Mycobacterium tuberculosis ClpC1 N-Terminal Domain Is Dispensable for Adaptor Protein-Dependent Allosteric Regulation



Abstract
:
1. Introduction
2. Results
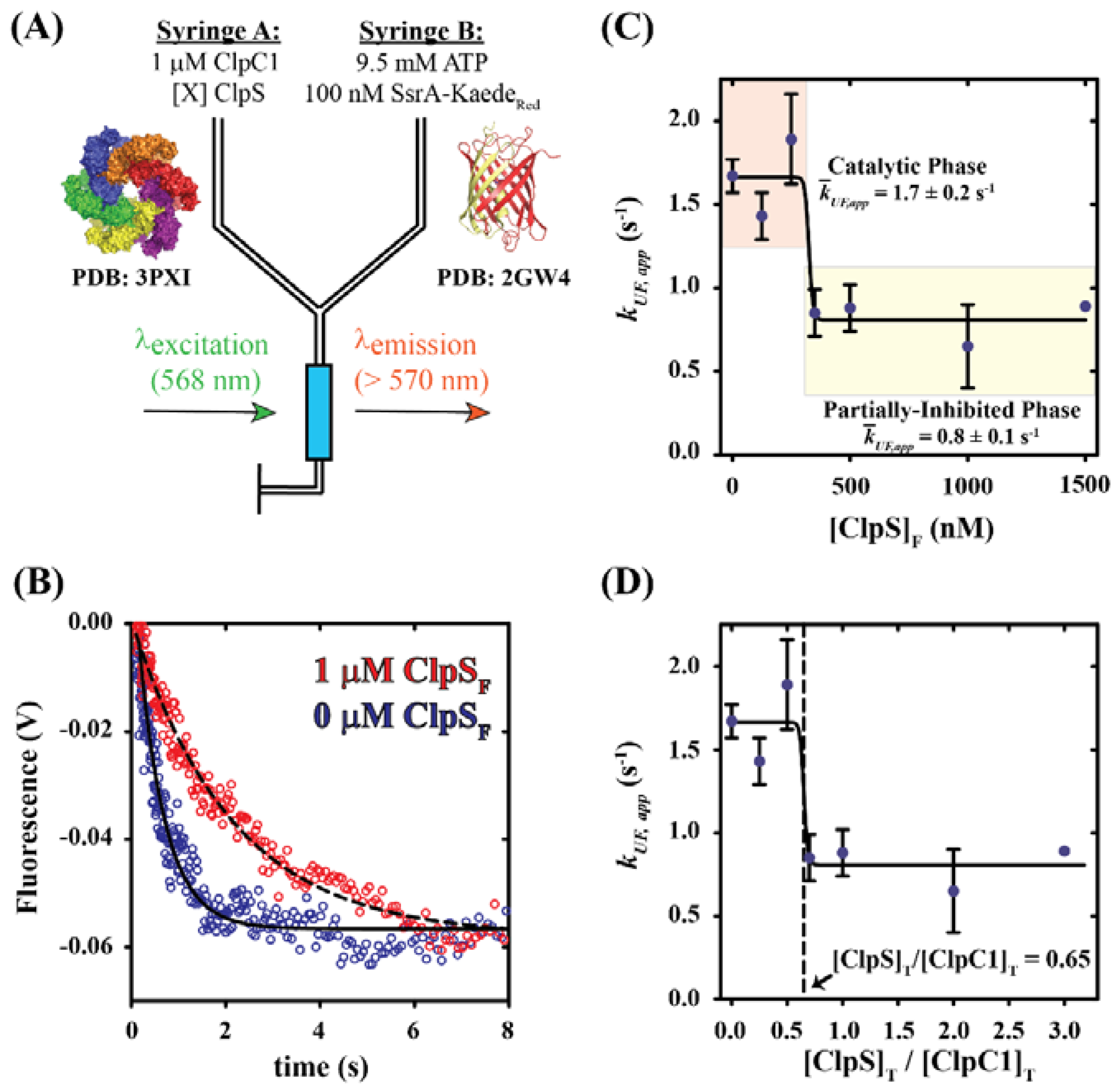
2.1. Dependence of Apparent Unfolding Rate Constant on [ClpS]
2.2. N-Terminal Domain of ClpC1 Is Dispensable for ClpS-Mediated Inhibition of Protein Unfolding
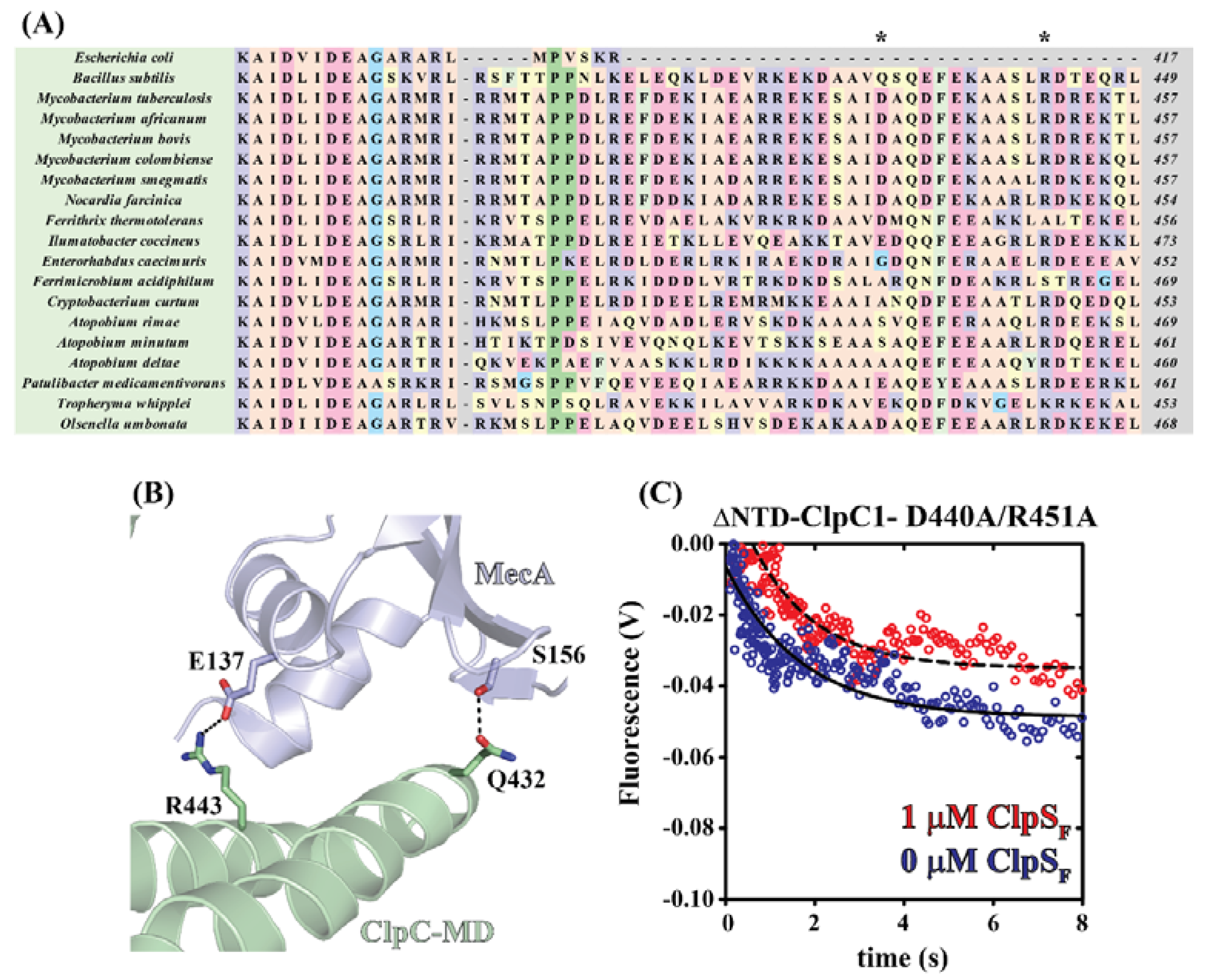
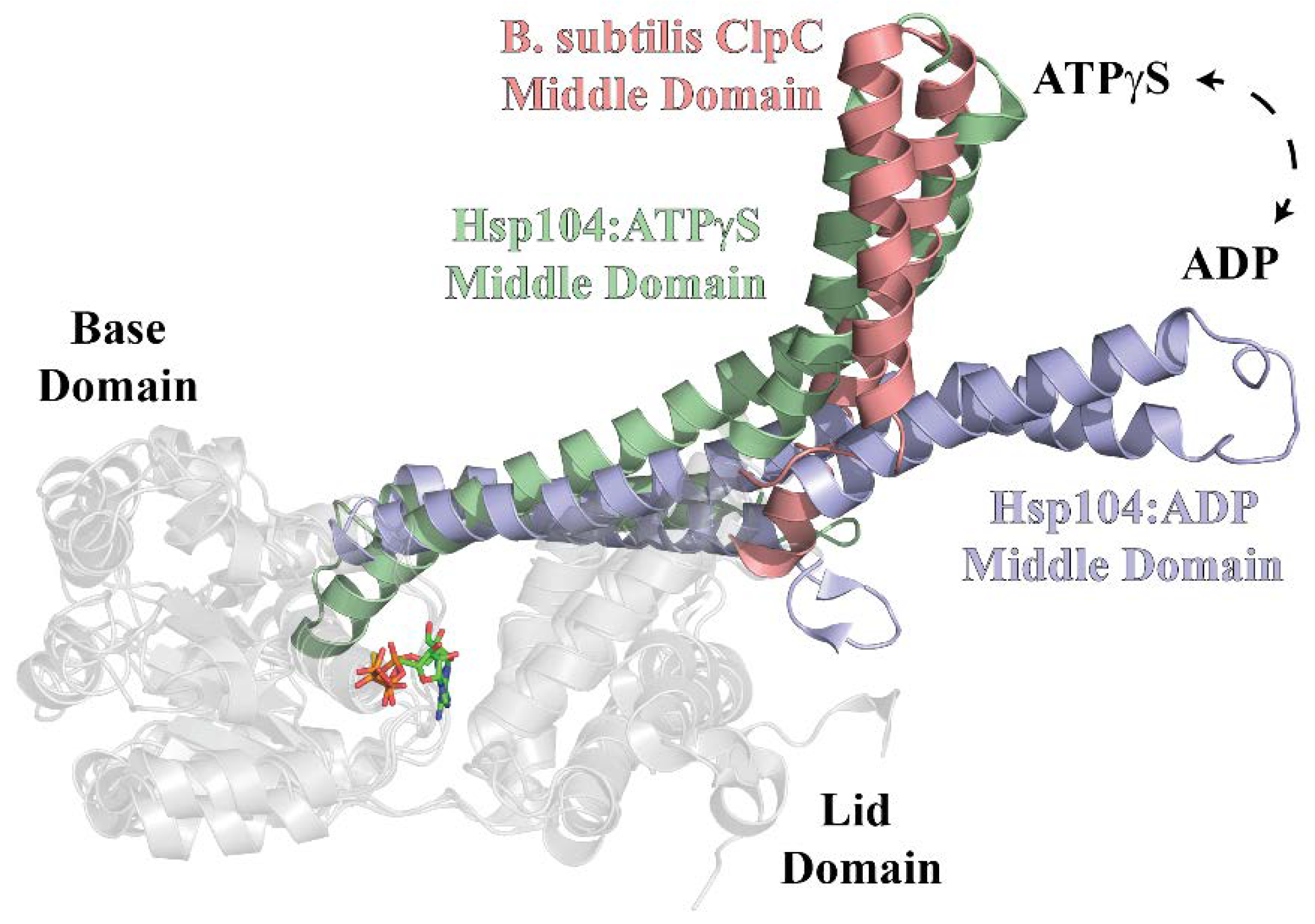
2.3. Mycobacterial ClpS Primary Sequence Analysis Reveals ClpC1-MD (Middle Domain) Binding Features
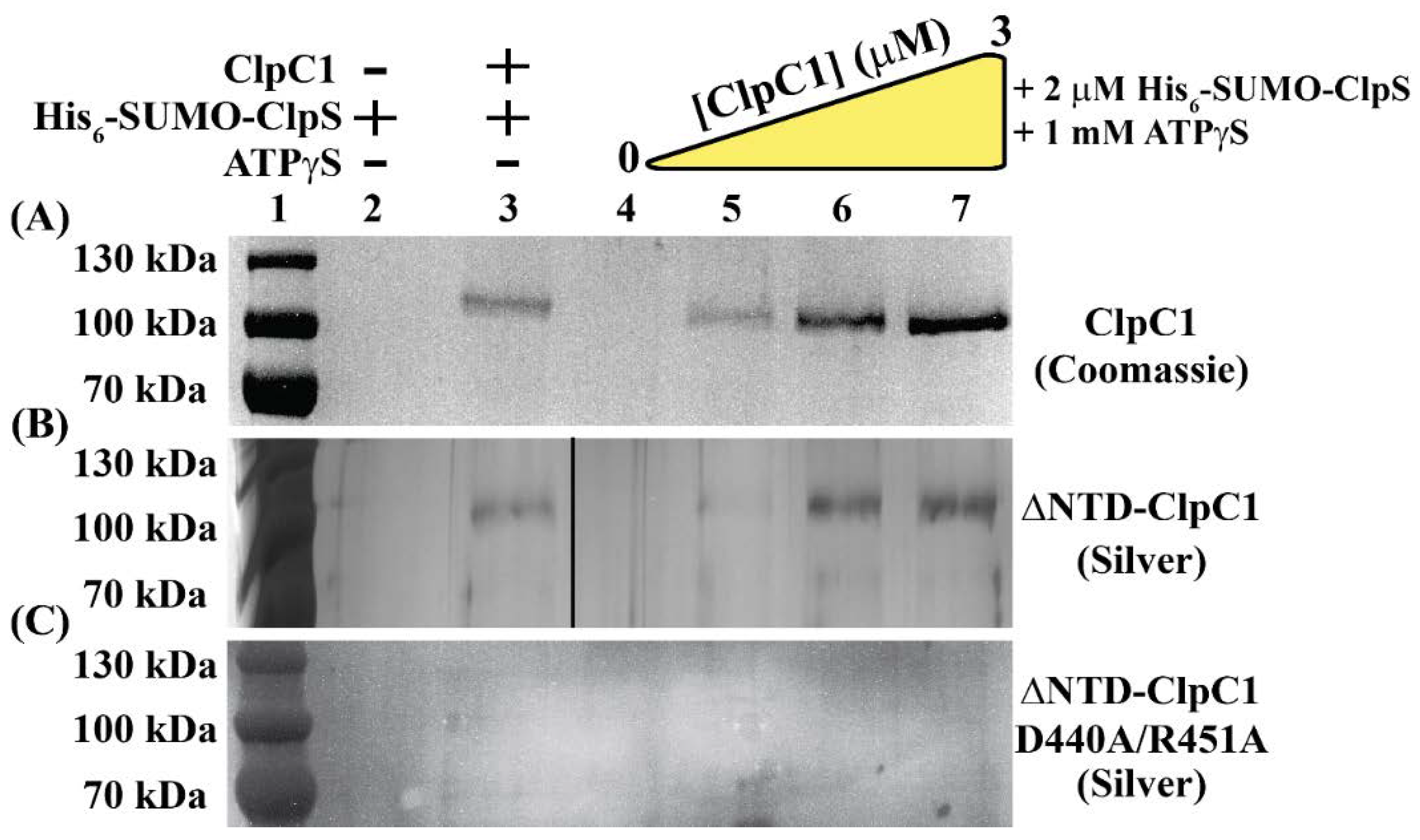
2.4. Identification of ClpC1 Surfaces Involved in Complex Formation
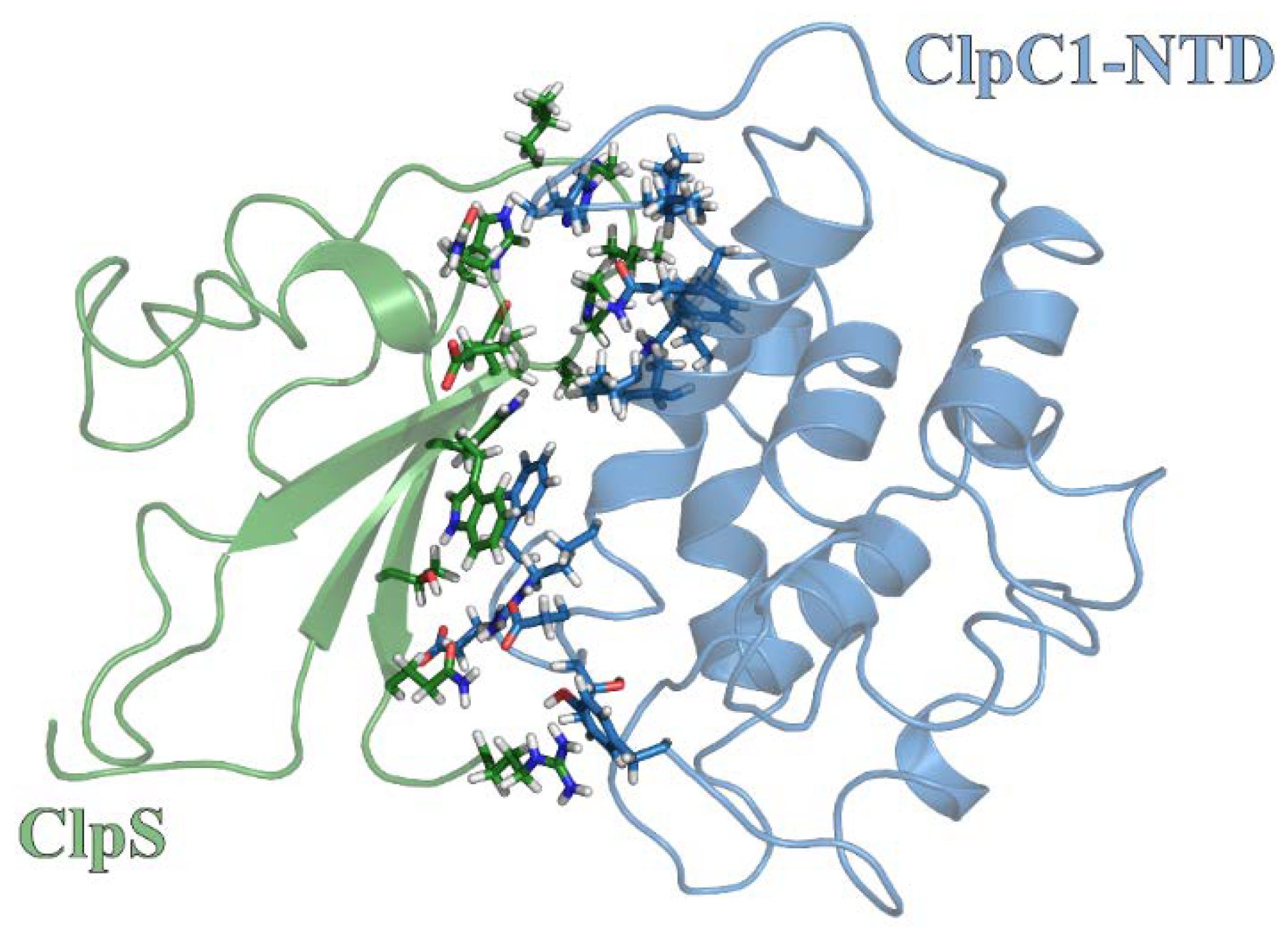
2.5. Molecular Dynamics Simulations Predict Unique ClpS Interface Involved in Complex Formation
3. Discussion
ClpC1 Binding of ClpS May Involve Multiple Binding Surfaces
4. Materials and Methods
4.1. Materials
4.2. Protein Expression and Purification
4.3. Methods
4.3.1. Stopped-Flow Fluorescence Assay
4.3.2. Ni-NTA Pulldown Experiments
4.3.3. Structure Preparation
4.3.4. Docking Process
4.3.5. Molecular Dynamics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NTD | N-terminal domain |
| MD | Middle domain |
| NTP | Nucleoside triphosphate |
| ATP | Adenosine triphosphate |
| AAA+ | ATPases associated with various cellular activities |
References
- Gottesman, S.; Maurizi, M.R. Regulation by proteolysis: Energy-dependent proteases and their targets. Microbiol. Rev. 1992, 56, 592–621. [Google Scholar] [PubMed]
- Gottesman, S.; Wickner, S.; Maurizi, M.R. Protein quality control: Triage by chaperones and proteases. Genes Dev. 1997, 11, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Baker, T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef] [PubMed]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol. 2016, 14, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Alhuwaider, A.A.H.; Dougan, D.A. AAA+ machines of protein destruction in mycobacteria. Front. Mol. Biosci. 2017, 4, 49. [Google Scholar] [CrossRef] [PubMed]
- Marina, A.F.; Laura, I.K.; Andrea, G.; Julia, S.G.; Hector, R.M.; María de, P.S.; Angel, A.C.; Fabiana, B. Virulence factors of the Mycobacterium tuberculosis complex. Virulence 2013, 4, 3–66. [Google Scholar] [CrossRef] [Green Version]
- Karzai, A.W.; Roche, E.D.; Sauer, R.T. The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nat. Struct. Biol. 2000, 7, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Lucius, A.L. Examination of the polypeptide substrate specificity for Escherichia coli ClpA. Biochemistry 2013, 52, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Dougan, D.A.; Reid, B.G.; Horwich, A.L.; Bukau, B. ClpS, a substrate modulator of the ClpAP machine. Mol. Cell 2002, 9, 673–683. [Google Scholar] [CrossRef]
- Zeth, K.; Ravelli, R.B.; Paal, K.; Cusack, S.; Bukau, B.; Dougan, D.A. Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA. Nat. Struct. Biol. 2002, 9, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.Y.; Sauer, R.T.; Baker, T.A. Distinct structural elements of the adaptor ClpS are required for regulating degradation by ClpAP. Nat. Struct. Mol. Biol. 2008, 15, 288–294. [Google Scholar] [CrossRef] [PubMed]
- De Donatis, G.M.; Singh, S.K.; Viswanathan, S.; Maurizi, M.R. A single ClpS monomer is sufficient to direct the activity of the ClpA hexamer. J. Biol. Chem. 2010, 285, 8771–8781. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, J.; Schlothauer, T.; Dougan, D.A.; Lilie, H.; Tischendorf, G.; Mogk, A.; Bukau, B.; Turgay, K. Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J. 2006, 25, 1481–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlothauer, T.; Mogk, A.; Dougan, D.A.; Bukau, B.; Turgay, K. MecA, an adaptor protein necessary for ClpC chaperone activity. Proc. Natl. Acad. Sci. USA 2003, 100, 2306–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroni, M.; Franke, K.B.; Maurer, M.; Jäger, J.; Hantke, I.; Gloge, F.; Linder, D.; Gremer, S.; Turgay, K.; Bukau, B.; et al. Regulatory coiled-coil domains promote head-to-head assemblies of AAA+ chaperones essential for tunable activity control. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trentini, D.B.; Suskiewicz, M.J.; Heuck, A.; Kurzbauer, R.; Deszcz, L.; Mechtler, K.; Clausen, T. Arginine phosphorylation marks proteins for degradation by a Clp protease. Nature 2016, 539, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Tryggvesson, A.; Stahlberg, F.M.; Mogk, A.; Zeth, K.; Clarke, A.K. Interaction specificity between the chaperone and proteolytic components of the cyanobacterial Clp protease. Biochem. J. 2012, 446, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tryggvesson, A.; Ståhlberg, F.M.; Töpel, M.; Tanabe, N.; Mogk, A.; Clarke, A.K. Characterization of ClpS2, an essential adaptor protein for the cyanobacterium Synechococcus elongatus. FEBS Lett. 2015, 589, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- Leodolter, J.; Warweg, J.; Weber-Ban, E. The Mycobacterium tuberculosis ClpP1P2 Protease Interacts Asymmetrically with Its ATPase Partners ClpX and ClpC1. PLoS ONE 2015, 10, e0125345. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.R.; Sauer, R.T. Substrate delivery by the AAA+ ClpX and ClpC1 unfoldases activates the mycobacterial ClpP1P2 peptidase. Mol. Microbiol. 2014, 93, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, N.P.; Sikriwal, D.; Rath, P.; Choudhary, R.K.; Batra, J.K. Mycobacterium tuberculosis ClpC1: Characterization and role of the N-terminal domain in its function. FEBS J. 2008, 275, 6149–6158. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Chaudhary, H.; Marsee, J.D. Phylogenetic analysis predicts structural divergence for proteobacterial ClpC proteins. J. Struct. Biol. 2018, 201, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, I.; Mizuno, H.; Tong, K.I.; Furuta, T.; Tanaka, F.; Yoshimura, M.; Miyawaki, A.; Ikura, M. Crystallographic evidence for water-assisted photo-induced peptide cleavage in the stony coral fluorescent protein Kaede. J. Mol. Biol. 2007, 372, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Mal, T.K.; Tong, K.I.; Ando, R.; Furuta, T.; Ikura, M.; Miyawaki, A. Photo-induced peptide cleavage in the green-to-red conversion of a fluorescent protein. Mol. Cell 2003, 12, 1051–1058. [Google Scholar] [CrossRef]
- Ando, R.; Hama, H.; Yamamoto-Hino, M.; Mizuno, H.; Miyawaki, A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 12651–12656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glynn, S.E.; Nager, A.R.; Baker, T.A.; Sauer, R.T. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat. Struct. Mol. Biol. 2012, 19, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendar, B.; Lucius, A.L. Molecular mechanism of polypeptide translocation catalyzed by the Escherichia coli ClpA protein translocase. J. Mol. Biol. 2010, 399, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Lucius, A.L.; Miller, J.M.; Rajendar, B. Application of the sequential n-step kinetic mechanism to polypeptide translocases. Methods Enzymol. 2011, 488, 239–264. [Google Scholar] [CrossRef] [PubMed]
- Van Holde, K.E.; Johnson, W.C.; Ho, P.S. Principles of Physical Biochemistry; Prentice Hall: Upper Saddle River, NJ, USA, 1998; p. 657. [Google Scholar]
- Vasudevan, D.; Rao, S.P.; Noble, C.G. Structural basis of mycobacterial inhibition by cyclomarin A. J. Biol. Chem. 2013, 288, 30883–30891. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Mei, Z.; Qi, Y.; Yan, C.; Hu, Q.; Wang, J.; Shi, Y. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature 2011, 471, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Esser, L.; Singh, S.K.; Maurizi, M.R.; Xia, D. Crystal structure of the heterodimeric complex of the adaptor, ClpS, with the N-domain of the AAA+ chaperone, ClpA. J. Biol. Chem. 2002, 277, 46753–46762. [Google Scholar] [CrossRef] [PubMed]
- Gates, S.N.; Yokom, A.L.; Lin, J.; Jackrel, M.E.; Rizo, A.N.; Kendsersky, N.M.; Buell, C.E.; Sweeny, E.A.; Mack, K.L.; Chuang, E.; et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 2017, 357, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moolenaar, G.F.; Franken, K.L.; Dijkstra, D.M.; Thomas-Oates, J.E.; Visse, R.; van de Putte, P.; Goosen, N. The C-terminal region of the UvrB protein of Escherichia coli contains an important determinant for UvrC binding to the preincision complex but not the catalytic site for 3’-incision. J. Biol. Chem. 1995, 270, 30508–30515. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, G.F.; Franken, K.L.; van de Putte, P.; Goosen, N. Function of the homologous regions of the Escherichia coli DNA excision repair proteins UvrB and UvrC in stabilization of the UvrBC-DNA complex and in 3’-incision. Mutat. Res. 1997, 385, 195–203. [Google Scholar] [CrossRef]
- Xia, D.; Esser, L.; Singh, S.K.; Guo, F.; Maurizi, M.R. Crystallographic investigation of peptide binding sites in the N-domain of the ClpA chaperone. J. Struct. Biol. 2004, 146, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.K.; Riwanto, M.; Sambandamurthy, V.; Roggo, S.; Miault, C.; Zwingelstein, C.; Krastel, P.; Noble, C.; Beer, D.; Rao, S.P.; et al. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem. Int. Ed. Engl. 2011, 50, 5889–5891. [Google Scholar] [CrossRef] [PubMed]
- Gavrish, E.; Sit, C.S.; Cao, S.; Kandror, O.; Spoering, A.; Peoples, A.; Ling, L.; Fetterman, A.; Hughes, D.; Bissell, A.; et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 2014, 21, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Suh, J.W. Anti-tuberculosis lead molecules from natural products targeting Mycobacterium tuberculosis ClpC1. J. Ind. Microbiol. Biotechnol. 2016, 43, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Mossessova, E.; Lima, C.D. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 2000, 5, 865–876. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed]
- Schuenemann, V.J.; Kralik, S.M.; Albrecht, R.; Spall, S.K.; Truscott, K.N.; Dougan, D.A.; Zeth, K. Structural basis of N-end rule substrate recognition in Escherichia coli by the ClpAP adaptor protein ClpS. EMBO Rep. 2009, 10, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE). Available online: https://www.chemcomp.com/MOE-Molecular_Operating_Environment.htm (accessed on 13 November 2018).
- Protonate 3D: Assignment of Macromolecular Protonation State and Geometry. Available online: https://www.chemcomp.com/journal/proton.htm (accessed on 13 November 2018).
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Kalé, L.S.R.; Bh, M.; Brunner, R.; Gursoy, A.; Krawetz, N.; Phillips, J.; Shinozaki, A.; Varadarajan, K.; Schulten, K. NAMD2: Greater Scalability for Parallel Molecular Dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar] [CrossRef] [Green Version]
- Lamoureux, G.H.E.; Vorobyov, I.V.; Roux, B.; MacKerell, A.D. A Polarizable Model of Water for Molecular Dynamics Simulations of Biomolecules. Chem. Phys. Lett. 2006, 418, 245–249. [Google Scholar] [CrossRef]
- Martyna, G.J.; Douglas, J.T.; Klein, M.L. Constant-pressure molecular-dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.Z.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant-pressure molecular-dynamics simulation: The langevin piston method. J. Chem. Phys. 1995, 103, 4613. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An N.Log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [ClpS]F (nM) | kUF,app (s−1) | |
|---|---|---|
| Full-Length ClpC1 | ∆NTD-ClpC1 | |
| 0 | 1.7 ± 0.1 | 2.4 ± 1.1 |
| 125 | 1.4 ± 0.1 | |
| 250 | 1.9 ± 0.3 | 1.4 ± 0.1 |
| 350 | 0.9 ± 0.1 | |
| 500 | 0.9 ± 0.1 | |
| 1000 | 0.7 ± 0.3 | 0.9 ± 0.2 |
| 1500 | 0.89 ± 0.03 | |
| ClpC1 | ClpS | Energy | Dist |
|---|---|---|---|
| Phe2 | Trp33 | −4.802 | 3.95 |
| Phe2 | Asp34 | −0.118 | 4.26 |
| Phe2 | Trp94 | −2.193 | 3.95 |
| Phe2 | Thr96 | −0.877 | 4.19 |
| Glu3 | Thr96 | −1.604 | 4.16 |
| Thr6 | Arg101 | −0.128 | 4.21 |
| Asp7 | Trp33 | −0.252 | 3.87 |
| Asp7 | Gln98 | −3.051 | 3.99 |
| Asp7 | Arg101 | 0.018 | 4.20 |
| Arg10 | Trp33 | −5.015 | 3.93 |
| Arg10 | Gln98 | −1.263 | 3.52 |
| Val13 | Asp35 | −0.257 | 4.34 |
| Val13 | Pro36 | −1.65 | 4.09 |
| Val14 | Glu68 | −0.466 | 4.35 |
| Val14 | Gly69 | −0.605 | 4.02 |
| Gln17 | Pro36 | 1.15 | 4.19 |
| Gly76 | His66 | −0.244 | 3.89 |
| Gly76 | Asn67 | 0.098 | 3.92 |
| His77 | Pro36 | 1.52 | 3.94 |
| His77 | Val37 | 0.391 | 4.21 |
| His77 | Asn38 | −0.334 | 4.02 |
| His77 | Leu39 | 0.291 | 4.29 |
| His77 | His66 | 1.173 | 3.83 |
| Ile78 | Pro36 | −0.065 | 3.94 |
| Pro79 | Val37 | −0.877 | 3.71 |
| Pro79 | Asn38 | 0.5 | 4.47 |
| Phe80 | Pro36 | −1.774 | 4.02 |
| Phe80 | Val37 | −2.005 | 3.99 |
| Lys85 | Asp35 | −22.528 | 3.47 |
| Lys85 | Pro36 | −7.72 | 3.62 |
| Lys85 | Val37 | −1.107 | 4.02 |
| Tyr145 | Arg101 | −0.077 | 3.98 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marsee, J.D.; Ridings, A.; Yu, T.; Miller, J.M. Mycobacterium tuberculosis ClpC1 N-Terminal Domain Is Dispensable for Adaptor Protein-Dependent Allosteric Regulation. Int. J. Mol. Sci. 2018, 19, 3651. https://doi.org/10.3390/ijms19113651
Marsee JD, Ridings A, Yu T, Miller JM. Mycobacterium tuberculosis ClpC1 N-Terminal Domain Is Dispensable for Adaptor Protein-Dependent Allosteric Regulation. International Journal of Molecular Sciences. 2018; 19(11):3651. https://doi.org/10.3390/ijms19113651
Chicago/Turabian StyleMarsee, Justin D., Amy Ridings, Tao Yu, and Justin M. Miller. 2018. "Mycobacterium tuberculosis ClpC1 N-Terminal Domain Is Dispensable for Adaptor Protein-Dependent Allosteric Regulation" International Journal of Molecular Sciences 19, no. 11: 3651. https://doi.org/10.3390/ijms19113651
APA StyleMarsee, J. D., Ridings, A., Yu, T., & Miller, J. M. (2018). Mycobacterium tuberculosis ClpC1 N-Terminal Domain Is Dispensable for Adaptor Protein-Dependent Allosteric Regulation. International Journal of Molecular Sciences, 19(11), 3651. https://doi.org/10.3390/ijms19113651