and 0.075 mL 5% sodium nitrite (NaNO
2). After 6 min, 0.15 mL 10% AlCl
3 were vortexed together, and then 0.5 mL 1 M sodium hydroxide was added to the mixture. The intensity of the red color was measured at λ = 510 nm.
4.5. Total Phenols Quantification
Total phenolic content (TPC) of the extract was estimated according to the method described by Mircea et al. [
50]. The sample was prepared by dissolving 6.8 mg in 1 mL methanol and homogenized. Briefly, 400 μL of the sample was mixed with 0.2 mL Folin–Ciocalteu reagent, and 3.16 mL distilled water. After 5 min incubation, 0.6 mL 20% sodium carbonate solution (Na
2CO
3) was added and incubated for 2 h in the dark, at room temperature. The blank was prepared in the same manner, using water to replace the chamomile extract. The absorbance was determined using a UV–visible spectrophotometer at 765 nm. The standard curve of gallic acid was obtained using the same procedure.
4.6. Radical Scavenger Capacity
Radical scavenger capacity was assessed by two different tests: (a) DPPH (2,2-diphenyl-1-picryl-hydrazyl-hydrate) free radical method is an antioxidant assay based on electron transfer that produces a violet solution in ethanol. A color change is noted if antioxidants are present in the analyzed sample at 517 nm; (b) Reduction of blue-green ABTS radical colored solution by hydrogen-donating antioxidants was measured by the suppression of its characteristic long wave (734nm) absorbance spectrum. Both methods were previously described by Cioanca et al. [
51]. Positive controls were obtained from standard quercetin and gallic acid of high purity grade. The scavenging activity (A%) was calculated according to the formula A% = 100 × (Econtrol − Esample)/(Econtrol), where Econtrol is the absorbance of the control solution, whereas Esample is the absorbance of the solution measured in the presence of plant extracts or standards.
4.7. Iron Chelation Potential
The intensity given by ferrozine-Fe2+ complexes in the presence of iron chelating agents from plant extract is measured at 562 nm. Briefly, 2 mM iron chloride solution was added to a test tube containing a variable amount of investigated extracts (0.078125–10 mg/mL), DMSO (400 µL), and 5 mM ferrozine (80 µL). The standard was represented by caffeic acid of high purity grade (Sigma). By linearly plotting the inhibition percentage against sample concentration, we obtained EC50 (sample concentration providing 50% inhibition of ferrozine).
4.8. Lipoxygenase Inhibition Activity
An aliquot (0.05 mL) of 15-lipoxygenase in borate buffer (pH 9) was mixed with 0.05 mL of the sample solution in DMSO (various concentrations); 10 min later, 2 mL of 0.16 mM linoleic acid borate buffer were added, and the absorbances were registered at 234 nm for 90 s [
26]. Inhibition of 15-lipoxygenase was established with the formula: % inhibition = (AEFI − AECI) × 100/AEFI; AEFI is the difference of the enzyme absorbance without inhibitor at 90 and 30 s, while AECI represents the same difference of the enzyme–inhibitor mixture.
4.9. Cell Culture and Preparation
The A375 human melanoma cell line (ECACC; Sigma Aldrich origin Japan stored UK) was cultured in minimal essential medium supplemented with 10% fetal bovine serum, 1% antibiotic/antimycotic (penicillin/streptomycin) mixture, and 1% non-essential amino acids, in humidified air containing 5% CO
2 at 37 °C. All the medium and supplements were purchased from Lonza Group Ltd. (Basel, Switzerland). Human dendritic cells were differentiated from isolated PBMCs from buffy coats according to Nair et al. [
52]. In brief, density centrifugation was performed using Ficoll (GE Healthcare, Uppsala, Sweden). Isolated PBMCs have been plated at a density of 2 × 10
8 cells per dish, and supernatant has been discarded upon 2 h of plastic adherence. Subsequently, cells were differentiated in RPMI 1640 GlutaMax medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FCS, 100 IU/mL penicillin, 100 µg/mL streptomycin, 10 mM HEPES (Sigma-Aldrich, Steinheim, Germany), 1 mM sodium pyruvate, and 50 µM 2-β-ME (Thermo Fisher Scientific, Massachusetts, MA, USA) supplemented with 40 ng/mL recombinant human GM-CSF (Peprotech, Rocky Hill, NJ, USA), and human IL4 (Peprotech, Rocky Hill, NJ, USA), with an additional medium exchange after 4 days. Differentiated cells were harvested by cell scraping, and transferred to 6-well plates for further experiments, or to tissue-treated 8-well chambered coverslides (Ibidi, Martinsried, Germany) for fluorescence microscopy staining. The supernatants were analyzed by ELISAs for TNF-alpha, IL-10, and IL-6 (R&D Systems, Wiesbaden, Germany), according to the manufacturer’s manual.
4.10. Antiproliferative Activity Assays
The growth-inhibitory activity of the extracts against A375 cells was determined by the well-known MTT assay. Experiments were carried out in the same way as previously described [
53]. Briefly, cells were plated into 96-well plates at a density of 5000 cells/well, and incubated with selected concentrations of extracts (10, 30, 60 µg/mL) for 72 h. After this period, 5 mg/mL MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution was added, followed by another 4 h of incubation. Dimethyl sulfoxide (DMSO) was used in order to dissolve the crystals of precipitated formazan. The absorbance was measured using a microplate reader at 545 nm. Wells with vehicle-treated cells were used as control [
54].
The XTT Cell Viability Assay (Thermo Fischer Scientific) was used, according to the manufacturer’s manual, on human dendritic cells. In brief, the final XTT solution was added to cell culture wells with cells, or medium only as a control. Upon 45 min incubation time at 5% CO2 and 37 °C, aliquots of the cells were analyzed in flat 96-well plates (Greiner, Frickenhausen, Germany) at 460, and normalized to 650 nm. In advance, cells have been stimulated with vehicle, or tested extracts in the presence or absence of LPS for 48 h at 60 µg/mL. Cell number was obtained after XTT assay and normalized to 104 cells.
4.11. Cell Cycle Analysis by Flow Cytometry
In order to measure the distribution of DNA between the phases of the cell cycle following incubation with selected extracts, flow cytometric analysis was performed [
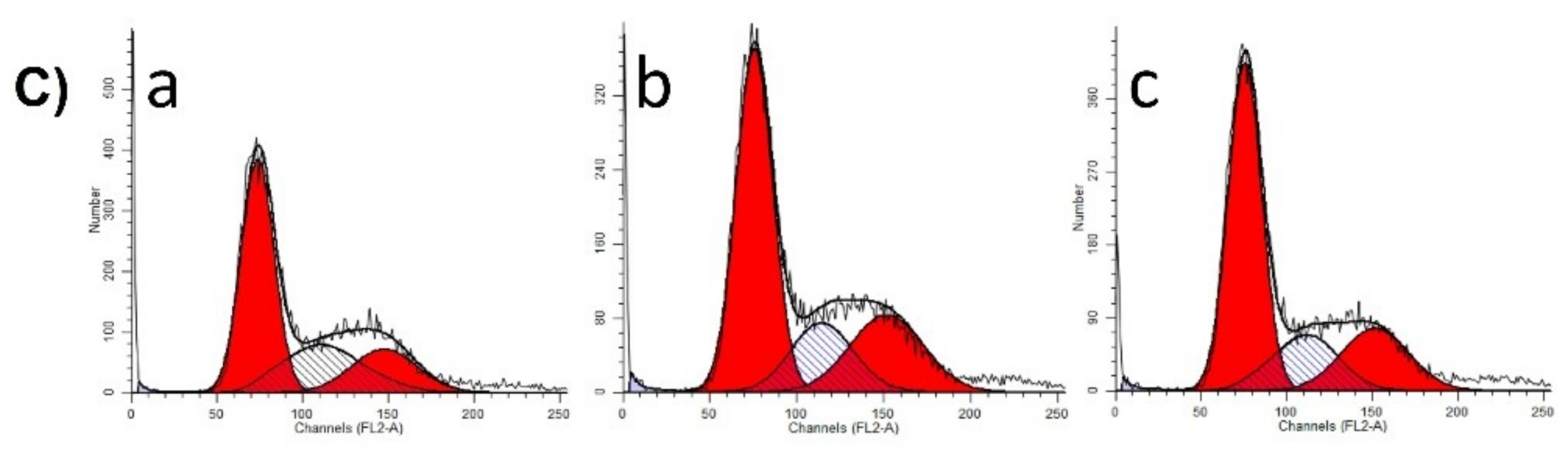
55]. A375 human melanoma cells were seeded into 6-well plates at a density of 300,000–400,000 cells/well. After 24 h of treatment with selected extract concentrations (30 or 60 µg/mL), the cells were washed twice with cold phosphate-buffered saline (PBS), harvested with trypsin, and centrifuged at 1500 rpm for 10 min. After a washing step, the cells were fixed in 1 mL 70% ethanol, at −20 °C, for 30 min. Samples were stained with 1.0 mL dye solution containing 0.02 mg/mL RNase A, 0.1 mg/mL PI, 0.003 mL/mL Triton-X, and 1.0 mg/ mL sodium citrate in distilled water, and the mixture was incubated in the dark for 60 min at room temperature. Cells were analyzed by a Partec CyFlow instrument (Partec GmbH, Münster, Germany). In each analysis, 20,000 events were recorded, and the percentages of the cells in the different cell cycle phases (subG1, G1, S and G2/M) were determined by using ModFit Software (Version 5 Verity Software House, Topsham, ME, USA).
4.12. Determination of the In Situ Caspase Activity
For the determination of the in situ caspase 3 activity, a commercially available colorimetric assay (Casp-3-C, Sigma-Aldrich, Budapest, Hungary) was performed. Cells (107) were seeded in tissue culture flasks for control, and 1012 cells were seeded for treatment. On the second day, the cells were treated with selected concentrations of extracts (30 or 60 µg/mL). After incubation for 48 h, cells were counted, centrifuged, and washed with PBS. The cells were resuspended in kit lysis buffer at a concentration of 107 cells per 100 mL, and incubated on ice for 20 min. The lysate was centrifuged, and the supernatant was used. The protein concentration of the lysate was measured with the Pierce BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA), and equal amounts of protein were used for the measurement. In accordance with the manufacturer’s protocol, 5.0 µL portions of treated and untreated cell lysates were incubated with 10 µL selective caspase 3 substrate (acetyl–Asp–Glu–Val–Asp–p-nitroanilide) in a final volume of 100 µL in assay buffer, in 96-well plates. After an overnight incubation at 37 °C, the absorbance of p-nitroaniline was measured at 405 nm with a microplate reader. Comparison of the absorbances of the p-nitroaniline from treated and untreated samples allowed determination of the fold increase in caspase 3 activity.
4.13. Annexin V/PI Assay
The evaluation of early and late apoptosis, as well as necrosis, was conducted with selected concentrations of extracts (30, 60 µg/mL). Cells at 5 × 105 cells/well were seeded into 6-well plate (Greiner bio-one, Gmbh, Germany) and left overnight, in order to attach to the bottom of the plate. After 24 h, the cultured medium (DMEM) was removed, and a fresh medium containing the tested extracts was added. Untreated cells were used as control; cells treated with DMSO were used as solvent control. After 72 h, cells were trypsinized. Annexin V-FITC combined with propidium iodide (PI) kit (Invitrogen, ThermoFisher, Vienna, Austria) was used in cell death flow cytometric studies (apoptosis) following the manufacturer’s protocol. Briefly, 2–5 × 105 cells were washed twice in 1 × Annexin V Binding Buffer, centrifuged at 1500 rpm for 5 min, resuspended in the binding buffer and incubated with 5 μL of Annexin V-FITC for 15 min in the dark. After washing the cells with 200 μL specific binding buffer and centrifugation, the cell pellet was resuspended in 190 μL binding buffer, and 10 μL of PI solution was added immediately prior to analysis by flow cytometry.
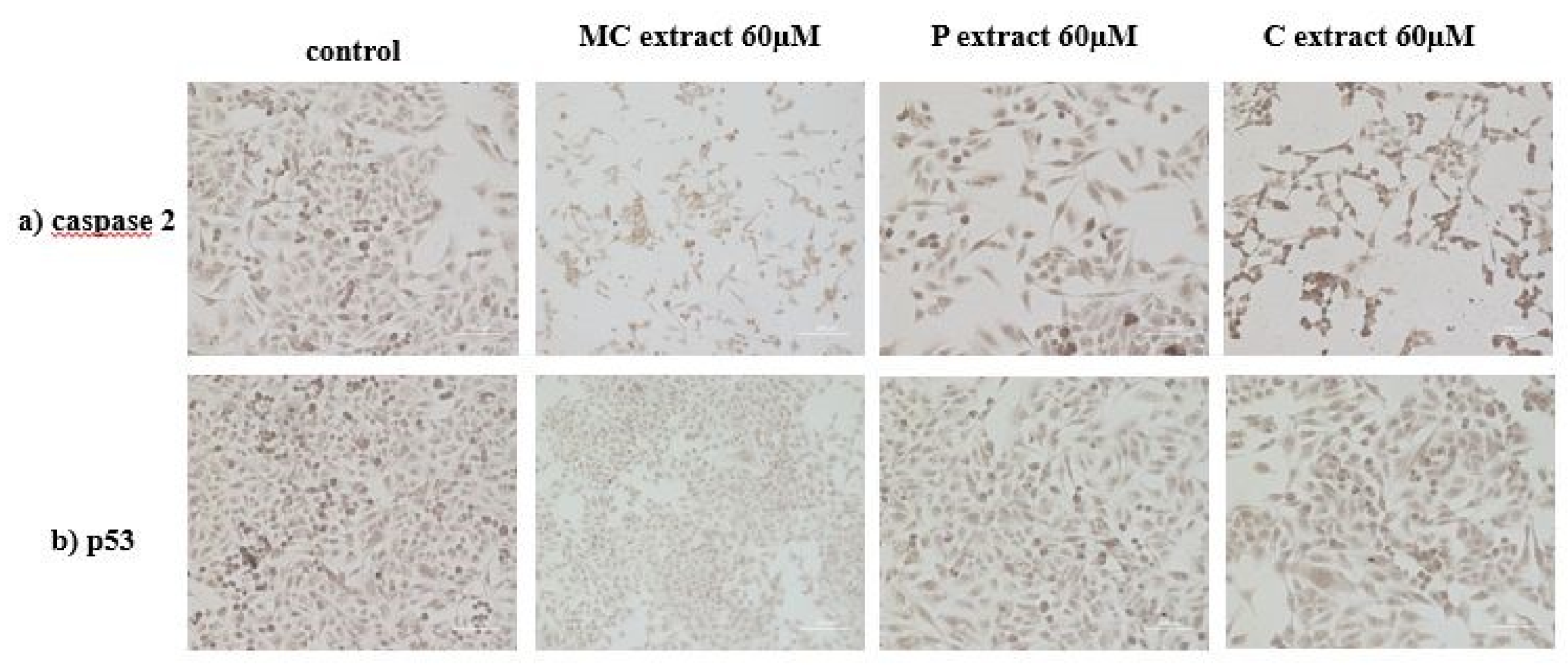
4.14. Immunocytochemistry
Immunocytochemistry was performed in order to evaluate the expression of protein caspase 2 and cellular tumor antigen p5. Cells were plated at a density of 10,000 cells/cm2 in 4-well glass chamber slides (Nalgene Nunc International, New York, NY, USA) and expanded for 24 h in culture medium. After 24 h, the tested extracts were added. Untreated cells were used as control. After a 72 h period of incubation with tested extracts, cells were prepared for immunocytochemical staining. The culture medium was removed, and cells were washed, and fixed and permeabilized for 10 min with methanol (Sigma-Aldrich) at −20 °C. After fixation and permeabilization, cells were analyzed for the expression of the proteins of interest, anti-h/m caspase 2 (mCaspase 2 affinity purified rabbit IgG) (R&D Systems, Abingdon, UK) and p53 (monoclonal mouse anti-human) (Dako, Carpinteria, CA, USA). The staining protocol used the secondary biotinylated antibody binding and substrate addition (AEC) (Dako EnVision™ + System-HRP, Dako, Carpinteria, CA, USA) following the manufacturer’s protocol. After counterstaining with hematoxylin solution (Hematoxylin, Mayer’s Lillie’s Modification, Dako, Carpinteria, CA, USA) for 10 min, and washing with tap water, the slides were mounted in an aqueous mounting media (Crystal/Mount™, Biomeda, Foster City, CA, USA). Microscopy analysis was performed on a Zeiss Axio Observer D1 microscope.
4.15. Fluorescence Microscopy
Cells were grown on tissue-treated 8-well chambered coverslides (Ibidi, Martinsried, Germany), cultured, and fixed as described before [
56]. DAPI (4′,6-diamidine-2′-phenylindole dihydrochloride) solution (Roche Diagnostics, Mannheim, Germany) and phalloidin Alexa Fluor 488 solution (Thermo Fisher) was applied for 1 h. Confocal laser scanning microscopy was performed with a Zeiss LSM510 Meta system equipped with an inverted Observer Z1 microscope and a Plan-Apochromat 63 ×/1.4 oil immersion objective (Carl Zeiss MicroImaging GmbH, Göttingen, Germany).
4.16. Statistics
The Prism software package GraphPad Prism 5.01 (GraphPad Software, San Diego, CA, USA) was used for data collection and presentation. The data ranged from three to five separate experiments, and are presented as mean ± SD. Statistical significance was assessed by one-way ANOVA with Newman–Keuls post-test for comparison of multiple groups, where *, **, ***, and **** indicate p < 0.05, p < 0.01, p < 0.001, and p < 0.0001, respectively, compared to the control group.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}