Alternative mRNA Splicing in the Pathogenesis of Obesity

Abstract
1. Introduction
2. Mis-Splicing of Metabolic Factors in Obesity
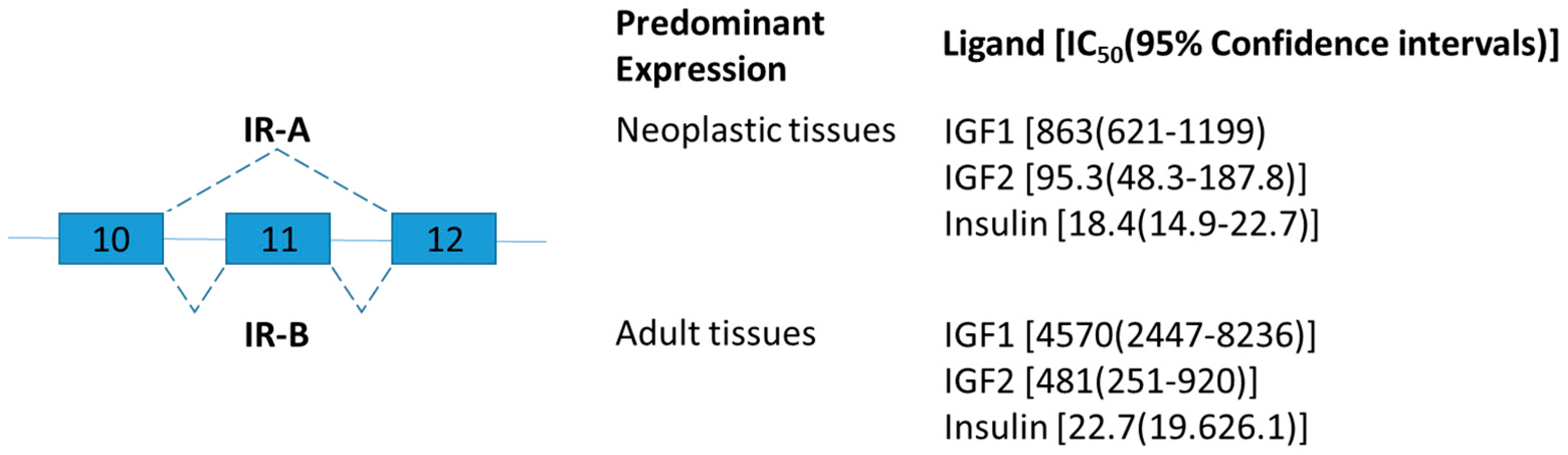
2.1. Insulin Receptor
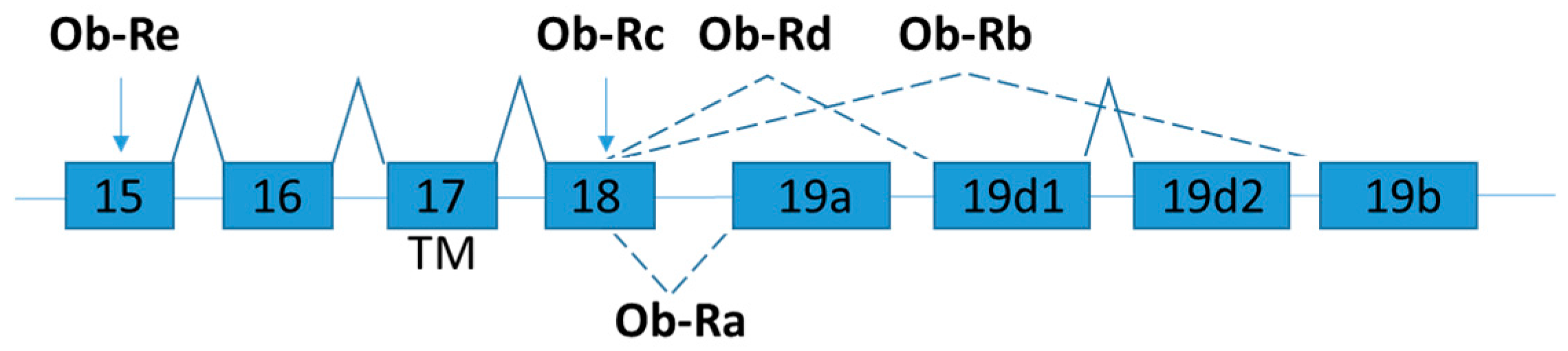
2.2. Leptin Receptor
2.3. Nuclear Receptor Corepressor
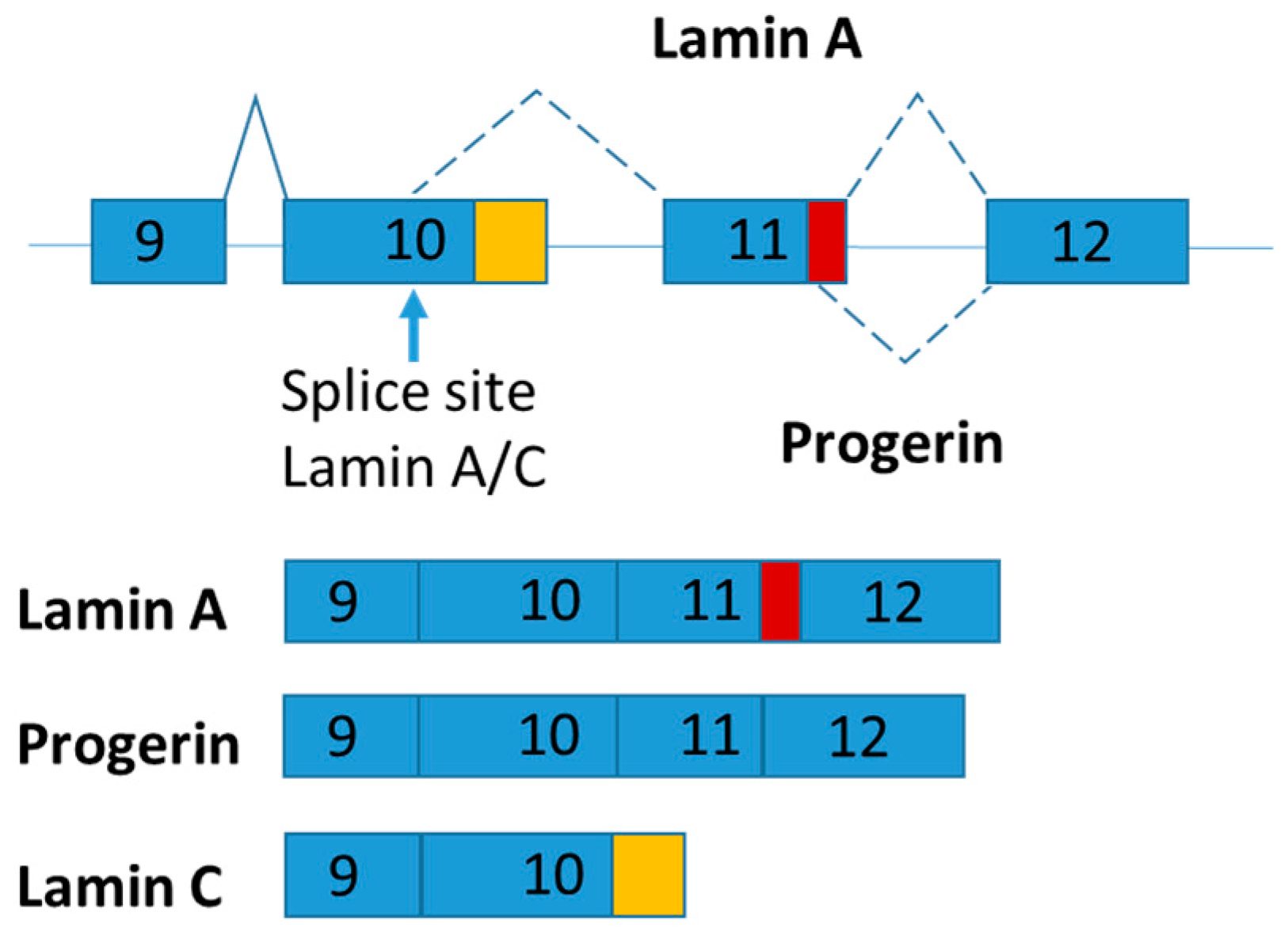
2.4. LMNA
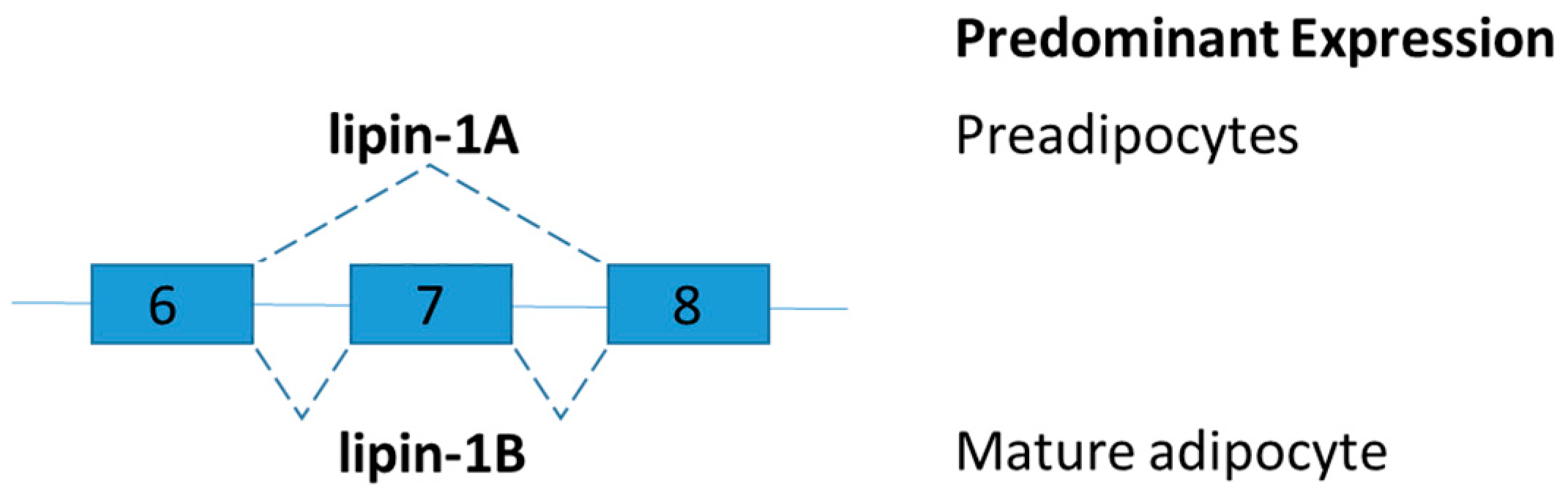
2.5. Lipin-1
3. The Expression Level of Splicing Factors Altered in Obese Subjects
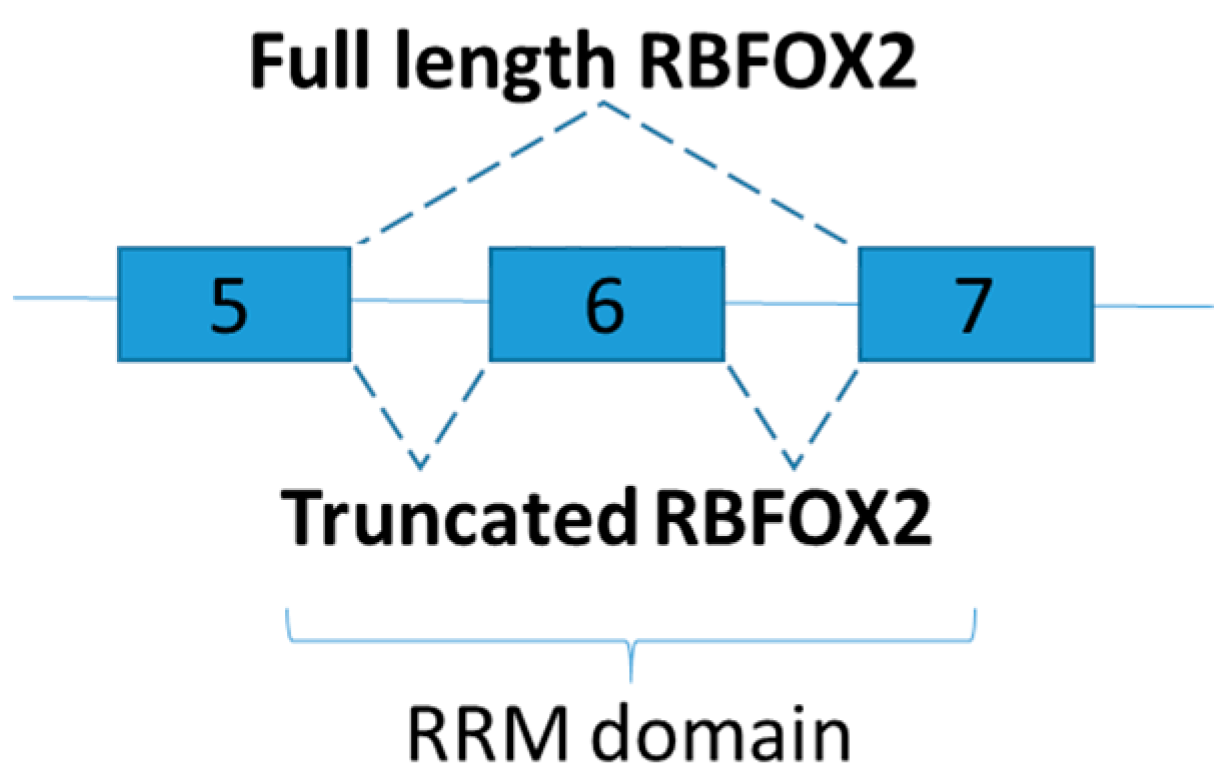
3.1. RNA Binding Protein, Fox-1 Homolog 2
3.2. Neuro-Oncological Ventral Antigen (NOVA) Splicing Factors
4. Alternative Splicing as a Therapeutic Target for Obesity
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 5-HTR2c | Serotonin 2C receptor |
| HFD | High fat diet |
| IR | Insulin receptor |
| KO | Knockout |
| NCoR | Nuclear Receptor Corepressor |
| NOVA | Neuro-Oncological Ventral Antigen |
| nt | Nucleotides |
| Ob-R | Obesity receptor |
| RBFOX2 | RNA Binding Protein, Fox-1 Homolog 2 |
| SIRT1 | Sirtuin 1 |
| SMA | Spinal muscular atrophy |
| SMN | Survival motor neuron |
| SR | serine/arginine |
| SRSF1 | Serine and Arginine Rich Splicing Factor 1 |
| SRSF3 | Serine and Arginine Rich Splicing Factor 3 |
| LDL | Low-density lipoprotein |
References
- Black, D.L. Protein diversity from alternative splicing: A challenge for bioinformatics and post-genome biology. Cell 2000, 103, 367–370. [Google Scholar] [CrossRef]
- Piovesan, A.; Caracausi, M.; Antonaros, F.; Pelleri, M.C.; Vitale, L. GeneBase 1.1: A tool to summarize data from NCBI gene datasets and its application to an update of human gene statistics. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.I.; Sanchez-Pulido, L.; Haerty, W.; Ponting, C.P. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wieringa, B.; Hofer, E.; Weissmann, C. A minimal intron length but no specific internal sequence is required for splicing the large rabbit beta-globin intron. Cell 1984, 37, 915–925. [Google Scholar] [CrossRef]
- Shimada, M.K.; Sasaki-Haraguchi, N.; Mayeda, A. Identification and Validation of Evolutionarily Conserved Unusually Short Pre-mRNA Introns in the Human Genome. Int. J. Mol. Sci. 2015, 16, 10376–10388. [Google Scholar] [CrossRef] [PubMed]
- Abebrese, E.L.; Ali, S.H.; Arnold, Z.R.; Andrews, V.M.; Armstrong, K.; Burns, L.; Crowder, H.R.; Day, R.T., Jr.; Hsu, D.G.; Jarrell, K.; et al. Identification of human short introns. PLoS ONE 2017, 12, e0175393. [Google Scholar] [CrossRef] [PubMed]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome Profiling in Human Diseases: New Advances and Perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef] [PubMed]
- Tress, M.L.; Abascal, F.; Valencia, A. Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem. Sci. 2017, 42, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Ezkurdia, I.; Rodriguez, J.M.; Carrillo-de Santa Pau, E.; Vazquez, J.; Valencia, A.; Tress, M.L. Most highly expressed protein-coding genes have a single dominant isoform. J. Proteom. Res. 2015, 14, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Yabas, M.; Elliott, H.; Hoyne, G.F. The Role of Alternative Splicing in the Control of Immune Homeostasis and Cellular Differentiation. Int. J. Mol. Sci. 2015, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Colantoni, A.; Bianchi, V.; Gherardini, P.F.; Tomba, G.S.; Ausiello, G.; Helmer-Citterich, M.; Ferre, F. Alternative splicing tends to avoid partial removals of protein-protein interaction sites. BMC Genom. 2013, 14, 379. [Google Scholar] [CrossRef] [PubMed]
- Jo, B.S.; Choi, S.S. Introns: The Functional Benefits of Introns in Genomes. Genom. Inform. 2015, 13, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Lee, J.T.; Huang, Z.; Wong, C.M. Coupling and coordination in gene expression processes with pre-mRNA splicing. Int. J. Mol. Sci. 2015, 16, 5682–5696. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Sterne-Weiler, T.; Sanford, J.R. Exon identity crisis: Disease-causing mutations that disrupt the splicing code. Genome Biol. 2014, 15, 201. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Chabot, B.; Shkreta, L. Defective control of pre-messenger RNA splicing in human disease. J. Cell Biol. 2016, 212, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Ussar, S.; Fujisaka, S.; Kahn, C.R. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Mol. Metab. 2016, 5, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, S.H.; Lee, M.S.; Kim, M.S. Epigenetic modification by dietary factors: Implications in metabolic syndrome. Mol. Asp. Med. 2017, 54, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Watza, D.; Findley, A.; Alazizi, A.; Wen, X.; Pai, A.A.; Pique-Regi, R.; Luca, F. Environmental perturbations lead to extensive directional shifts in RNA processing. PLoS Genet. 2017, 13, e1006995. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, V.E.; Kwo, J. Obesity: Physiologic changes and implications for preoperative management. BMC Anesthesiol. 2015, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Holly, A.C.; Melzer, D.; Pilling, L.C.; Fellows, A.C.; Tanaka, T.; Ferrucci, L.; Harries, L.W. Changes in splicing factor expression are associated with advancing age in man. Mech. Ageing Dev. 2013, 134, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.P.; Pilling, L.C.; Emond, F.; Flurkey, K.; Harrison, D.E.; Yuan, R.; Peters, L.L.; Kuchel, G.A.; Ferrucci, L.; Melzer, D.; et al. Changes in the expression of splicing factor transcripts and variations in alternative splicing are associated with lifespan in mice and humans. Aging Cell 2016, 15, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Harries, L.W. Splicing regulatory factors, ageing and age-related disease. Ageing Res. Rev. 2017, 36, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Pihlajamaki, J.; Lerin, C.; Itkonen, P.; Boes, T.; Floss, T.; Schroeder, J.; Dearie, F.; Crunkhorn, S.; Burak, F.; Jimenez-Chillaron, J.C.; et al. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab. 2011, 14, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Vernia, S.; Edwards, Y.J.; Han, M.S.; Cavanagh-Kyros, J.; Barrett, T.; Kim, J.K.; Davis, R.J. An alternative splicing program promotes adipose tissue thermogenesis. Elife 2016, 5, e17672. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, D.; Kakela, P.; Nikkola, E.; Venesmaa, S.; Ilves, I.; Herzig, K.H.; Kolehmainen, M.; Karhunen, L.; Kuusisto, J.; Gylling, H.; et al. Regulation of alternative splicing in human obesity loci. Obesity 2016, 24, 2033–2037. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.H.; Masiarz, F.; Kan, Y.W.; Goldfine, I.D.; et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell 1985, 40, 747–758. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Norgaard-Pedersen, D.; Brandt, J.; Pettersson, I.; Slaaby, R. IGF1 and IGF2 specificities to the two insulin receptor isoforms are determined by insulin receptor amino acid 718. PLoS ONE 2017, 12, e0178885. [Google Scholar] [CrossRef] [PubMed]
- Sesti, G.; D’Alfonso, R.; Vargas Punti, M.D.; Frittitta, L.; Trischitta, V.; Liu, Y.Y.; Borboni, P.; Longhi, R.; Montemurro, A.; Lauro, R. Peptide-based radioimmunoassay for the two isoforms of the human insulin receptor. Diabetologia 1995, 38, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Sell, S.M.; Reese, D.; Ossowski, V.M. Insulin-inducible changes in insulin receptor mRNA splice variants. J. Biol. Chem. 1994, 269, 30769–30772. [Google Scholar] [PubMed]
- Mosthaf, L.; Vogt, B.; Haring, H.U.; Ullrich, A. Altered expression of insulin receptor types A and B in the skeletal muscle of non-insulin-dependent diabetes mellitus patients. Proc. Natl. Acad. Sci. USA 1991, 88, 4728–4730. [Google Scholar] [CrossRef] [PubMed]
- Sbraccia, P.; D’Adamo, M.; Leonetti, F.; Caiola, S.; Iozzo, P.; Giaccari, A.; Buongiorno, A.; Tamburrano, G. Chronic primary hyperinsulinaemia is associated with altered insulin receptor mRNA splicing in muscle of patients with insulinoma. Diabetologia 1996, 39, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Bodkin, N.L.; Ortmeyer, H.K.; Hansen, B.C.; Shuldiner, A.R. Hyperinsulinemia is associated with altered insulin receptor mRNA splicing in muscle of the spontaneously obese diabetic rhesus monkey. J. Clin. Investig. 1994, 94, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Bodkin, N.L.; Ortmeyer, H.K.; Zenilman, M.E.; Webster, N.J.; Hansen, B.C.; Shuldiner, A.R. Altered insulin receptor messenger ribonucleic acid splicing in liver is associated with deterioration of glucose tolerance in the spontaneously obese and diabetic rhesus monkey: Analysis of controversy between monkey and human studies. J. Clin. Endocrinol. Metab. 1996, 81, 1552–1556. [Google Scholar] [PubMed]
- Kaminska, D.; Hamalainen, M.; Cederberg, H.; Kakela, P.; Venesmaa, S.; Miettinen, P.; Ilves, I.; Herzig, K.H.; Kolehmainen, M.; Karhunen, L.; et al. Adipose tissue INSR splicing in humans associates with fasting insulin level and is regulated by weight loss. Diabetologia 2014, 57, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Besic, V.; Shi, H.; Stubbs, R.S.; Hayes, M.T. Aberrant liver insulin receptor isoform a expression normalises with remission of type 2 diabetes after gastric bypass surgery. PLoS ONE 2015, 10, e0119270. [Google Scholar] [CrossRef] [PubMed]
- Escribano, O.; Beneit, N.; Rubio-Longas, C.; Lopez-Pastor, A.R.; Gomez-Hernandez, A. The Role of Insulin Receptor Isoforms in Diabetes and Its Metabolic and Vascular Complications. J. Diabetes Res. 2017, 2017, 1403206. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Talukdar, I.; Webster, N.J. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol. Cell. Biol. 2009, 29, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Jumaa, H.; Webster, N.J. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 2013, 4, 1336. [Google Scholar] [CrossRef] [PubMed]
- Gorska, E.; Popko, K.; Stelmaszczyk-Emmel, A.; Ciepiela, O.; Kucharska, A.; Wasik, M. Leptin receptors. Eur. J. Med. Res. 2010, 15, 50–54. [Google Scholar] [PubMed]
- Fei, H.; Okano, H.J.; Li, C.; Lee, G.H.; Zhao, C.; Darnell, R.; Friedman, J.M. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc. Natl. Acad. Sci. USA 1997, 94, 7001–7005. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Proenca, R.; Montez, J.M.; Carroll, K.M.; Darvishzadeh, J.G.; Lee, J.I.; Friedman, J.M. Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996, 379, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Uotani, S.; Bjorbaek, C.; Tornoe, J.; Flier, J.S. Functional properties of leptin receptor isoforms: Internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes 1999, 48, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: Role of short-form leptin receptors and osteopontin. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ioffe, E.; Fidahusein, N.; Connolly, E.; Friedman, J.M. Absence of soluble leptin receptor in plasma from dbPas/dbPas and other db/db mice. J. Biol. Chem. 1998, 273, 10078–10082. [Google Scholar] [CrossRef] [PubMed]
- Aubert, R.; Herzog, J.; Camus, M.C.; Guenet, J.L.; Lemonnier, D. Description of a new model of genetic obesity: The dbPas mouse. J. Nutr. 1985, 115, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Van Dielen, F.M.; van’t Veer, C.; Buurman, W.A.; Greve, J.W. Leptin and soluble leptin receptor levels in obese and weight-losing individuals. J. Clin. Endocrinol. Metab. 2002, 87, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Maamra, M.; Bidlingmaier, M.; Postel-Vinay, M.C.; Wu, Z.; Strasburger, C.J.; Ross, R.J. Generation of human soluble leptin receptor by proteolytic cleavage of membrane-anchored receptors. Endocrinology 2001, 142, 4389–4393. [Google Scholar] [CrossRef] [PubMed]
- Clement, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998, 392, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Cornelis, M.C.; Kraft, P.; Qi, L.; van Dam, R.M.; Girman, C.J.; Laurie, C.C.; Mirel, D.B.; Gong, H.; Sheu, C.C.; et al. Genome-wide association study identifies polymorphisms in LEPR as determinants of plasma soluble leptin receptor levels. Hum. Mol. Genet. 2010, 19, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Schaab, M.; Kratzsch, J. The soluble leptin receptor. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ceccarini, G.; Eisenstein, M.; Tan, K.; Friedman, J.M. Phenotypic effects of an induced mutation of the ObRa isoform of the leptin receptor. Mol. Metab. 2013, 2, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol. 2001, 21, 6091–6101. [Google Scholar] [CrossRef] [PubMed]
- Goodson, M.L.; Mengeling, B.J.; Jonas, B.A.; Privalsky, M.L. Alternative mRNA splicing of corepressors generates variants that play opposing roles in adipocyte differentiation. J. Biol. Chem. 2011, 286, 44988–44999. [Google Scholar] [CrossRef] [PubMed]
- Goodson, M.L.; Young, B.M.; Snyder, C.A.; Schroeder, A.C.; Privalsky, M.L. Alteration of NCoR corepressor splicing in mice causes increased body weight and hepatosteatosis without glucose intolerance. Mol. Cell. Biol. 2014, 34, 4104–4114. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.A.; Goodson, M.L.; Schroeder, A.C.; Privalsky, M.L. Regulation of corepressor alternative mRNA splicing by hormonal and metabolic signaling. Mol. Cell. Endocrinol. 2015, 413, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, T.; Meyers, K.; Mullican, S.E.; Leitner, K.; Adeniji-Adele, A.; Avila, J.; Bucan, M.; Ahima, R.S.; Kaestner, K.H.; Lazar, M.A. Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature 2008, 456, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Astapova, I.; Vella, K.R.; Ramadoss, P.; Holtz, K.A.; Rodwin, B.A.; Liao, X.H.; Weiss, R.E.; Rosenberg, M.A.; Rosenzweig, A.; Hollenberg, A.N. The nuclear receptor corepressor (NCoR) controls thyroid hormone sensitivity and the set point of the hypothalamic-pituitary-thyroid axis. Mol. Endocrinol. 2011, 25, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Williams, E.G.; Mouchiroud, L.; Canto, C.; Fan, W.; Downes, M.; Heligon, C.; Barish, G.D.; Desvergne, B.; Evans, R.M.; et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 2011, 147, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fan, W.; Xu, J.; Lu, M.; Yamamoto, H.; Auwerx, J.; Sears, D.D.; Talukdar, S.; Oh, D.; Chen, A.; et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 2011, 147, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [PubMed]
- Revechon, G.; Viceconte, N.; McKenna, T.; Sola Carvajal, A.; Vrtacnik, P.; Stenvinkel, P.; Lundgren, T.; Hultenby, K.; Franco, I.; Eriksson, M. Rare progerin-expressing preadipocytes and adipocytes contribute to tissue depletion over time. Sci. Rep. 2017, 7, 4405. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Comai, L. Recent advances in understanding the role of lamins in health and disease. F1000Research 2016, 5, 2536. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Courvalin, J.C. How do mutations in lamins A and C cause disease? J. Clin. Investig. 2004, 113, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Chacon, M.R.; Gutierrez, C.; Vilarrasa, N.; Gomez, J.M.; Caubet, E.; Megia, A.; Vendrell, J. LMNA mRNA expression is altered in human obesity and type 2 diabetes. Obesity 2008, 16, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kieckhaefer, J.E.; Cao, K. Mouse models of laminopathies. Aging Cell 2013, 12, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mejia, I.C.; de Toledo, M.; Chavey, C.; Lapasset, L.; Cavelier, P.; Lopez-Herrera, C.; Chebli, K.; Fort, P.; Beranger, G.; Fajas, L.; et al. Antagonistic functions of LMNA isoforms in energy expenditure and lifespan. EMBO Rep. 2014, 15, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Peterfy, M.; Phan, J.; Reue, K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J. Biol. Chem. 2005, 280, 32883–32889. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.; Reue, K. Lipin, a lipodystrophy and obesity gene. Cell Metab. 2005, 1, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hu, M.; Liang, X.; Ajmo, J.M.; Li, X.; Bataller, R.; Odena, G.; Stevens, S.M., Jr.; You, M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014, 146, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Kurylowicz, A.; Owczarz, M.; Polosak, J.; Jonas, M.I.; Lisik, W.; Jonas, M.; Chmura, A.; Puzianowska-Kuznicka, M. SIRT1 and SIRT7 expression in adipose tissues of obese and normal-weight individuals is regulated by microRNAs but not by methylation status. Int. J. Obes. 2016, 40, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.L. Alternative splicing of mRNA as a mode of endocrine regulation. Trends Endocrinol. Metab. TEM 1997, 8, 405–413. [Google Scholar] [CrossRef]
- Verma, S.K.; Deshmukh, V.; Liu, P.; Nutter, C.A.; Espejo, R.; Hung, M.L.; Wang, G.S.; Yeo, G.W.; Kuyumcu-Martinez, M.N. Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling. J. Biol. Chem. 2013, 288, 35372–35386. [Google Scholar] [CrossRef] [PubMed]
- Nutter, C.A.; Jaworski, E.A.; Verma, S.K.; Deshmukh, V.; Wang, Q.; Botvinnik, O.B.; Lozano, M.J.; Abass, I.J.; Ijaz, T.; Brasier, A.R.; et al. Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes. Cell Rep. 2016, 15, 2200–2213. [Google Scholar] [CrossRef] [PubMed]
- Kuroyanagi, H. Fox-1 family of RNA-binding proteins. Cell. Mol. Life Sci. CMLS 2009, 66, 3895–3907. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Ou, J.; Zhu, L.J.; Green, M.R. Global Promotion of Alternative Internal Exon Usage by mRNA 3' End Formation Factors. Mol. Cell 2015, 58, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Arya, A.D.; Wilson, D.I.; Baralle, D.; Raponi, M. RBFOX2 protein domains and cellular activities. Biochem. Soc. Trans. 2014, 42, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Lu, Y.H.; Liu, Y.R.; Lin, Y.J. RBM4a-regulated splicing cascade modulates the differentiation and metabolic activities of brown adipocytes. Sci. Rep. 2016, 6, 20665. [Google Scholar] [CrossRef] [PubMed]
- Schlein, C.; Heeren, J. Implications of thermogenic adipose tissues for metabolic health. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Plutzky, J. Brown Fat and Browning for the Treatment of Obesity and Related Metabolic Disorders. Diabetes Metab. J. 2016, 40, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhong, L.; Lee, J.T.H.; Zhang, J.; Wu, D.; Geng, L.; Wang, Y.; Wong, C.M.; Xu, A. The FGF21-CCL11 Axis Mediates Beiging of White Adipose Tissues by Coupling Sympathetic Nervous System to Type 2 Immunity. Cell Metab. 2017, 26, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Preussner, M.; Goldammer, G.; Neumann, A.; Haltenhof, T.; Rautenstrauch, P.; Muller-McNicoll, M.; Heyd, F. Body Temperature Cycles Control Rhythmic Alternative Splicing in Mammals. Mol. Cell 2017, 67, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Sune-Pou, M.; Prieto-Sanchez, S.; Boyero-Corral, S.; Moreno-Castro, C.; El Yousfi, Y.; Sune-Negre, J.M.; Hernandez-Munain, C.; Sune, C. Targeting Splicing in the Treatment of Human Disease. Genes 2017, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.G.; Wood, M.J. RNA splicing: Disease and therapy. Brief. Funct. Genom. 2011, 10, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Le, K.Q.; Prabhakar, B.S.; Hong, W.J.; Li, L.C. Alternative splicing as a biomarker and potential target for drug discovery. Acta Pharmacol. Sin. 2015, 36, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Effenberger, K.A.; Urabe, V.K.; Jurica, M.S. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Misteli, T. Small Molecule Modulators of Pre-mRNA Splicing in Cancer Therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Morris, J.C.; Oltean, S.; Donaldson, L.F. Pharmacology of Modulators of Alternative Splicing. Pharmacol. Rev. 2017, 69, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Younis, I.; Berg, M.; Kaida, D.; Dittmar, K.; Wang, C.; Dreyfuss, G. Rapid-response splicing reporter screens identify differential regulators of constitutive and alternative splicing. Mol. Cell. Biol. 2010, 30, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Laustriat, D.; Gide, J.; Barrault, L.; Chautard, E.; Benoit, C.; Auboeuf, D.; Boland, A.; Battail, C.; Artiguenave, F.; Deleuze, J.F.; et al. In Vitro and In Vivo Modulation of Alternative Splicing by the Biguanide Metformin. Mol. Ther. Nucleic Acids 2015, 4, e262. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Foretz, M. Revisiting the mechanisms of metformin action in the liver. Ann. d’Endocrinol. 2013, 74, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Krainer, A.R. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol. Cancer Res. MCR 2014, 12, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Santo, J.; Lopez-Herrera, C.; Apolit, C.; Bareche, Y.; Lapasset, L.; Chavey, C.; Capozi, S.; Mahuteau-Betzer, F.; Najman, R.; Fornarelli, P.; et al. Pharmacological modulation of LMNA SRSF1-dependent splicing abrogates diet-induced obesity in mice. Int. J. Obes. 2017, 41, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Bakkour, N.; Lin, Y.L.; Maire, S.; Ayadi, L.; Mahuteau-Betzer, F.; Nguyen, C.H.; Mettling, C.; Portales, P.; Grierson, D.; Chabot, B.; et al. Small-molecule inhibition of HIV pre-mRNA splicing as a novel antiretroviral therapy to overcome drug resistance. PLoS Pathog. 2007, 3, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Isles, A.R. Htr2c Splice Variants and 5HT2CR-Mediated Appetite. Trends Endocrinol. Metab. TEM 2017, 28, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.B.; Ramond, F.; Farrington, D.T.; Aguiar, A.S., Jr.; Chevarin, C.; Berthiau, A.S.; Caussanel, S.; Lanfumey, L.; Herrick-Davis, K.; Hamon, M.; et al. RNA splicing and editing modulation of 5-HT(2C) receptor function: Relevance to anxiety and aggression in VGV mice. Mol. Psychiatry 2013, 18, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Halford, J.C.; Harrold, J.A. 5-HT(2C) receptor agonists and the control of appetite. Handb. Exp. Pharmacol. 2012, 209, 349–356. [Google Scholar]
- Kishore, S.; Stamm, S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 2006, 311, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Galiveti, C.R.; Raabe, C.A.; Konthur, Z.; Rozhdestvensky, T.S. Differential regulation of non-protein coding RNAs from Prader-Willi Syndrome locus. Sci. Rep. 2014, 4, 6445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shen, M.; Gresch, P.J.; Ghamari-Langroudi, M.; Rabchevsky, A.G.; Emeson, R.B.; Stamm, S. Oligonucleotide-induced alternative splicing of serotonin 2C receptor reduces food intake. EMBO Mol. Med. 2016, 8, 878–894. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Stevenson, P.L.; Carter, R.N.; Maccoll, G.; French, K.L.; Simons, J.P.; Al-Shawi, R.; Kelly, V.; Chapman, K.E.; Holmes, M.C. Overexpression of 5-HT2C receptors in forebrain leads to elevated anxiety and hypoactivity. Eur. J. Neurosci. 2009, 30, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Hache, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J. Child Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Paton, D.M. Nusinersen: Antisense oligonucleotide to increase SMN protein production in spinal muscular atrophy. Drugs Today 2017, 53, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Liu, Y.H.; Sahashi, K.; Rigo, F.; Bennett, C.F.; Krainer, A.R. Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev. 2015, 29, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nature commun. 2017, 8, 1476. [Google Scholar] [CrossRef] [PubMed]
- Jangi, M.; Fleet, C.; Cullen, P.; Gupta, S.V.; Mekhoubad, S.; Chiao, E.; Allaire, N.; Bennett, C.F.; Rigo, F.; Krainer, A.R.; et al. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E2347–E2356. [Google Scholar] [CrossRef] [PubMed]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [PubMed]
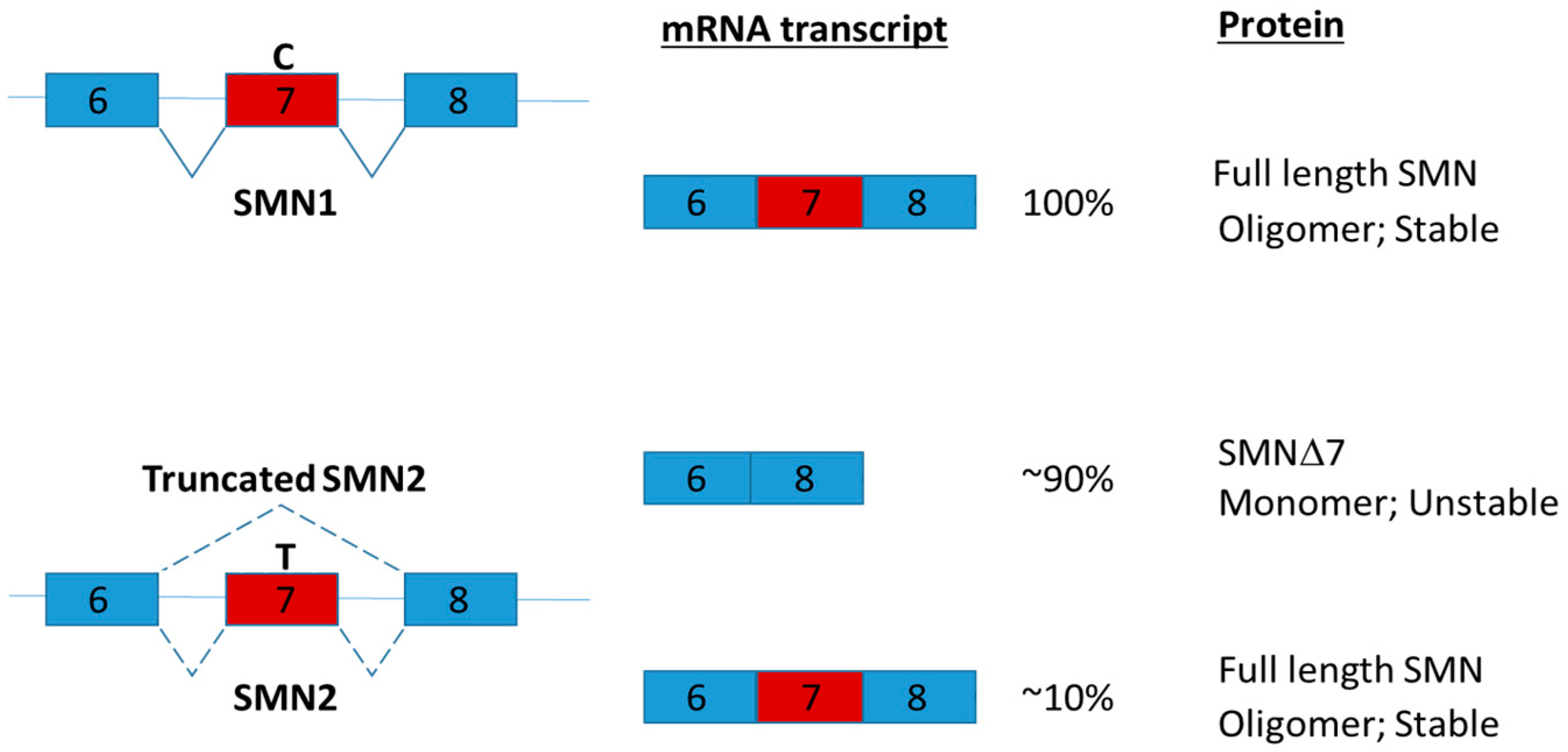
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.G.; Munoz, E.; Tandon, A.; Kwon, D.Y.; Sumner, C.J.; Fischbeck, K.H. Regulation of SMN protein stability. Mol. Cell. Biol. 2009, 29, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Maharshi, V.; Hasan, S. Nusinersen: The First Option beyond Supportive Care for Spinal Muscular Atrophy. Clin. Drug Investig. 2017, 37, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, K.J.; Prior, T.W.; Scott, C.B.; McNaught, T.P.; Wride, M.C.; Reyna, S.P.; Bromberg, M.B. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann. Neurol. 2005, 57, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Scoto, M.; Finkel, R.S.; Mercuri, E.; Muntoni, F. Therapeutic approaches for spinal muscular atrophy (SMA). Gene Ther. 2017, 24, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Tein, I.; Sloane, A.E.; Donner, E.J.; Lehotay, D.C.; Millington, D.S.; Kelley, R.I. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: Primary or secondary defect(s)? Pediatr. Neurol. 1995, 12, 21–30. [Google Scholar] [CrossRef]
- Crawford, T.O.; Sladky, J.T.; Hurko, O.; Besner-Johnston, A.; Kelley, R.I. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann. Neurol. 1999, 45, 337–343. [Google Scholar] [CrossRef]
- Bowerman, M.; Swoboda, K.J.; Michalski, J.P.; Wang, G.S.; Reeks, C.; Beauvais, A.; Murphy, K.; Woulfe, J.; Screaton, R.A.; Scott, F.W.; et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol. 2012, 72, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Michalski, J.P.; Beauvais, A.; Murray, L.M.; DeRepentigny, Y.; Kothary, R. Defects in pancreatic development and glucose metabolism in SMN-depleted mice independent of canonical spinal muscular atrophy neuromuscular pathology. Hum. Mol. Genet. 2014, 23, 3432–3444. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.; Ramirez, A.; Bucchia, M.; Rinchetti, P.; Rideout, H.; Papadimitriou, D.; Re, D.B.; Corti, S. Is spinal muscular atrophy a disease of the motor neurons only: Pathogenesis and therapeutic implications? Cell. Mol. Life Sci. 2016, 73, 1003–1020. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- Reautschnig, P.; Vogel, P.; Stafforst, T. The notorious R.N.A. in the spotlight-drug or target for the treatment of disease. RNA Biol. 2017, 14, 651–668. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Model | Key Metabolic Phenotypes | Proposed Mechanism | Reference |
|---|---|---|---|
| DADm: single amino acid substitution (Y478A) in the Ncor1 DAD domain that is unable to associate with or activate Hdac3 | Reduced weight and whole-body fat, Increased oxygen consumption and heat production Increased insulin sensitivity | Increased lipid consumption and obesity-resistant metabolic phenotype | [64] |
| NCoR ID : contains only 1 RID–N1 and thus would be unable to interact with the thyroid hormone receptor | Reduced body weight with a tendency for lower body fat content Increased oxygen consumption | Increased peripheral sensitivity to thyroid hormone | [65] |
| Muscle-specific KO | Increased of both muscle mass and of mitochondrial number and activity Reduced LDL cholesterol Improved insulin sensitivity Decreased in the respiratory exchange ratio | Increased muscle quantity and oxidative profile | [66] |
| Adipocyte-specific KO | Increased adipocyte hyperplasia Reduced inflammation in adipose tissue Increased insulin sensitivity in major metabolic organs (liver, fat and muscle) | Decreased inflammation contributing to the enhancement of insulin sensitivity | [67] |
| NCoRω−/−: NCoRω splice-specific knockout | Increased glucose tolerance Enhanced insulin resistance Increased the size of adipocytes Enhanced liver steatosis Elevated total serum cholesterol level and LDL complexes Reduced in the levels of circulating triglycerides and free fatty acids | Retention of the NCoRω splice variant counteracts prodiabetic physiology in the animals on the HFD | [62] |
| Gene | Changes of Variant Level in Pathological Conditions of Obese Subject | Key Changes in Metabolic Phenotypes | References |
|---|---|---|---|
| Insulin receptor (IR) | Increase IR-A (skipping of exon 11) in the liver | Correlate with fasting plasma glucose and insulin level | [41,42,43] |
| Leptin receptor (OB-R) | Decrease soluble OB-R * | Correlate with body mass index | [52,53,54] |
| LMNA | Increase Lamin C in subcutaneous adipose tissue | Correlate with type 2 diabetes | [72] |
| Lipin-1 | Increase Lipin 1-A (skipping of 7) in liver | Cause alcoholic fatty liver disease | [76] |
| RNA Binding Protein, Fox-1 Homolog 2 (RBFOX2) | Increase truncated RBFOX2 (skipping of exon 6) in heart | Lower calcium handling in diabetic heart | [81] |
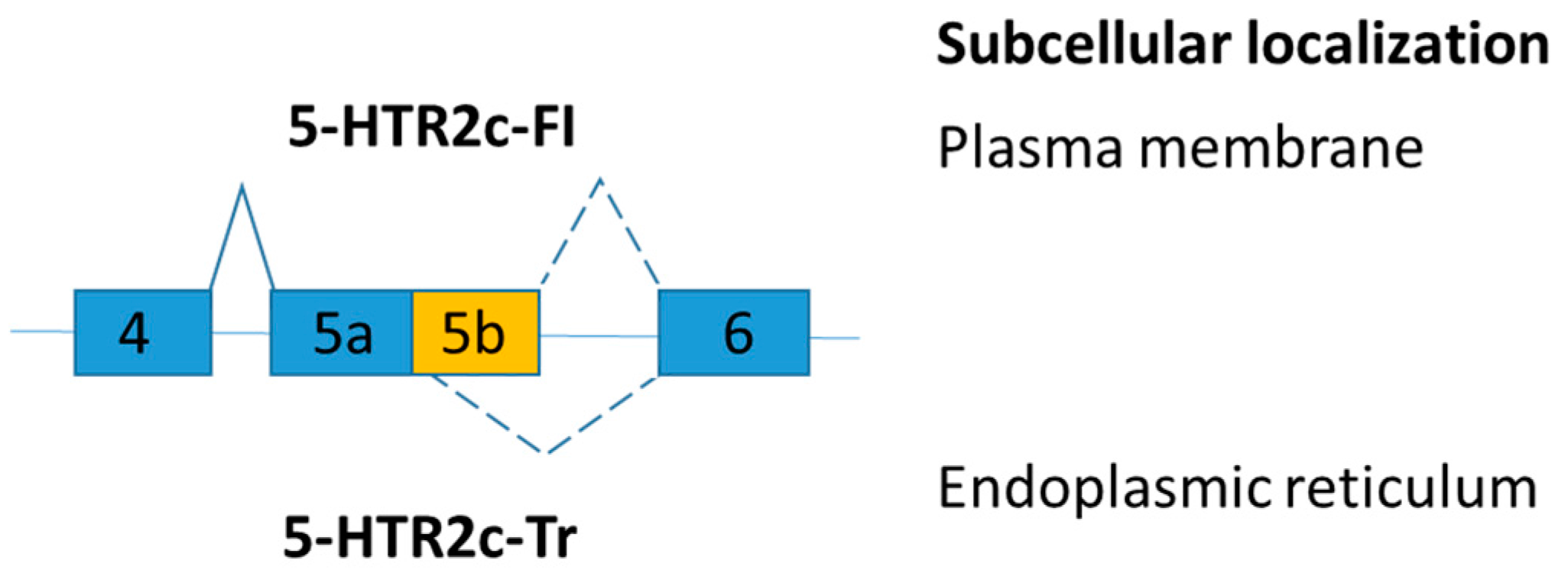
| Serotonin 2C receptor (5-HTR2c) | Increase truncated 5-HTR2c (skipping of exon 5b) in brain | Reduce satiety and enhance food intake | [109] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, C.-M.; Xu, L.; Yau, M.Y.-C. Alternative mRNA Splicing in the Pathogenesis of Obesity. Int. J. Mol. Sci. 2018, 19, 632. https://doi.org/10.3390/ijms19020632
Wong C-M, Xu L, Yau MY-C. Alternative mRNA Splicing in the Pathogenesis of Obesity. International Journal of Molecular Sciences. 2018; 19(2):632. https://doi.org/10.3390/ijms19020632
Chicago/Turabian StyleWong, Chi-Ming, Lu Xu, and Mabel Yin-Chun Yau. 2018. "Alternative mRNA Splicing in the Pathogenesis of Obesity" International Journal of Molecular Sciences 19, no. 2: 632. https://doi.org/10.3390/ijms19020632
APA StyleWong, C.-M., Xu, L., & Yau, M. Y.-C. (2018). Alternative mRNA Splicing in the Pathogenesis of Obesity. International Journal of Molecular Sciences, 19(2), 632. https://doi.org/10.3390/ijms19020632