Hsp90 Stabilizes SIRT1 Orthologs in Mammalian Cells and C. elegans


Abstract
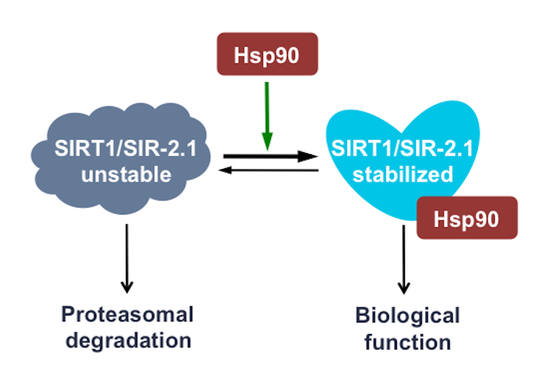
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
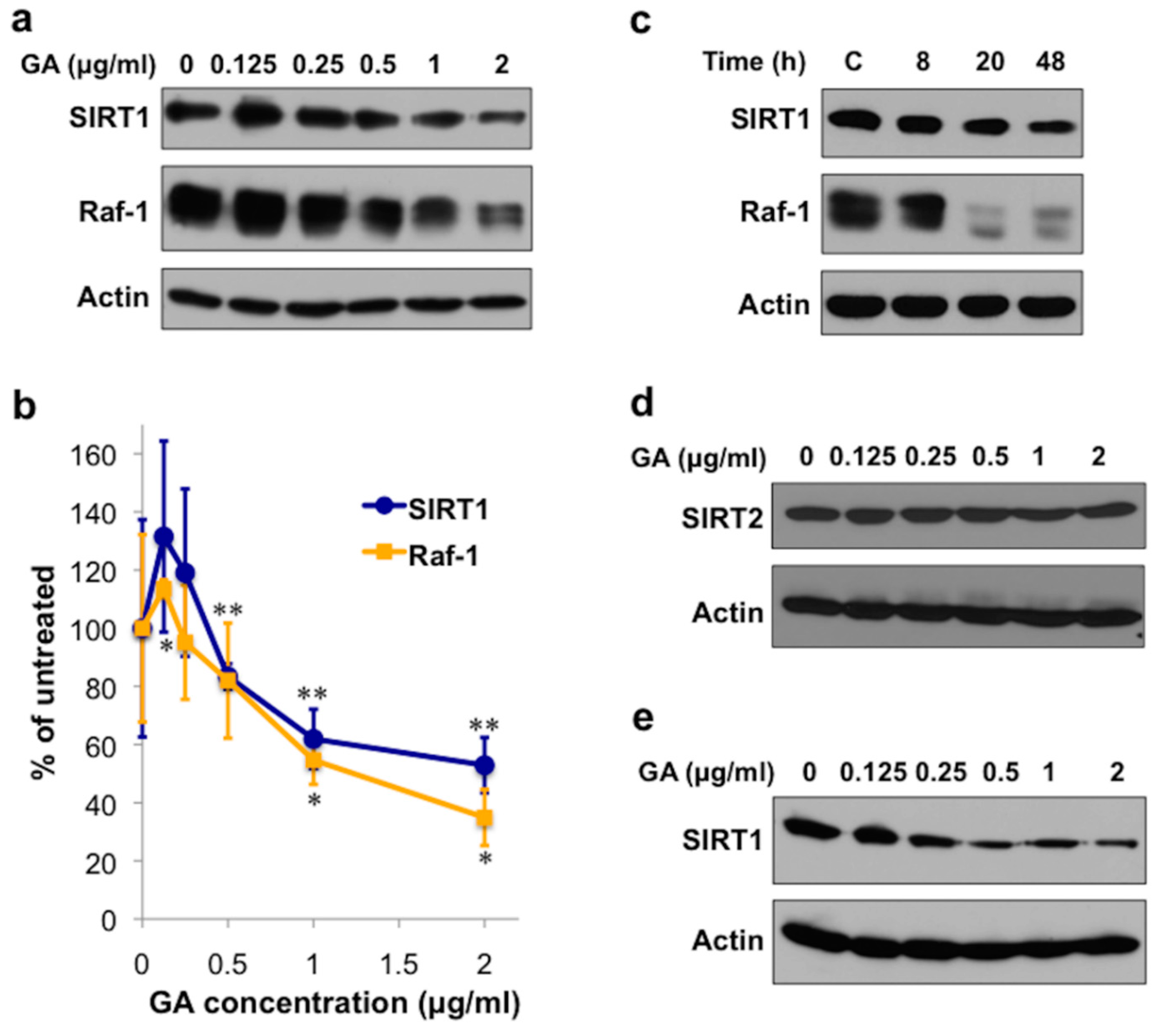
2.1. The Hsp90 Inhibitor Geldanamycin Depletes SIRT1 Protein in Mammalian Cells
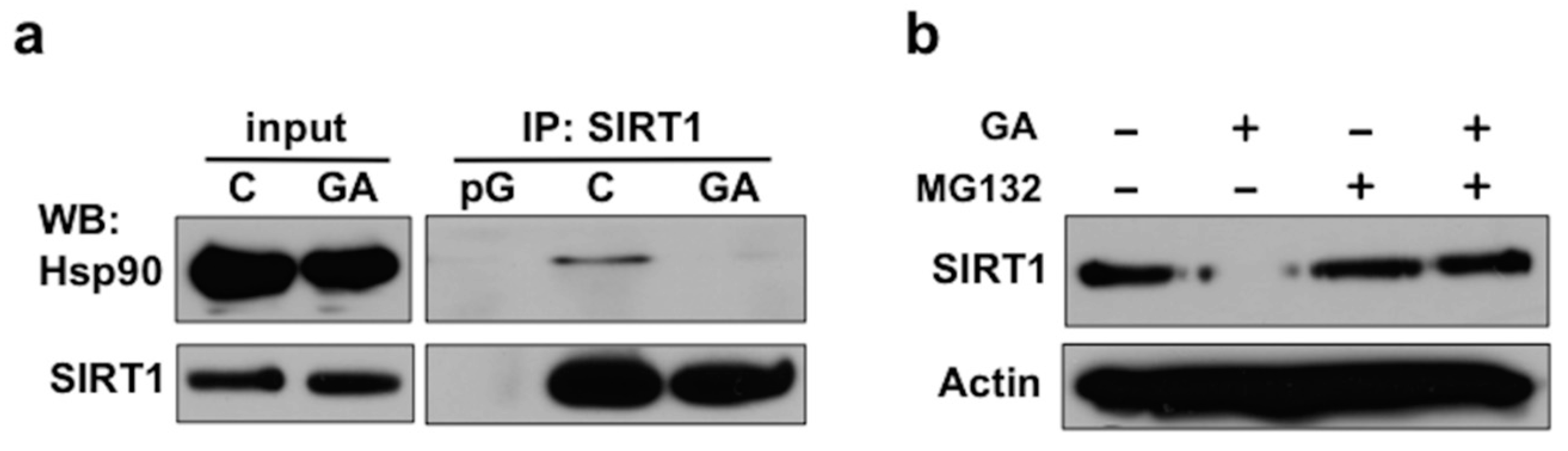
2.2. Disruption of the Hsp90-SIRT1 Interaction Leads to Destabilization and Proteasomal Degradation of SIRT1
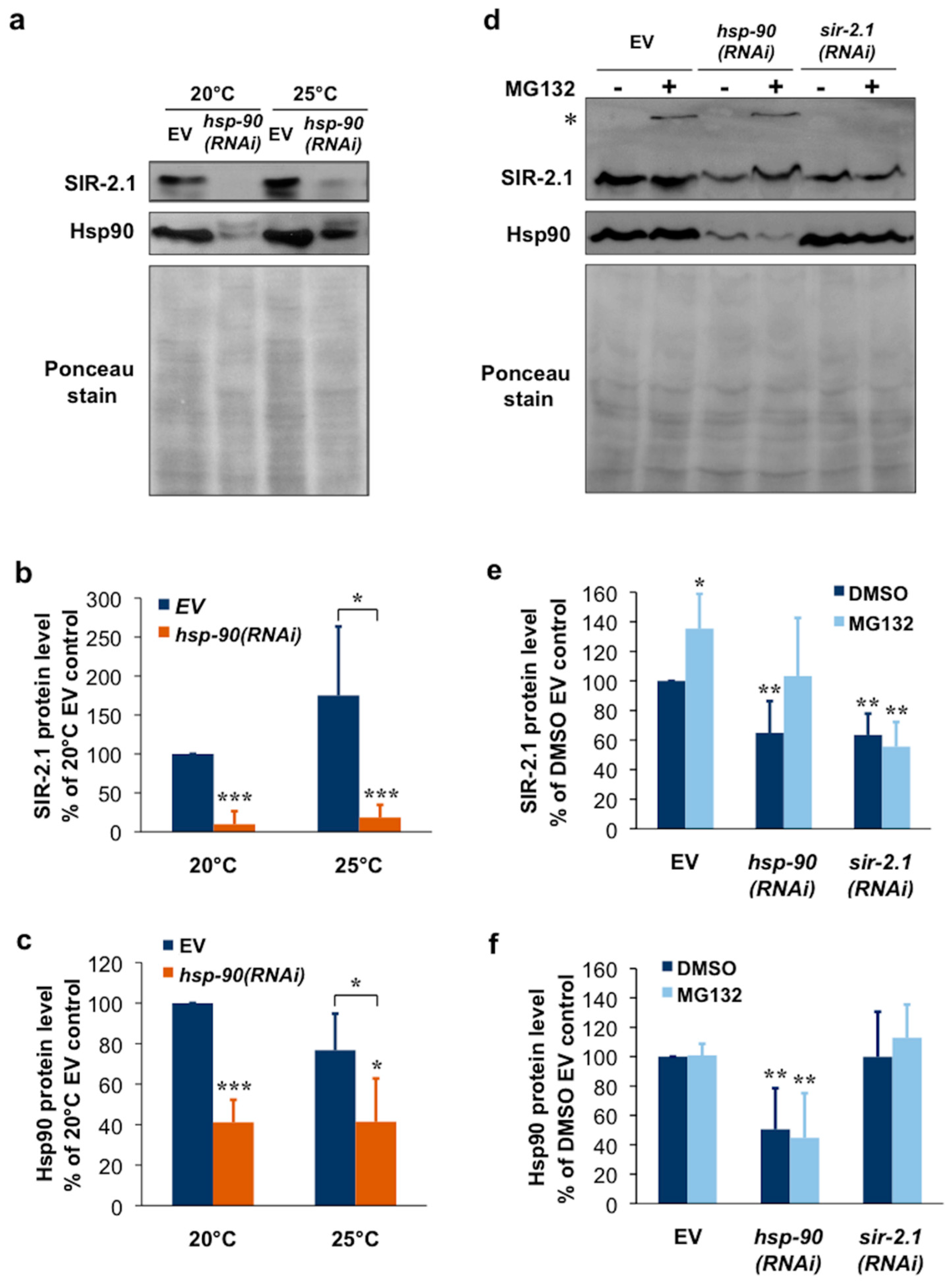
2.3. Hsp90 Is Required for SIR-2.1 Protein Stability in Caenorhabditis elegans
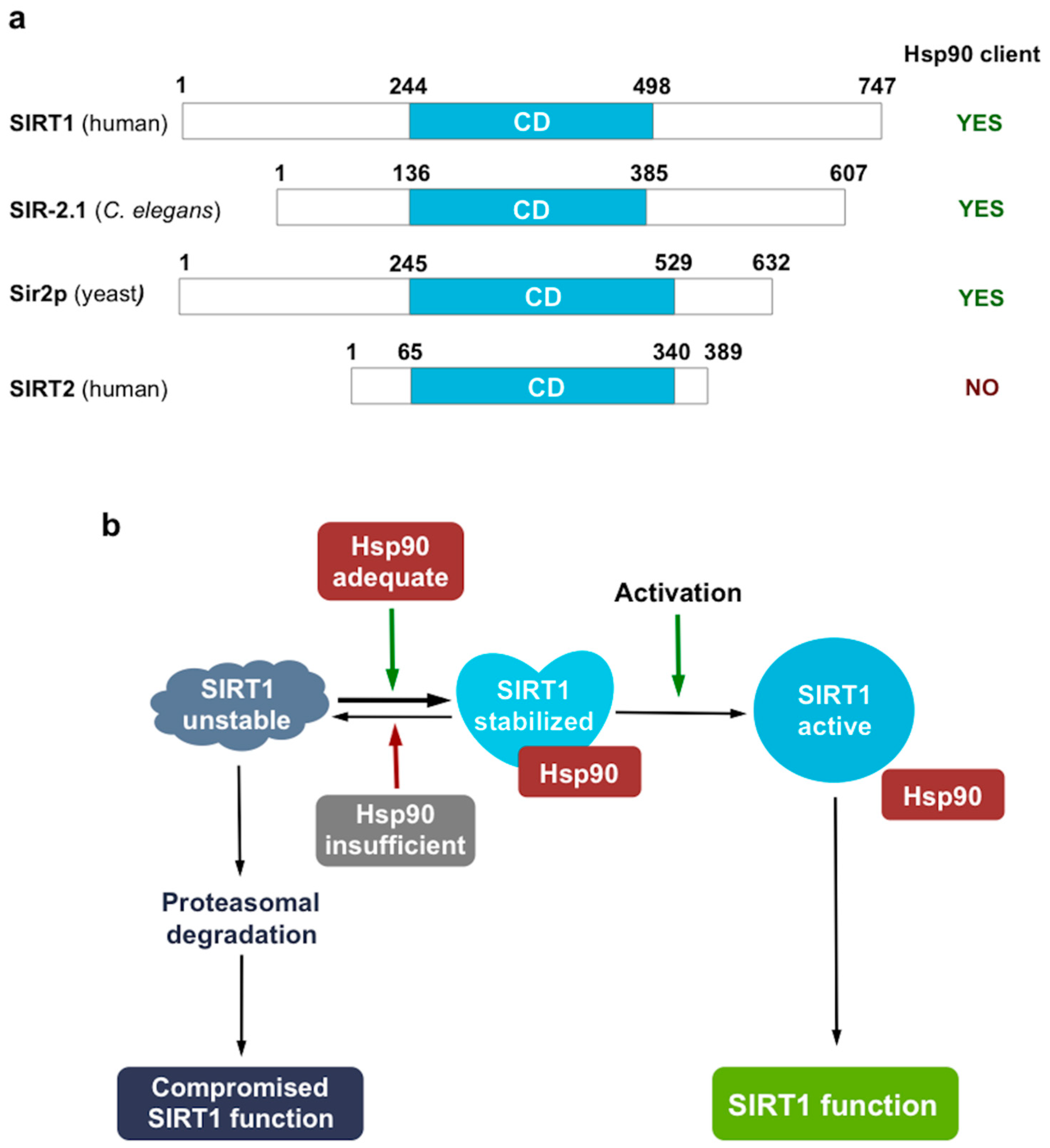
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Mammalian Cell Lysis
4.4. Western Blotting
4.5. Immunoprecipitation
4.6. C. elegans Strains and Maintenance
4.7. RNA Interference
4.8. C. elegans Lysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Ak thymoma kinase, protein kinase B |
| ATCC | American Type Culture Collection |
| BMAL1 | brain and muscle ARNT-like 1 |
| CDK4 | cyclin dependent kinase 4 |
| COS-7 | CV-1 in origin SV40 transformed cells |
| DYRK | dual-specificity tyrosine phosphorylation-regulated kinase |
| ECL | enhanced chemiluminescence |
| eNOS | endothelial nitric oxide synthase |
| FOXO | forkhead box O transcription factor |
| GA | geldanamycin |
| HepG2 | hepatoma G2 |
| HSF1 | heat shock transcription factor 1 |
| Hsp90 | heat shock protein 90 |
| hsp-90 | C. elegans heat shock protein 90 gene |
| JNK | Jun N-terminal kinase |
| LKB1 | liver kinase B1 |
| NAD+ | nicotinamide adenine dinucleotide |
| NF-κB | nuclear factor κB |
| PGC1γ | PPARγ co-activator 1α |
| PPARγ | peroxisome proliferator-activated receptor-γ |
| Raf-1 | rapidly accelerated fibrosarcoma-1 kinase |
| SIRT1 | sirtuin 1, Mammalian silent information regulator ortholog 1 |
| Src | Rous sarcoma virus thyrosine kinase |
| STAC | small molecular sirtuin activating compound |
References
- Blander, G.; Guarente, L. The Sir2 Family of Protein Deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Greiss, S.; Gartner, A. Sirtuin/Sir2 phylogeny, evolutionary considerations and structural conservation. Mol. Cells 2009, 28, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Madsen, A.S.; Olsen, C.A.; Hirschey, M.D. Metabolic control by sirtuins and other enzymes that sense NAD+, NADH, or their ratio. Biochim. Biophys. Acta 2017, 1858, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Burnett, C.; Valentini, S.; Cabreiro, F.; Goss, M.; Somogyvári, M.; Piper, M.D.; Hoddinott, M.; Sutphin, G.L.; Leko, V.; McElwee, J.J.; et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 2011, 477. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Muñoz-Martin, M.; Cañamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 Extends Life Span and Delays Aging in Mice through the Regulation of Nk2 Homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [PubMed]
- McBurney, M.W.; Yang, X.; Jardine, K.; Hixon, M.; Boekelheide, K.; Webb, J.R.; Lansdorp, P.M.; Lemieux, M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol. Cell. Biol. 2003, 23, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. Targeting Sirtuin 1 to Improve Metabolism: All You Need Is NAD+? Pharmacol. Rev. 2012, 64, 166–187. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.M.; Haigis, M.C. Sirtuins in Cancer: A Balancing Act between Genome Stability and Metabolism. Mol. Cells 2015, 38, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Ott, M. The ups and downs of SIRT1. Trends Biochem. Sci. 2008, 33, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.; Ahmed, S.; Allison, S.; Jiang, M.; Milner, J. JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle 2008, 7, 3091–3097. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhang, J.; Kheterpal, I.; Kennedy, N.; Davis, R.J.; Ye, J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal Kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J. Biol. Chem. 2011, 286, 22227–22234. [Google Scholar] [CrossRef] [PubMed]
- Caito, S.; Rajendrasozhan, S.; Cook, S.; Chung, S.; Yao, H.; Friedman, A.E.; Brookes, P.S.; Rahman, I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010, 24, 3145–3159. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yan, B.; Liao, D.; Huang, S.; Qiu, Y. Acetylation of HDAC1 and degradation of SIRT1 form a positive feedback loop to regulate p53 acetylation during heat-shock stress. Cell Death Dis. 2015, 6, e1747-10. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Kesimer, M.; Tolun, G.; Zheng, X.; Xu, Q.; Lu, J.; Sheehan, J.K.; Griffith, J.D.; Li, X. The NAD+-dependent protein deacetylase activity of SIRT1 is regulated by its oligomeric status. Sci. Rep. 2012, 2, 640. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohászka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998, 79. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.; Naim, H.; Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative Analysis of Hsp90-Client Interactions Reveals Principles of Substrate Recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Karras, G.I.; Yi, S.; Sahni, N.; Fischer, M.; Xie, J.; Vidal, M.; D’Andrea, A.D.; Whitesell, L.; Lindquist, S. HSP90 Shapes the Consequences of Human Genetic Variation. Cell 2017, 168, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Csermely, P.; Sodblacti, C. Hsp90 chaperones PPARγ and regulates differentiation and survival of 3T3-L1 adipocytes. Cell Death Differ. 2013, 20, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Aligue, R.; Akhavan-Niak, H.; Russell, P. A role for Hsp90 in cell cycle control: Wee1 tyrosine kinase activity requires interaction with Hsp90. EMBO J. 1994, 13, 6099–6106. [Google Scholar] [CrossRef] [PubMed]
- Birnby, D.A.; Link, E.M.; Vowels, J.J.; Tian, H.; Colacurcio, P.L.; Thomas, J.H. A transmembrane guanylyl cyclase (DAF-11) and Hsp90 (DAF-21) regulate a common set of chemosensory behaviors in Caenorhabditis elegans. Genetics 2000, 155, 85–104. [Google Scholar] [PubMed]
- Inoue, T.; Hirata, K.; Kuwana, Y.; Fujita, M.; Miwa, J.; Roy, R.; Yamaguchi, Y. Cell cycle control by daf-21/Hsp90 at the first meiotic prophase/metaphase boundary during oogenesis in Caenorhabditis elegans. Dev. Growth Differ. 2006, 48, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Gaiser, A.M.; Kaiser, C.J.O.; Haslbeck, V.; Richter, K. Downregulation of the Hsp90 system causes defects in muscle cells of Caenorhabditis elegans. PLoS ONE 2011, 6, e25485. [Google Scholar] [CrossRef] [PubMed]
- Somogyvári, M.; Gecse, E.; Sőti, C. DAF-21/Hsp90 is required for C. elegans longevity by ensuring DAF-16/FOXO isoform A function. Sci. Rep. 2018, 8, 12048. [Google Scholar] [CrossRef] [PubMed]
- Radli, M.; Rüdiger, S.G.D. Dancing with the Diva: Hsp90-Client Interactions. J. Mol. Biol. 2018, 430, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Kajtar, J.; Hollosi, M.; Jalsovszky, G.; Holly, S.; Kahn, C.R.; Gergely, P., Jr.; Soti, C.; Mihaly, K.; Somogyi, J. ATP induces a conformational change of the 90-kDa heat shock protein (hsp90). J. Biol. Chem. 1993, 268, 1901–1907. [Google Scholar] [PubMed]
- Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef]
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.W.; An, W.G.; Neckers, L.M. Geldanamycin-Induced Destabilization of Raf-1 Involves the Proteasome. Biochem. Biophys. Res. Commun. 1997, 239, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef]
- Woodford, M.R.; Dunn, D.M.; Ciciarelli, J.G.; Beebe, K.; Neckers, L.; Mollapour, M. Targeting Hsp90 in Non-Cancerous Maladies. Curr. Top. Med. Chem. 2016, 16, 2792–2804. [Google Scholar] [CrossRef] [PubMed]
- Devaney, E.; Gillan, V. Hsp90 Inhibitors in Parasitic Nematodes: Prospects and Challenges. Curr. Top. Med. Chem. 2016, 16, 2805–2811. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Fleming Outeiro, T.; Cavadas, C. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Gillan, V.; Maitland, K.; McCormack, G.; Him, N.AI.I.N.; Devaney, E. Functional genomics of hsp-90 in parasitic and free-living nematodes. Int. J. Parasitol. 2009, 39, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.; Kim, S.K.; Berdichevsky, A.; Guarente, L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev. Cell 2005, 9, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Sanders, B.D.; Jackson, B.; Marmorstein, R. Structural basis for sirtuin function: What we know and what we don’t. Biochim. Biophys. Acta 2010, 1804, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Laskar, S.; Bhattacharyya, M.K.; Shankar, R.; Bhattacharyya, S. HSP90 controls SIR2 mediated gene silencing. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wei, W.; Jiang, Y.; Peng, H.; Cai, J.; Mao, C.; Dai, H.; Choy, W.; Bemis, J.E.; Jirousek, M.R.; et al. Crystal Structures of Human SIRT3 Displaying Substrate-induced Conformational Changes. J. Biol. Chem. 2009, 284, 24394–24405. [Google Scholar] [CrossRef] [PubMed]
- Pannek, M.; Simic, Z.; Fuszard, M.; Meleshin, M.; Rotili, D.; Mai, A.; Schutkowski, M.; Steegborn, C. Crystal structures of the mitochondrial deacylase Sirtuin 4 reveal isoform-specific acyl recognition and regulation features. Nat. Commun. 2017, 8, 1513. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.W.; Feldman, J.L.; Devries, M.K.; Dong, A.; Edwards, A.M.; Denu, J.M. Structure and biochemical functions of SIRT6. J. Biol. Chem. 2011, 286, 14575–14587. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, A.; Min, J.; Antoshenko, T.; Wang, C.-L.; Allali-Hassani, A.; Dong, A.; Loppnau, P.; Vedadi, M.; Bochkarev, A.; Sternglanz, R.; et al. Structural Basis of Inhibition of the Human NAD+-Dependent Deacetylase SIRT5 by Suramin. Structure 2007, 15, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Allison, D.; Condon, B.; Zhang, F.; Gheyi, T.; Zhang, A.; Ashok, S.; Russell, M.; MacEwan, I.; Qian, Y.; et al. The 2.5 Å Crystal Structure of the SIRT1 Catalytic Domain Bound to Nicotinamide Adenine Dinucleotide (NAD+) and an Indole (EX527 Analogue) Reveals a Novel Mechanism of Histone Deacetylase Inhibition. J. Med. Chem. 2013, 56, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Morishima, Y.; Osawa, Y. The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. J. Biol. Chem. 2008, 283, 22885–22889. [Google Scholar] [CrossRef] [PubMed]
- Verba, K.A.; Wang, R.Y.-R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.M.; Huber, F.M.; Hoelz, A. Structural and functional analysis of human SIRT1. J. Mol. Biol. 2014, 426, 526–541. [Google Scholar] [CrossRef] [PubMed]
- Edkins, A.L.; Price, J.T.; Pockley, A.G.; Blatch, G.L. Heat shock proteins as modulators and therapeutic targets of chronic disease: An integrated perspective. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160521. [Google Scholar] [CrossRef] [PubMed]
- Devaney, E.; O’Neill, K.; Harnett, W.; Whitesell, L.; Kinnaird, J.H. Hsp90 is essential in the filarial nematode Brugia pahangi. Int. J. Parasitol. 2005, 35, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Greiss, S.; Hall, J.; Ahmed, S.; Gartner, A.C. C. elegans SIR-2.1 translocation is linked to a proapoptotic pathway parallel to cep-1/p53 during DNA damage-induced apoptosis. Genes Dev. 2008, 22, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef] [PubMed]
- Kamath, R.S.; Martinez-Campos, M.; Zipperlen, P.; Fraser, A.G.; Ahringer, J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001, 2. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, M.T.; Somogyvári, M.; Sőti, C. Hsp90 Stabilizes SIRT1 Orthologs in Mammalian Cells and C. elegans. Int. J. Mol. Sci. 2018, 19, 3661. https://doi.org/10.3390/ijms19113661
Nguyen MT, Somogyvári M, Sőti C. Hsp90 Stabilizes SIRT1 Orthologs in Mammalian Cells and C. elegans. International Journal of Molecular Sciences. 2018; 19(11):3661. https://doi.org/10.3390/ijms19113661
Chicago/Turabian StyleNguyen, Minh Tu, Milán Somogyvári, and Csaba Sőti. 2018. "Hsp90 Stabilizes SIRT1 Orthologs in Mammalian Cells and C. elegans" International Journal of Molecular Sciences 19, no. 11: 3661. https://doi.org/10.3390/ijms19113661
APA StyleNguyen, M. T., Somogyvári, M., & Sőti, C. (2018). Hsp90 Stabilizes SIRT1 Orthologs in Mammalian Cells and C. elegans. International Journal of Molecular Sciences, 19(11), 3661. https://doi.org/10.3390/ijms19113661