Endothelial Function: A Short Guide for the Interventional Cardiologist

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
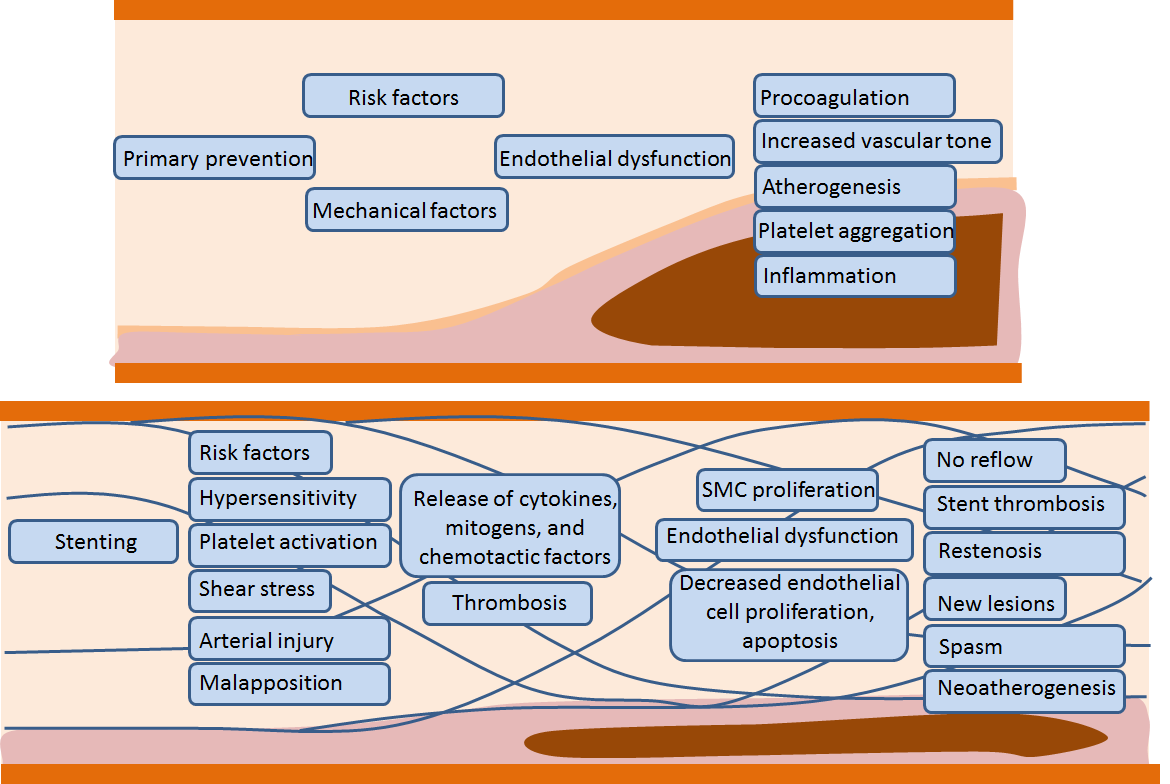
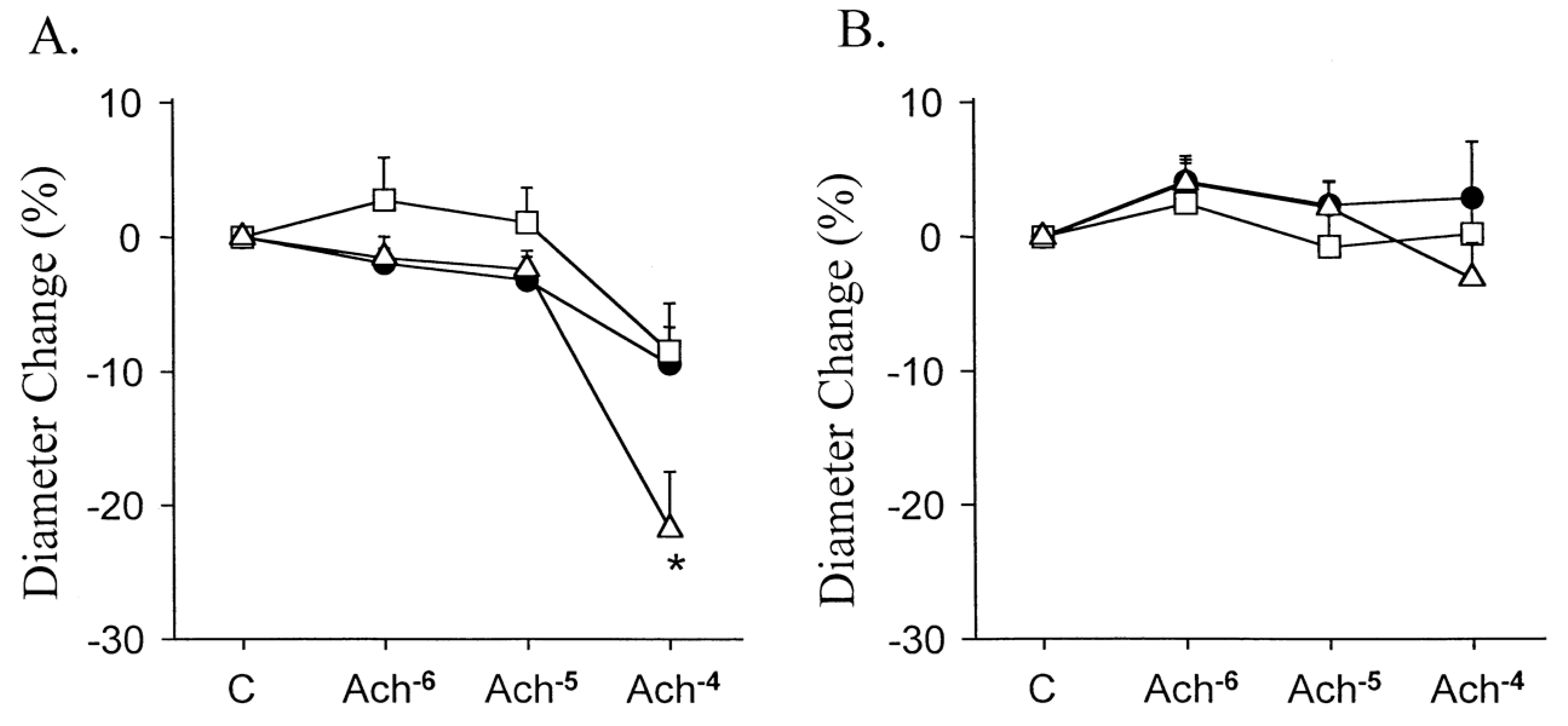
2. Endothelial Dysfunction as a Mechanism and Diagnostic/Prognostic Marker of Coronary Atherosclerosis
3. Endothelial Dysfunction after Coronary Artery Stenting
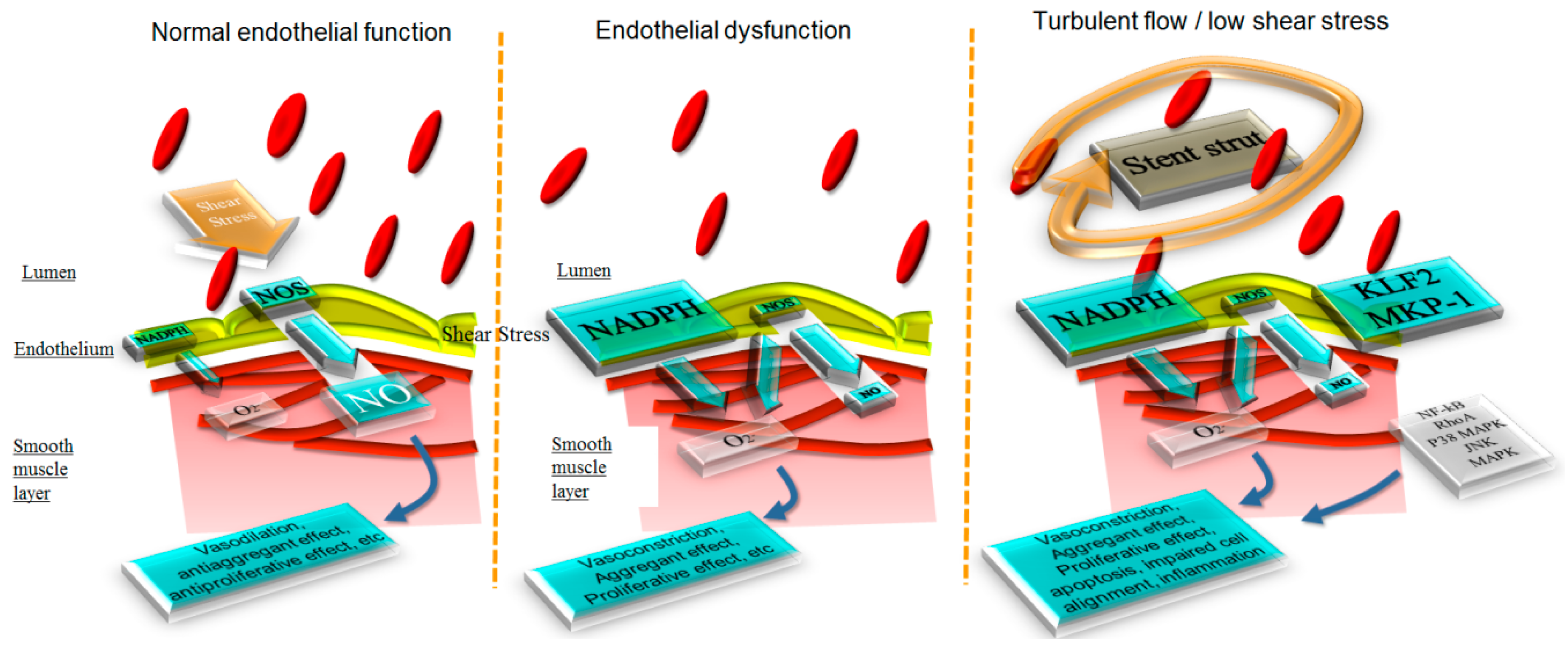
3.1. Impact of Stenting on Fluid Dynamics and Shear Stress
3.2. Impact of the Stent on Endothelial Function
3.2.1. The Effect of Strain
3.2.2. Biological Effect of the Drug
3.2.3. Hypersensitivity Reactions
3.3. Impact of Stenting on Platelet Function
4. Endothelial Regrowth after Stenting
5. Assessment of Endothelial Function in the Secondary Prevention
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| DES | Drug eluting stents |
| NO | Nitric oxide |
| PCI | Percutaneous coronary intervention |
References
- de Agostini, A.I.; Watkins, S.C.; Slayter, H.S.; Youssoufian, H.; Rosenberg, R.D. Localization of anticoagulantly active heparan sulfate proteoglycans in vascular endothelium: Antithrombin binding on cultured endothelial cells and perfused rat aorta. J. Cell. Biol. 1990, 111, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- van Hinsbergh, V.W. Endothelial permeability for macromolecules. Mechanistic aspects of pathophysiological modulation. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Munzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications. Ann. Med. 2011, 43, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Gori, T.; Burstein, J.M.; Ahmed, S.; Miner, S.E.; Al-Hesayen, A.; Kelly, S.; Parker, J.D. Folic acid prevents nitroglycerin-induced nitric oxide synthase dysfunction and nitrate tolerance: A human in vivo study. Circulation 2001, 104, 1119–1123. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Boden, W.E.; O’Rourke, R.A.; Teo, K.K.; Hartigan, P.M.; Maron, D.J.; Kostuk, W.; Knudtson, M.; Dada, M.; Casperson, P.; Harris, C.L.; et al. Design and rationale of the Clinical Outcomes Utilizing Revascularization and Aggressive DruG Evaluation (COURAGE) trial Veterans Affairs Cooperative Studies Program no. 424. Am. Heart J. 2006, 151, 1173–1179. [Google Scholar] [CrossRef]
- Ong, P.; Aziz, A.; Hansen, H.S.; Prescott, E.; Athanasiadis, A.; Sechtem, U. Structural and Functional Coronary Artery Abnormalities in Patients With Vasospastic Angina Pectoris. Circ. J. 2015, 79, 1431–1438. [Google Scholar] [CrossRef]
- Libby, P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 2001, 104, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Rioufol, G.; Finet, G.; Ginon, I.; Andre-Fouet, X.; Rossi, R.; Vialle, E.; Desjoyaux, E.; Convert, G.; Huret, J.F.; Tabib, A. Multiple atherosclerotic plaque rupture in acute coronary syndrome: A three-vessel intravascular ultrasound study. Circulation 2002, 106, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Schachinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Forconi, S.; Gori, T. Endothelium and hemorheology. Clin. Hemorheol. Microcirc. 2013, 53, 3–10. [Google Scholar] [PubMed]
- Gori, T.; Munzel, T. Endothelial dysfunction after stenting and scaffolding of coronary arteries. Clin. Hemorheol. Microcirc. 2014, 58, 175–181. [Google Scholar] [PubMed]
- Reddy, K.G.; Nair, R.N.; Sheehan, H.M.; Hodgson, J.M. Evidence that selective endothelial dysfunction may occur in the absence of angiographic or ultrasound atherosclerosis in patients with risk factors for atherosclerosis. J. Am. Coll. Cardiol. 1994, 23, 833–843. [Google Scholar] [CrossRef]
- Vita, J.A.; Treasure, C.B.; Nabel, E.G.; McLenachan, J.M.; Fish, R.D.; Yeung, A.C.; Vekshtein, V.I.; Selwyn, A.P.; Ganz, P. Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation 1990, 81, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.J.; Lerman, A.; Bech, J.W.; De Bruyne, B.; Eeckhout, E.; Fearon, W.F.; Higano, S.T.; Lim, M.J.; Meuwissen, M.; Piek, J.J.; et al. Physiological assessment of coronary artery disease in the cardiac catheterization laboratory: A scientific statement from the American Heart Association Committee on Diagnostic and Interventional Cardiac Catheterization, Council on Clinical Cardiology. Circulation 2006, 114, 1321–1341. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.; Hansen, H.S.; Sechtem, U.; Prescott, E.; Ong, P. Sex-Related Differences in Vasomotor Function in Patients With Angina and Unobstructed Coronary Arteries. J. Am. Coll. Cardiol. 2017, 70, 2349–2358. [Google Scholar] [CrossRef]
- Gori, T.; Muxel, S.; Damaske, A.; Radmacher, M.C.; Fasola, F.; Schaefer, S.; Schulz, A.; Jabs, A.; Parker, J.D.; Munzel, T. Endothelial function assessment: Flow-mediated dilation and constriction provide different and complementary information on the presence of coronary artery disease. Eur. Heart J. 2012, 33, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; von Henning, U.; Muxel, S.; Schaefer, S.; Fasola, F.; Vosseler, M.; Schnorbus, B.; Binder, H.; Parker, J.D.; Munzel, T. Both flow-mediated dilation and constriction are associated with changes in blood flow and shear stress: Two complementary perspectives on endothelial function. Clin. Hemorheol. Microcirc. 2016, 64, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, J.; Crouse, J.R.; Hsu, F.C.; Burke, G.L.; Herrington, D.M. Brachial flow-mediated dilation predicts incident cardiovascular events in older adults: The Cardiovascular Health Study. Circulation 2007, 115, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J.; Charbonneau, F.; Title, L.M.; Buithieu, J.; Rose, M.S.; Conradson, H.; Hildebrand, K.; Fung, M.; Verma, S.; Lonn, E.M. Microvascular function predicts cardiovascular events in primary prevention: Long-term results from the Firefighters and Their Endothelium (FATE) study. Circulation 2011, 123, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Lind, L.; Berglund, L.; Larsson, A.; Sundstrom, J. Endothelial function in resistance and conduit arteries and 5-year risk of cardiovascular disease. Circulation 2011, 123, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Muxel, S.; Fasola, F.; Radmacher, M.C.; Jabs, A.; Munzel, T.; Gori, T. Endothelial functions: Translating theory into clinical application. Clin. Hemorheol. Microcirc. 2010, 45, 109–115. [Google Scholar] [PubMed]
- Cragg, A.; Einzig, S.; Castaneda-Zuniga, W.; Amplatz, K.; White, J.G.; Rao, G.H. Vessel wall arachidonate metabolism after angioplasty: Possible mediators of postangioplasty vasospasm. Am. J. Cardiol. 1983, 51, 1441–1445. [Google Scholar] [CrossRef]
- Wilentz, J.R.; Sanborn, T.A.; Haudenschild, C.C.; Valeri, C.R.; Ryan, T.J.; Faxon, D.P. Platelet accumulation in experimental angioplasty: Time course and relation to vascular injury. Circulation 1987, 75, 636–642. [Google Scholar] [CrossRef]
- Thyberg, J.; Hedin, U.; Sjolund, M.; Palmberg, L.; Bottger, B.A. Regulation of differentiated properties and proliferation of arterial smooth muscle cells. Arteriosclerosis 1990, 10, 966–990. [Google Scholar] [CrossRef]
- Otsuka, F.; Finn, A.V.; Yazdani, S.K.; Nakano, M.; Kolodgie, F.D.; Virmani, R. The importance of the endothelium in atherothrombosis and coronary stenting. Nat. Rev. Cardiol. 2012, 9, 439–453. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Kolandaivelu, K.; Swaminathan, R.; Gibson, W.J.; Kolachalama, V.B.; Nguyen-Ehrenreich, K.L.; Giddings, V.L.; Coleman, L.; Wong, G.K.; Edelman, E.R. Stent thrombogenicity early in high-risk interventional settings is driven by stent design and deployment and protected by polymer-drug coatings. Circulation 2011, 123, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Gijsen, F.J.; Oortman, R.M.; Wentzel, J.J.; Schuurbiers, J.C.; Tanabe, K.; Degertekin, M.; Ligthart, J.M.; Thury, A.; de Feyter, P.J.; Serruys, P.W.; et al. Usefulness of shear stress pattern in predicting neointima distribution in sirolimus-eluting stents in coronary arteries. Am. J. Cardiol. 2003, 92, 1325–1328. [Google Scholar] [CrossRef]
- Wentzel, J.J.; Krams, R.; Schuurbiers, J.C.; Oomen, J.A.; Kloet, J.; van Der Giessen, W.J.; Serruys, P.W.; Slager, C.J. Relationship between neointimal thickness and shear stress after Wallstent implantation in human coronary arteries. Circulation 2001, 103, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Foin, N.; Gutierrez-Chico, J.L.; Nakatani, S.; Torii, R.; Bourantas, C.V.; Sen, S.; Nijjer, S.; Petraco, R.; Kousera, C.; Ghione, M.; et al. Incomplete stent apposition causes high shear flow disturbances and delay in neointimal coverage as a function of strut to wall detachment distance: Implications for the management of incomplete stent apposition. Circ. Cardiovasc. Interv. 2014, 7, 180–189. [Google Scholar] [CrossRef]
- Fledderus, J.O.; Boon, R.A.; Volger, O.L.; Hurttila, H.; Yla-Herttuala, S.; Pannekoek, H.; Levonen, A.L.; Horrevoets, A.J. KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1339–1346. [Google Scholar] [CrossRef]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–1870. [Google Scholar] [CrossRef]
- Mott, R.E.; Helmke, B.P. Mapping the dynamics of shear stress-induced structural changes in endothelial cells. Am. J. Physiol. Cell Physiol. 2007, 293, C1616–1626. [Google Scholar] [CrossRef]
- Cuhlmann, S.; Van der Heiden, K.; Saliba, D.; Tremoleda, J.L.; Khalil, M.; Zakkar, M.; Chaudhury, H.; Luong, L.A.; Mason, J.C.; Udalova, I.; et al. Disturbed blood flow induces RelA expression via c-Jun N-terminal kinase 1: A novel mode of NF-kappaB regulation that promotes arterial inflammation. Circ. Res. 2011, 108, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, H.; Zakkar, M.; Boyle, J.; Cuhlmann, S.; van der Heiden, K.; Luong, L.A.; Davis, J.; Platt, A.; Mason, J.C.; Krams, R.; et al. c-Jun N-terminal kinase primes endothelial cells at atheroprone sites for apoptosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Chien, S.; Shyy, J.Y. Regulation of endothelial cell cycle by laminar versus oscillatory flow: Distinct modes of interactions of AMP-activated protein kinase and Akt pathways. Circ. Res. 2007, 100, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Caramori, P.R.; Lima, V.C.; Seidelin, P.H.; Newton, G.E.; Parker, J.D.; Adelman, A.G. Long-term endothelial dysfunction after coronary artery stenting. J. Am. Coll. Cardiol. 1999, 34, 1675–1679. [Google Scholar] [CrossRef]
- Sabate, M.; Kay, I.P.; van Der Giessen, W.J.; Cequier, A.; Ligthart, J.M.; Gomez-Hospital, J.A.; Carlier, S.G.; Coen, V.L.; Marijnissen, J.P.; Wardeh, A.J.; et al. Preserved endothelium-dependent vasodilation in coronary segments previously treated with balloon angioplasty and intracoronary irradiation. Circulation 1999, 100, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Togni, M.; Windecker, S.; Wenaweser, P.; Tueller, D.; Kaisaier, A.; Maier, W.; Meier, B.; Hess, O.M. Deleterious effect of coronary brachytherapy on vasomotor response to exercise. Circulation 2004, 110, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Fuke, S.; Maekawa, K.; Kawamoto, K.; Saito, H.; Sato, T.; Hioka, T.; Ohe, T. Impaired endothelial vasomotor function after sirolimus-eluting stent implantation. Circ. J. 2007, 71, 220–225. [Google Scholar] [CrossRef]
- Kim, J.W.; Seo, H.S.; Park, J.H.; Na, J.O.; Choi, C.U.; Lim, H.E.; Kim, E.J.; Rha, S.W.; Park, C.G.; Oh, D.J. A prospective, randomized, 6-month comparison of the coronary vasomotor response associated with a zotarolimus- versus a sirolimus-eluting stent: Differential recovery of coronary endothelial dysfunction. J. Am. Coll. Cardiol. 2009, 53, 1653–1659. [Google Scholar] [CrossRef]
- Kim, J.W.; Suh, S.Y.; Choi, C.U.; Na, J.O.; Kim, E.J.; Rha, S.W.; Park, C.G.; Seo, H.S.; Oh, D.J. Six-month comparison of coronary endothelial dysfunction associated with sirolimus-eluting stent versus Paclitaxel-eluting stent. JACC Cardiovasc. Interv. 2008, 1, 65–71. [Google Scholar] [CrossRef]
- Lim, S.H.; Flammer, A.J.; Yoon, M.H.; Lennon, R.J.; Gulati, R.; Mathew, V.; Rihal, C.S.; Lerman, L.O.; Lerman, A. The long-term effect of coronary stenting on epicardial and microvascular endothelial function. Circ. Cardiovasc. Interv. 2012, 5, 523–529. [Google Scholar] [CrossRef]
- Serruys, P.W.; Ormiston, J.A.; Onuma, Y.; Regar, E.; Gonzalo, N.; Garcia-Garcia, H.M.; Nieman, K.; Bruining, N.; Dorange, C.; Miquel-Hebert, K.; et al. A bioabsorbable everolimus-eluting coronary stent system (ABSORB): 2-year outcomes and results from multiple imaging methods. Lancet 2009, 373, 897–910. [Google Scholar] [CrossRef]
- Togni, M.; Windecker, S.; Cocchia, R.; Wenaweser, P.; Cook, S.; Billinger, M.; Meier, B.; Hess, O.M. Sirolimus-eluting stents associated with paradoxic coronary vasoconstriction. J. Am. Coll. Cardiol. 2005, 46, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ariotti, S.; Adamo, M.; Costa, F.; Patialiakas, A.; Briguori, C.; Thury, A.; Colangelo, S.; Campo, G.; Tebaldi, M.; Ungi, I.; et al. Is Bare-Metal Stent Implantation Still Justifiable in High Bleeding Risk Patients Undergoing Percutaneous Coronary Intervention?: A Pre-Specified Analysis From the ZEUS Trial. JACC Cardiovasc. Interv. 2016, 9, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Ando, J.; Yamamoto, K. Effects of shear stress and stretch on endothelial function. Antioxid. Redox Signal. 2011, 15, 1389–1403. [Google Scholar] [CrossRef] [PubMed]
- Finn, A.V.; Kolodgie, F.D.; Harnek, J.; Guerrero, L.J.; Acampado, E.; Tefera, K.; Skorija, K.; Weber, D.K.; Gold, H.K.; Virmani, R. Differential response of delayed healing and persistent inflammation at sites of overlapping sirolimus- or paclitaxel-eluting stents. Circulation 2005, 112, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Guagliumi, G.; Farb, A.; Musumeci, G.; Valsecchi, O.; Tespili, M.; Motta, T.; Virmani, R. Images in cardiovascular medicine. Sirolimus-eluting stent implanted in human coronary artery for 16 months: Pathological findings. Circulation 2003, 107, 1340–1341. [Google Scholar] [CrossRef] [PubMed]
- Jabs, A.; Okamoto, E.; Vinten-Johansen, J.; Bauriedel, G.; Wilcox, J.N. Sequential patterns of chemokine- and chemokine receptor-synthesis following vessel wall injury in porcine coronary arteries. Atherosclerosis 2007, 192, 75–84. [Google Scholar] [CrossRef]
- Joner, M.; Finn, A.V.; Farb, A.; Mont, E.K.; Kolodgie, F.D.; Ladich, E.; Kutys, R.; Skorija, K.; Gold, H.K.; Virmani, R. Pathology of drug-eluting stents in humans: Delayed healing and late thrombotic risk. J. Am. Coll. Cardiol. 2006, 48, 193–202. [Google Scholar] [CrossRef]
- Hwang, C.W.; Wu, D.; Edelman, E.R. Physiological transport forces govern drug distribution for stent-based delivery. Circulation 2001, 104, 600–605. [Google Scholar] [CrossRef]
- Nakazawa, G.; Finn, A.V.; Vorpahl, M.; Ladich, E.R.; Kolodgie, F.D.; Virmani, R. Coronary responses and differential mechanisms of late stent thrombosis attributed to first-generation sirolimus- and paclitaxel-eluting stents. J. Am. Coll. Cardiol. 2011, 57, 390–398. [Google Scholar] [CrossRef]
- Curcio, A.; Torella, D.; Cuda, G.; Coppola, C.; Faniello, M.C.; Achille, F.; Russo, V.G.; Chiariello, M.; Indolfi, C. Effect of stent coating alone on in vitro vascular smooth muscle cell proliferation and apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H902–908. [Google Scholar] [CrossRef] [PubMed]
- Guagliumi, G.; Sirbu, V.; Musumeci, G.; Gerber, R.; Biondi-Zoccai, G.; Ikejima, H.; Ladich, E.; Lortkipanidze, N.; Matiashvili, A.; Valsecchi, O.; et al. Examination of the in vivo mechanisms of late drug-eluting stent thrombosis: Findings from optical coherence tomography and intravascular ultrasound imaging. JACC Cardiovasc. Interv. 2012, 5, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Camenzind, E.; Steg, P.G.; Wijns, W. Stent thrombosis late after implantation of first-generation drug-eluting stents: A cause for concern. Circulation 2007, 115, 1440–1455. [Google Scholar] [CrossRef] [PubMed]
- McFadden, E.P.; Stabile, E.; Regar, E.; Cheneau, E.; Ong, A.T.; Kinnaird, T.; Suddath, W.O.; Weissman, N.J.; Torguson, R.; Kent, K.M.; et al. Late thrombosis in drug-eluting coronary stents after discontinuation of antiplatelet therapy. Lancet 2004, 364, 1519–1521. [Google Scholar] [CrossRef]
- Anadol, R.; Gori, T. The mechanisms of late scaffold thrombosis. Clin. Hemorheol. Microcirc. 2017, 67, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Cheng, Q.; Mori, H.; Lipinski, M.J.; Acampado, E.; Perkins, L.E.L.; Hossainy, S.F.; Pacetti, S.D.; Kolodgie, F.D.; Virmani, R.; et al. Acute Thrombogenicity of Fluoropolymer-Coated Versus Biodegradable and Polymer Free Stents. EuroIntervention 2018. [Google Scholar] [CrossRef]
- Otsuka, F.; Cheng, Q.; Yahagi, K.; Acampado, E.; Sheehy, A.; Yazdani, S.K.; Sakakura, K.; Euller, K.; Perkins, L.E.L.; Kolodgie, F.D.; et al. Acute Thrombogenicity of a Durable Polymer Everolimus-Eluting Stent Relative to Contemporary Drug-Eluting Stents With Biodegradable Polymer Coatings Assessed Ex Vivo in a Swine Shunt Model. JACC Cardiovasc. Interv. 2015, 8, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Jabs, A.; Gobel, S.; Wenzel, P.; Kleschyov, A.L.; Hortmann, M.; Oelze, M.; Daiber, A.; Munzel, T. Sirolimus-induced vascular dysfunction. Increased mitochondrial and nicotinamide adenosine dinucleotide phosphate oxidase-dependent superoxide production and decreased vascular nitric oxide formation. J. Am. Coll. Cardiol. 2008, 51, 2130–2138. [Google Scholar] [CrossRef]
- Schober, A. Chemokines in vascular dysfunction and remodeling. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1950–1959. [Google Scholar] [CrossRef]
- Schober, A.; Zernecke, A. Chemokines in vascular remodeling. Thromb Haemost 2007, 97, 730–737. [Google Scholar]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef] [PubMed]
- Nording, H.M.; Seizer, P.; Langer, H.F. Platelets in inflammation and atherogenesis. Front. Immunol. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- Davi, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Schulz, R.; Nylander, S. Adenosine-mediated effects of ticagrelor: Evidence and potential clinical relevance. J. Am. Coll. Cardiol. 2014, 63, 2503–2509. [Google Scholar] [CrossRef] [PubMed]
- Bonello, L.; Frere, C.; Cointe, S.; Laine, M.; Mancini, J.; Thuny, F.; Kerbaul, F.; Lemesle, G.; Paganelli, F.; Guieu, R.; et al. Ticagrelor increases endothelial progenitor cell level compared to clopidogrel in acute coronary syndromes: A prospective randomized study. Int. J. Cardiol. 2015, 187, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Lombo, B.; Diez, J.G. Ticagrelor: The evidence for its clinical potential as an oral antiplatelet treatment for the reduction of major adverse cardiac events in patients with acute coronary syndromes. Core Evid. 2011, 6, 31–42. [Google Scholar] [PubMed]
- Kim, H.K.; Jeong, M.H.; Lim, K.S.; Kim, J.H.; Lim, H.C.; Kim, M.C.; Hong, Y.J.; Kim, S.S.; Park, K.H.; Chang, K.S. Effects of ticagrelor on neointimal hyperplasia and endothelial function, compared with clopidogrel and prasugrel, in a porcine coronary stent restenosis model. Int. J. Cardiol. 2017, 240, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, F.; Cardelli, J.A.; Martin, K.A.; Blenis, J.; Huang, S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene 2006, 25, 7029–7040. [Google Scholar] [CrossRef]
- Tsurumi, Y.; Murohara, T.; Krasinski, K.; Chen, D.; Witzenbichler, B.; Kearney, M.; Couffinhal, T.; Isner, J.M. Reciprocal relation between VEGF and NO in the regulation of endothelial integrity. Nat. Med. 1997, 3, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Vinals, F.; Chambard, J.C.; Pouyssegur, J. p70 S6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J. Biol. Chem. 1999, 274, 26776–26782. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Yakes, F.M.; Rojo, F.; Shin, N.Y.; Bakin, A.V.; Baselga, J.; Arteaga, C.L. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat. Med. 2002, 8, 1145–1152. [Google Scholar] [CrossRef]
- Asahara, T.; Masuda, H.; Takahashi, T.; Kalka, C.; Pastore, C.; Silver, M.; Kearne, M.; Magner, M.; Isner, J.M. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ. Res. 1999, 85, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Polimeni, A. Predictors of stent thrombosis and their implications for clinical practice. Nat. Cardiovasc. Rev. in press.
- van Beusekom, H.M.; Whelan, D.M.; Hofma, S.H.; Krabbendam, S.C.; van Hinsbergh, V.W.; Verdouw, P.D.; van der Giessen, W.J. Long-term endothelial dysfunction is more pronounced after stenting than after balloon angioplasty in porcine coronary arteries. J. Am. Coll. Cardiol. 1998, 32, 1109–1117. [Google Scholar] [CrossRef]
- Joner, M.; Nakazawa, G.; Finn, A.V.; Quee, S.C.; Coleman, L.; Acampado, E.; Wilson, P.S.; Skorija, K.; Cheng, Q.; Xu, X.; et al. Endothelial cell recovery between comparator polymer-based drug-eluting stents. J. Am. Coll. Cardiol. 2008, 52, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Sarno, G.; Girasis, C.; Vranckx, P.; de Vries, T.; Swart, M.; Bressers, M.; Garcia-Garcia, H.M.; van Es, G.A.; Raber, L.; et al. A patient-level pooled analysis assessing the impact of the SYNTAX (synergy between percutaneous coronary intervention with taxus and cardiac surgery) score on 1-year clinical outcomes in 6,508 patients enrolled in contemporary coronary stent trials. JACC Cardiovasc. Interv. 2011, 4, 645–653. [Google Scholar] [CrossRef]
- Nakazawa, G.; Nakano, M.; Otsuka, F.; Wilcox, J.N.; Melder, R.; Pruitt, S.; Kolodgie, F.D.; Virmani, R. Evaluation of polymer-based comparator drug-eluting stents using a rabbit model of iliac artery atherosclerosis. Circ. Cardiovasc. Interv. 2011, 4, 38–46. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Kwon, T.G.; Lennon, R.J.; Lerman, L.O.; Lerman, A. Prognostic Value of Flow-Mediated Vasodilation in Brachial Artery and Fingertip Artery for Cardiovascular Events: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2015, 4, e002270. [Google Scholar] [CrossRef]
- Karatzis, E.N.; Ikonomidis, I.; Vamvakou, G.D.; Papaioannou, T.G.; Protogerou, A.D.; Andreadou, I.; Voidonikola, P.T.; Karatzi, K.N.; Papamichael, C.M.; Lekakis, J.P. Long-term prognostic role of flow-mediated dilatation of the brachial artery after acute coronary syndromes without ST elevation. Am. J. Cardiol. 2006, 98, 1424–1428. [Google Scholar] [CrossRef]
- Shechter, M.; Matetzky, S.; Arad, M.; Feinberg, M.S.; Freimark, D. Vascular endothelial function predicts mortality risk in patients with advanced ischaemic chronic heart failure. Eur. J. Heart Fail. 2009, 11, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Guddeti, R.R.; Kwon, T.G.; Lerman, L.O.; Lerman, A. Treating coronary disease and the impact of endothelial dysfunction. Prog. Cardiovasc. Dis. 2015, 57, 431–442. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gori, T. Endothelial Function: A Short Guide for the Interventional Cardiologist. Int. J. Mol. Sci. 2018, 19, 3838. https://doi.org/10.3390/ijms19123838
Gori T. Endothelial Function: A Short Guide for the Interventional Cardiologist. International Journal of Molecular Sciences. 2018; 19(12):3838. https://doi.org/10.3390/ijms19123838
Chicago/Turabian StyleGori, Tommaso. 2018. "Endothelial Function: A Short Guide for the Interventional Cardiologist" International Journal of Molecular Sciences 19, no. 12: 3838. https://doi.org/10.3390/ijms19123838
APA StyleGori, T. (2018). Endothelial Function: A Short Guide for the Interventional Cardiologist. International Journal of Molecular Sciences, 19(12), 3838. https://doi.org/10.3390/ijms19123838