Abstract
The SWEET (sugars will eventually be exported transporters) proteins are sugar efflux transporters containing the MtN3_saliva domain, which affects plant development as well as responses to biotic and abiotic stresses. These proteins have not been functionally characterized in the tetraploid cotton, Gossypium hirsutum, which is a widely cultivated cotton species. In this study, we comprehensively analyzed the cotton SWEET gene family. A total of 55 putative G. hirsutum SWEET genes were identified. The GhSWEET genes were classified into four clades based on a phylogenetic analysis and on the examination of gene structural features. Moreover, chromosomal localization and an analysis of homologous genes in Gossypium arboreum, Gossypium raimondii, and G. hirsutum suggested that a whole-genome duplication, several tandem duplications, and a polyploidy event contributed to the expansion of the cotton SWEET gene family, especially in Clade III and IV. Analyses of cis-acting regulatory elements in the promoter regions, expression profiles, and artificial selection revealed that the GhSWEET genes were likely involved in cotton developmental processes and responses to diverse stresses. These findings may clarify the evolution of G. hirsutum SWEET gene family and may provide a foundation for future functional studies of SWEET proteins regarding cotton development and responses to abiotic stresses.
1. Introduction
In plants, sugars are an essential carbon and energy source for growth and development. Additionally, unicellular organisms use sugars as a principal carbon source for energy production and as nutrients supplements. Multicellular organisms acquire sugars, such as glucose or sucrose, which are transported from source to sink organs and are associated with the cellular exchange of carbon and energy [1,2]. Sugar transport is controlled by the SUT (sucrose transporter), SWEET (sugars will eventually be exported transporters; sugar effluxer) proteins and so on [3,4,5]. The SUT proteins have been detected in many species and are responsible for the loading sucrose into phloem across cell membranes [6]. Meanwhile, the SWEET proteins were identified as sugar effluxers that transport monosaccharides or disaccharides across intracellular or plasma membranes [7,8]. The SWEET proteins are characterized by seven conserved MtN3_saliva transmembrane domains, which comprise a pair of PQ-loop repeats. Members of the SWEET family have been identified in plants, human, protozoa, metazoa, fungi, bacteria, and archaea [9,10,11]. Recent investigations detected 17 and 21 SWEET genes in Arabidopsis thaliana and rice, respectively, and up to 52 in soybean [12,13,14]. A phylogenetic analysis revealed that these genes can be classified into four clades [12,14,15]. Clade members interact with each other to facilitate the efficient diffusion of sugars for required cellular activities, but are also critical for nectar production and seed, embryo, and pollen development [2,12,16,17]. For example, AtSWEET2, AtSWEET16 and AtSWEET17 not just collectively mediate vacuolar sugar transport, and that exhibit strong expression in root cortical cells [16,17,18].
The SWEET gene family has been investigated in A. thaliana [12], rice [13], grapevine [19], soybean [14], tomato [20], potato [15], cucumber [21] and wheat [22]. The first identified plant SWEET gene is AtSWEET1, which encodes a protein involved in supplying nutrients to the gametophyte or nectary [12]. Moreover, AtSWEET8 is required for pollen viability [12], while AtSWEET11 and AtSWEET12 are localized in the plasma membrane, where they function as the predominant regulators of sucrose, glucose and fructose fluxes and contribute to the response to cold stress and water deficit conditions [3,23]. The overexpression of AtSWEET16 results in enhanced freezing tolerance [24]. In rice, OsSWEET1a, OsSWEET2a, OsSWEET3a, OsSWEET4, OsSWEET5, and OsSWEET15 are highly expressed in flowers or panicles at different developmental stages [13]. An earlier study confirmed that OsSWEET14-silenced mutants produced relatively small seeds and exhibited delayed growth [13]. Additionally, the expression levels of most SlSWEET genes were observed to be upregulated several fold in response to salt and temperature stresses in tomato [20]. Furthermore, BnSWEET12 expression is positively induced by abscisic acid (ABA), gibberellin (GA) and brassinosteroid (BR) treatments in oilseed rape [25]. Therefore, SWEET genes are involved in diverse physiological and biochemical pathways [15,19,26].
Cotton is one of the most important fiber crops worldwide, and cottonseed oil can be used as biodiesel [27,28]. Gossypium hirsutum, a tetraploid cotton, is the most widely cultivated cotton species, that supplies about 98% of the produced textile fibers [29]. However, to date, there have been relatively few studies conducted on cotton SWEET proteins. A recent study confirmed that the expression of a certain SWEET gene in cotton is induced by a transcription activator-like effector, which influences Xanthomonas citri subsp. malvacearum pathogenicity [30]. The suppressed expression of GhSCP2D, which encodes a putative sterol carrier protein, activates a cohort of SWEET genes, resulting in the elongation of cotton fiber cells [31]. Thus, a comprehensive analysis of the G. hirsutum SWEET gene family is essential. The completion of the tetraploid upland cotton genome sequencing project has enabled a genome-wide identification and functional analysis of the SWEET gene family in cotton.
In the present study, we conducted a genome-wide analysis to identify 55 putative SWEET gene family members in G. hirsutum. We also analyzed their phylogenetic relationships, structures, localization, genetic variations, and expression profiles in different tissues and in response to cold, heat, salt, and drought stresses. Our results provide the basis for future investigations of the roles of the SWEET proteins in developing cotton plants.
2. Results
2.1. Identification and Characterization of the G. hirsutum SWEET Gene Family
A total of 58 putative G. hirsutum SWEET genes were detected, and their coding sequences were manually confirmed using the FGENESH gene prediction tool as well as cDNA sequences after performing PCR with gene-specific primers. Consequently, three genes whose genome sequences and coding sequences were very short were excluded, and the coding sequences of 22 SWEET genes were modified from their initial annotation in the CottonGen database. Finally, the 55 SWEET genes confirmed in the G. hirsutum genome were named GhSWEET1–GhSWEET55 according to their chromosomal position (Tables 1 and S2). The number of amino acids, Molecular weight (MW) and Isoelectric point (pI) were calculated on the basis of the predicted protein sequences. Furthermore, for comparative analyses, 31 and 30 SWEET genes were identified in the Gossypium raimondii and Gossypium arboreum genomes, respectively, using the same methods (Table S3).

Table 1.
Characteristics of the G. hirsutum SWEET genes.
2.2. Phylogenetic Analysis and Structural Features of the SWEET Gene Family
To investigate the evolutionary relationships among the SWEET genes from three Gossypium species, A. thaliana, and rice, an unrooted phylogenetic tree was constructed using the neighbor-joining method (Figure 1). The SWEET genes were clearly divided into four classes (Clades I, II, III, and IV), consistently with the reported results for A. thaliana and rice SWEET genes [12,14]. The largest clade was Clade III, which consisted of five OsSWEET genes, seven AtSWEET genes, and 45 cotton SWEET genes (12 G. raimondii, 12 G. arboreum, and 21 G. hirsutum SWEET genes). Clade I comprised six OsSWEET and three AtSWEET genes, while Clade II included seven OsSWEET and five AtSWEET genes. Clades I and II contained an equal number of cotton SWEET genes, with six G. raimondii, six G. arboreum, and eleven G. hirsutum SWEET genes. Clade IV consisted of only two A. thaliana and one rice SWEET genes, but seven G. raimondii, six G. arboreum, and 12 G. hirsutum SWEET genes. These results suggested that SWEET gene families expanded, especially in Clade III and IV, during cotton evolution.
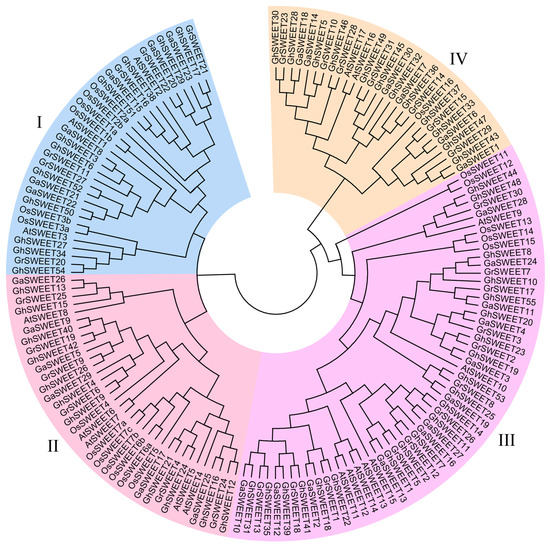
Figure 1.
Neighbor-joining phylogenetic tree of SWEET genes from three cotton species, A. thaliana, and rice. The SWEET genes were classified into four clades, which are indicated by different colors.
To elucidate the structural features of G. hirsutum SWEET genes, the exon-intron organization was analyzed. Gene structures generally exhibited a highly conserved distribution of exons and introns within the same clade or subclade (Figure 2B). Specifically, most Clade I genes (except GhSWEET38) and all Clade IV genes contained five introns. Meanwhile, Clade II was divided into two subclades, IIa and IIb. The six subclade IIa genes had five introns, while the subclade IIb genes contained four introns. Of the 21 Clade III genes, 19 had five introns, while GhSWEET10 comprised four introns and GhSWEET48, which was relatively short, contained three introns. These results revealed that introns were distributed consistently among members of the same phylogenetic clade.
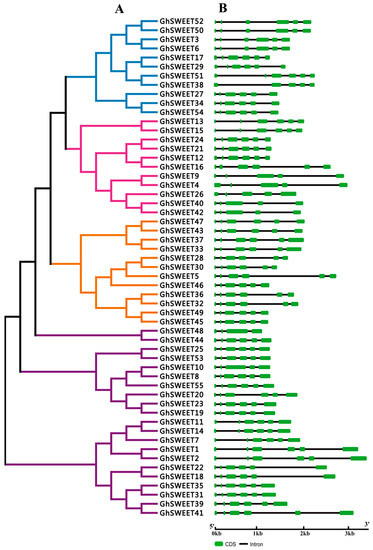
Figure 2.
Phylogenetic relationships and structural features of GhSWEET genes. (A) Neighbor-joining phylogenetic tree of GhSWEET genes. The SWEET genes were classified into four clades, and blue, pink, purple, and orange represent Clades I, II, III, and IV, respectively. (B) Exon-intron organization of GhSWEET genes. Green and black correspond to exons and introns, respectively.
2.3. Chromosomal Distribution and Homology of G. hirsutum SWEET Genes
G. hirsutum is a model for polyploid crop domestication, whose complex allotetraploid genome (AtDt, where “t” stands for tetraploid) was sequenced and oriented to 26 pseudochromosomes. Among the 55 G. hirsutum SWEET genes, 49 were localized on 12 of 13 At chromosomes and 11 of 13 Dt chromosomes (Figure 3). Six genes (GhSWEET50, GhSWEET51, GhSWEET52, GhSWEET53, GhSWEET54, and GhSWEET55) were distributed on scaffolds whose exact locations on chromosomes were not determined. Chromosomes At-chr7, At-chr11, Dt-chr11 and Dt-chr13 contained four genes, while chromosomes Dt-chr2, Dt-chr4, Dt-chr7, and Dt-chr3 included three genes each. Interestingly, 14 genes were clustered in seven tandem duplication regions at the chromosomes Dt-chr4, At-chr5, At-chr7, Dt-chr7, At-chr11, Dt-chr11, and Dt-chr13. Of these, similar clusters were identified between homoeologous chromosomes (At-chr7 and Dt-chr7 as well as At-chr11 and Dt-chr11).
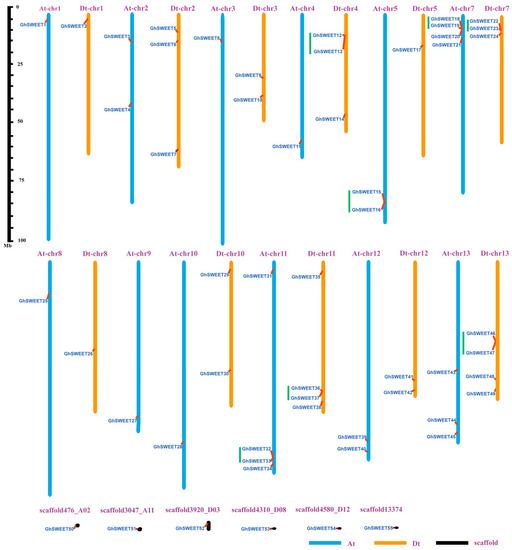
Figure 3.
Chromosomal distribution of GhSWEET genes. Blue, orange and black indicate the At subgenome chromosomes, Dt subgeome chromosomes, and scaffolds, respectively. Green lines indicate tandem duplicated genes. The scale is provided in megabases.
Most angiosperms have undergone at least one paleopolyploidy event during evolution [32,33]. The genome of G. hirsutum (AtDt), which is a typical allotetraploid, underwent a diploidization following the divergence of this cotton species from its diploid ancestors, G. arboreum (AA) and G. raimondii (DD). Firstly, we analyzed the SWEET paralogs between the At and Dt subgenomes (Figure 4). A total of 23 paralogous gene pairs were identified, with 18 of them anchored across eight homoeologous chromosomes pairs. To further clarify the divergence during cotton evolution, we analyzed the orthologous SWEET genes between the G. hirsutum Dt subgenome and the corresponding ancestral D diploid genome (G. raimondii), as well as between the G. hirsutum At subgenome and the corresponding ancestral A diploid genome (G. arboreum) (Figure S1). A total of 28 genes in the Dt subgenome had orthologs in the G. raimondii genome, while 23 genes in the At subgenome had orthologs in the G. arboreum genome. This revealed that the collinear relationship of the SWEET genes between the Dt subgenome and the G. raimondii genome was higher than that between the At subgenome and the G. arboreum genome, indicating either that more At subgenome SWEET genes were lost during evolution, or that genome sequencing was incomplete, leading to the identification of only a portion of the existing genes.
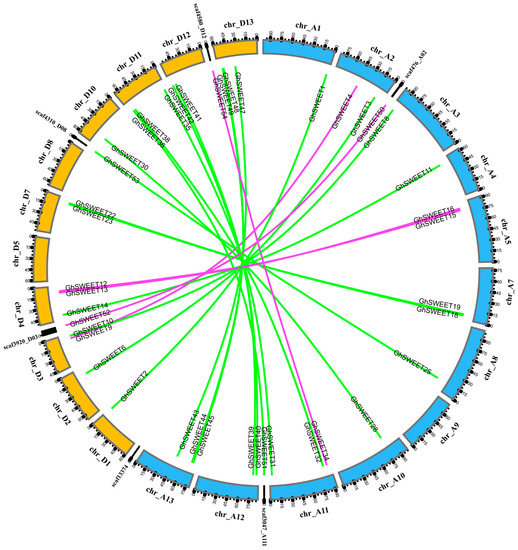
Figure 4.
Distribution of paralogous SWEET gene pairs between G. hirsutum At and Dt subgenomes. The 12 At subgenome chromosomes, 11 Dt subgenome chromosomes, and six scaffolds are indicated by different colors, with their names on the periphery. Green lines link the homologous genes located on homoeologous chromosomes in the At and Dt subgenomes. Purple lines link the homologous genes located on non-homoeologous chromosomes in the At and Dt subgenomes and scaffolds.
2.4. Analysis of Cis-Acting Regulatory Elements in the Putative GhSWEET Promoters
Plant SWEET genes are associated with responses to abiotic stresses [34,35,36]. Therefore, we analyzed the 1.5-kb sequence upstream of the ATG start codon using the PlantCARE database to detect putative cis-acting regulatory elements [37]. The GhSWEET promoters contained multiple conserved regulatory elements responsive to phytohormones and environmental stresses (Table S4). Elements related to defense stress, heat, GA, salicylic acid (SA), and drought were widely distributed across the promoter regions of 43, 42, 38, 36, and 35 SWEET genes, respectively. According to the regulatory elements in their promoters, 31 GhSWEET genes were responsive to methyl jasmonate (MeJA), while 25 genes were responsive to ABA and ethylene. In contrast, low temperatures and auxin appeared to affect a low number of GhSWEET genes. These findings implied that GhSWEET genes were involved in various regulatory mechanisms in cotton plants’ response to stresses.
2.5. G. hirsutum SWEET Gene Expression Profiles in Different Tissues
To investigate the roles of GhSWEET genes in different developmental processes, we used publicly available RNA-seq data to analyze SWEET gene expression patterns in seven different G. hirsutum tissues (root, stem, leaf, flower, ovule, seed, and fiber). The SWEET genes were clustered in four groups according to their expression profiles (Figure 5). The five Group I genes were highly expressed in the ovule and fiber. Meanwhile, the Group II genes, GhSWEET1, GhSWEET2, and GhSWEET13, were expressed specifically in the ovule, while most of the Group III genes were expressed in the root and flower. Moreover, the genes in the first clade of Group III (GhSWEET3, GhSWEET6, GhSWEET7, GhSWEET40, GhSWEET42, GhSWEET38, and GhSWEET51) were very highly expressed in the seed and ovule. Additionally, the Group IV genes exhibited diverse expression patterns, primarily in six tissues (root, stem, leaf, flower, ovule, and fiber). These results suggested that the SWEET genes were important for vegetative and reproductive growth.
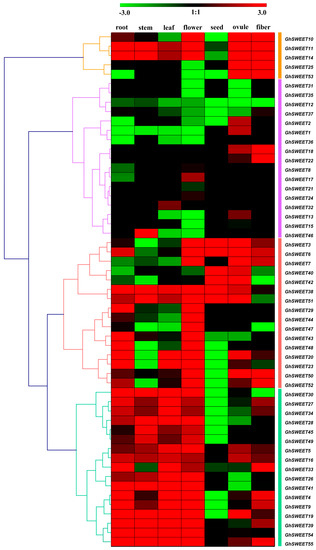
Figure 5.
Expression profiles of GhSWEET genes in different tissues. GhSWEET gene expression profiles are divided into four groups. The FPKM values were calculated by RNA-seq data, and shown as a heat map. The colored bar represents the scale of the relative expression levels.
2.6. Stress-Induced Expression Patterns of G. hirsutum SWEET Genes
Plants are often subjected to multiple abiotic stresses during growth and development. Thus, the expression patterns of the 55 GhSWEET genes were analyzed in plants exposed to different durations of cold, heat, salt, and drought stresses for different times by RNA-seq data downloaded from the public database. The expression of some SWEET genes was significantly affected by these treatments (Figure 6). The expression levels of six genes (GhSWEET5, GhSWEET20, GhSWEET45, GhSWEET49, GhSWEET51, and GhSWEET55) were increased by all four abiotic stresses. GhSWEET5, GhSWEET49, and GhSWEET55 expression levels were upregulated at 6 h but decreased quickly at 12 h under heat and salt stress conditions. The paralogous genes GhSWEET45 and GhSWEET49 exhibited similar expression patterns in response to multiple stresses. Overall, cold stress produced lower SWEET gene expression levels than the other types of stress. Additionally, some genes were differentially expressed at only one time point of a particular stress treatment. For example, GhSWEET11 expression was upregulated after 1 h of cold treatment, but was subsequently downregulated.

Figure 6.
Expression patterns of GhSWEET genes in response to various stresses. The ratios of FPKM between treatments (at 1, 3, 6 and 12 h) and controls (at 0 h) were calculated by RNA-seq data, and shown as a heat map. The colored bar represents the scale of the relative expression levels.
To further verify SWEET gene expression patterns in response to stresses, we selected five SWEET genes whose expression was induced by heat and drought conditions, and examined their expression profiles following exposure to 38 °C or 20% PEG 6000 (Figure 7). The qRT-PCR data revealed that the expression levels of the five genes were significantly upregulated by these two treatments. The GhSWEET20 and GhSWEET51 transcript levels quickly peaked after 1 h of heat treatment, whereas the increased expression of the GhSWEET5 transcript occurred after 3 h. Meanwhile, the GhSWEET49 and GhSWEET55 expression levels gradually increased, peaking after 6 h. In contrast, the drought-induced transcription of four SWEET genes (GhSWEET5, GhSWEET20, GhSWEET49 and GhSWEET55) peaked after 1 h of stress induction, but then rapidly decreased afterward. These results implied that GhSWEET genes might enhance the adaptability of plants to diverse abiotic stresses.
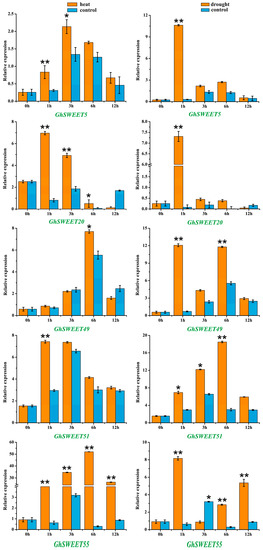
Figure 7.
Relative expression levels of the selected GhSWEET genes in response to heat and drought stresses. The GhHis3 was used as an internal control. Relative gene expression levels were calculated based on the 2−ΔΔCT method. The mean expression values were calculated on the basis of three independent replicates. Error bars indicate the standard deviations of three independent experiments. The relative expression levels of selected GhSWEET genes after treats were compared with the controls at the same time point. Significant differences were determined by t-test (* p < 0.05; ** p < 0.01).
2.7. Genetic Variations and Artificial Selection of GhSWEET Genes During Cotton Domestication
The increase in the available re-sequencing data for wild and cultivated cotton species has enabled the analysis of the changes in GhSWEET genes that occurred during cotton domestication. In this study, the single nucleotide polymorphisms (SNPs) in all GhSWEET genes were obtained for the wild and domesticated species using re-sequencing data from 31 wild and 321 cultivated cotton varieties (Table S5). A total of 16 genes lacked SNPs in the wild and domesticated cotton lines. In the wild species, 37 GhSWEET genes had at least one SNP, and the SNPs of 26 GhSWEET genes were detected in exons. In the domesticated species, 37 GhSWEET genes included SNPs, and the SNPs of 27 genes were present in exons. Additionally, the Dt subgenome comprised more SWEET genes with SNPs in exons than the At subgenome. Further analyses revealed that these genes with SNPs were distributed in the four clades of the phylogenetic tree. Specifically, Clade III members had 55 SNPs, i.e., more SNPs than the members of the other clades. Additionally, GhSWEET42 had 15 SNPs, i.e., more than any other gene. A subsequent calculation of the SNP density of all GhSWEET genes revealed that the SNP density of 10 SWEET genes (including eight At subgenome genes) in domesticated cotton was lower than in wild cotton (Figure 8). Moreover, GhSWEET19 and GhSWEET37 in domesticated upland cotton had no SNPs. Furthermore, the Fst values of the SNP loci during domestication indicated that 65.2% of the loci (88 of 135 SNP loci) were not under selection during evolution (Fst < 0.15). However, nine SNPs in seven G. hirsutum SWEET genes (GhSWEET14, GhSWEET16, GhSWEET32, GhSWEET37, GhSWEET42, GhSWEET45, and GhSWEET54) satisfied the Fst value cutoff of 0.45. These results indicated that domestication might have a critical effect on cotton SWEET genes, perhaps because of their importance in cotton development and response to various stresses.
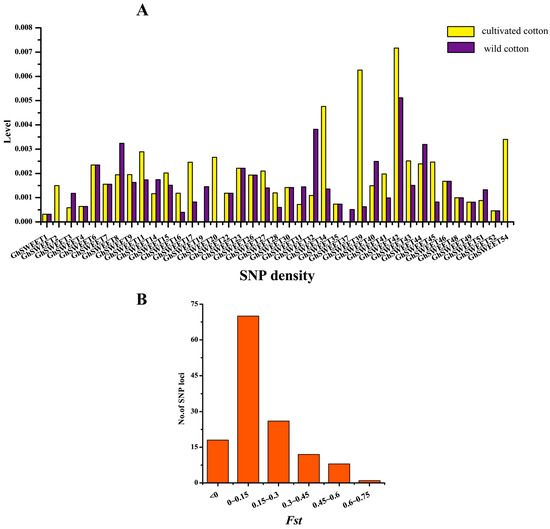
Figure 8.
Genetic variations in G. hirsutum SWEET genes. (A) SNP density in 39 GhSWEET genes. (B) Fst values of the SNP loci in 39 GhSWEET genes. The SNPs with a Fst value greater than 0.45 were considered to be loci under selection during domestication.
3. Discussion
The SWEET proteins have fundamental roles affecting sugar transport in plants, animals, and microorganisms. Genome-wide studies of the SWEET sugar transporter genes have been conducted for many plants [14,21]. However, a genome-wide characterization and functional analysis of cotton SWEET genes is still lacking. Additionally, there is increasing evidence that cotton SWEET proteins are crucial for stress responses and fiber development [30,31,38]. Thus, the comprehensive analysis of the identified G. hirsutum SWEET genes described herein may provide valuable information to elucidate the role of the corresponding gene family in cotton species.
3.1. Expansion of the Gossypium SWEET Gene Family
G. hirsutum is an allotetraploid species that resulted from the hybridization between an A-genome species resembling G. arboreum and a D-genome species resembling G. raimondii [29,39]. In the present study, 31, 30, and 55 SWEET genes were identified in the G. raimondii, G. arboreum, and G. hirsutum genomes, respectively. The fact that the SWEET genes are more abundant in cotton species than in A. thaliana (17), rice (21), grapevine (17), tomato (29), and cucumber (17) suggests that the cotton SWEET gene family expanded during evolution. Previous studies concluded that a Gossypium-specific whole-genome duplication (WGD) event occurred in the diploid cotton species G. raimondii and G. arboreum [33,40,41]. Therefore, the expansion of the SWEET gene family in G. raimondii and G. arboreum may have been due to an ancient WGD, with a subsequent polyploidization event leading to duplicate copies of genes in the G. hirsutum genome. Meanwhile, some SWEET gene clusters were detected in the G. hirsutum At and Dt subgenomes. Tandem duplication events have been important for the expansion of many gene families [42,43,44]. Therefore, we speculated that tandem duplications might have amplified some SWEET genes, leading to the emergence of SWEET gene clusters.
The SWEET genes are divided into four clades based on their phylogenetic relationships, with cotton presenting more Clade III and IV genes than other analyzed plant species. For example, the A. thaliana, rice, grapevine, tomato and cucumber genomes contain two [12], one [13], four [19], two [20] and three [21] Clade IV SWEET genes, respectively, i.e., fewer than the 7, 6, and 12 Clade IV SWEET genes present in the G. raimondii, G. arboreum, and G. hirsutum genomes, respectively. These differences suggested that Clade III and IV genes might be vital for the adaptation of cotton species to environmental conditions.
3.2. Expression Patterns of SWEET Gene Family
The spatiotemporal expression profiles of the SWEET genes have been reported for some species, including A. thaliana, rice, soybean, and cucumber. Most soybean SWEET genes are highly expressed in developing flowers and seeds [14]. Additionally, the eight tested CsSWEET genes from the four evolutionary clades were expressed at very high levels in male and female flowers [21]. In this study, we analyzed the GhSWEET transcript levels in seven tissues. According to our analyses of phylogenetic clusters (Figure 2A), similar exon-intron structures among most members of the same subfamily resulted in similar expression patterns in different tissues. More than half of the examined SWEET genes were predominantly expressed in the root, flower, ovule, and fiber, with relatively low expression levels in the stem and seed. Physiologically, the root, which absorbs nutrients from the soil, is the most fundamental organ with essential roles related to the maintenance of normal plant growth and development. Additionally, the flower, ovule, and fiber of cotton plants are associated with organogenesis, signal transduction pathways, and various metabolic processes. Our results implied that SWEET proteins exhibited diverse functions affecting cotton development and growth.
Previous studies revealed that SWEET genes not only influence plant growth, but they are also important for controlling plant responses to environmental stresses and phytohormones [15,20]. Thus, we investigated G. hirsutum SWEET gene expression patterns in response to different stresses (Figure 6). We observed that the expression levels of six GhSWEET genes were significantly upregulated by all four of the chosen treatments. A qRT-PCR analysis of five SWEET genes of them revealed that the expression levels of five of these SWEET genes were significantly up-regulated under heat and drought stress conditions (Figure 7). Specifically, two genes (GhSWEET20 and GhSWEET51) and four genes (GhSWEET5, GhSWEET20, GhSWEET49, and GhSWEET55) were considerably increased within 1 h of heat treatment and drought treatment, respectively. Therefore, we deduced that these genes have important roles in cotton stress resistance. In the future, these genes could be useful targets for breeding new cotton varieties with enhanced stress tolerance. Additionally, several hormone-responsive regulatory elements were identified in GhSWEET promoters (Table S4). This observation indicated that some SWEET genes might be expressed as part of phytohormone signaling pathways.
3.3. Functional Divergence of the Gossypium hirsutum SWEET Gene Family
Duplicated genes generally face one of three fates: non-functionalization, sub-functionalization, or neo-functionalization [45,46]. In this study, almost half of the duplicated gene pairs exhibited no differences in expression. These genes may have retained some essential functions based on gene dosage during evolution [47,48]. However, some paralogous SWEET gene pairs were associated with tissue-dependent diverse functions during plant growth and development. We observed that GhSWEET16 and GhSWEET33 were highly expressed in the root, leaf, and fiber, whereas their paralogous counterparts, GhSWEET12 and GhSWEET37, were silenced in these tissues (Figure 5). Thus, we speculated that the non-functionalization of one member of a duplicated gene pair might have been due to the maintenance of an appropriate gene dosage during cotton evolution. Additionally, one member of some duplicated gene pairs underwent neo-functionalization via WGD and polyploidy events. For example, AtSWEET1 is crucial for the regulation of floral development in A. thaliana [12]. In this study, its cotton orthologs (GhSWEET50 and GhSWEET52) influenced flowering, but were also abundantly expressed in the fiber, suggesting that these genes may contribute to cotton fiber development. In contrast, GhSWEET44 was specifically expressed in the flower, while its paralogous counterpart, GhSWEET48, was highly expressed in the root and flower. The variability in the expression profiles of paralogs in G. hirsutum may have been caused by several complex regulatory activities following gene duplication events.
Tandem duplications may have significantly affected the divergence of gene functions during evolution [49,50]. We examined the expression patterns of 14 tandem duplicated genes from seven clusters (Figure 5). As a result, all tandem duplicated pairs were differentially expressed. For example, GhSWEET32 was characteristically expressed in the leaf, while its tandem duplicated gene, GhSWEET33, was predominately expressed in the root, leaf and fiber. GhSWEET46 was specifically expressed in the stem, while its tandem duplicated gene, GhSWEET47, was abundantly expressed in developing flowers. This implied that tandem duplicated genes in the tetraploid cotton species might have exhibited functional redundancies and evolved new functions.
To date, the functions of some A. thaliana and rice SWEET proteins have been determined [12,13]. However, there is a relative lack of information regarding the functions of cotton SWEET proteins. In the present study, we provide evolutionary and functional insights into the roles of cotton SWEET gene family which will help future functional studies of the SWEET genes during cotton development and stress responses.
4. Materials and Methods
4.1. Identification and Characterization of G. hirsutum SWEET Genes
The G. hirsutum genome sequence and annotation files [39] were downloaded from the CottonGen website (available online: https://www.cottongen.org). Additionally, the related sequences of published A. thaliana and rice SWEET genes [13,14] were obtained from the TAIR 10 database (available online: http://www.arabidopsis.org) and the Rice Genome Database (http://www.ricedata.cn/gene/), respectively and their encoded protein sequences were used as queries in BLASTP searches of the G. hirsutum genome data. Next, putative SWEET proteins were filtered on the basis of the presence of a conserved MtN3_saliva domain using Pfam database analyses (E-value cut-off of 1.0; http://pfam.xfam.org/search#searchBatchBlock). The candidate SWEET genes were subsequently aligned. Meanwhile, inconsistencies in the sequence alignment were resolved and absent sequences were predicted using FGENESH software [51] and cloned to determine the complete sequences. The G. raimondii [33] and G. arboreum [40] SWEET genes were identified using similar methods.
The SWEET genes were named according to their chromosomal locations in G. hirsutum. Other related biological details, including the number of amino acids, molecular weight (MW), and isoelectric point (pI), were calculated using the DNAMAN (version 9.0) software (Lynnon Biosoft, Quebec City, QC, Canada).
4.2. Phylogenetic Analysis
Phylogenetic trees were constructed using full-length SWEET protein sequences and the neighbor-joining method of the MEGA (version 7.0) (Tokyo Metropolitan University, Tokyo, Japan) program with the pairwise gap deletion option [52]. The bootstrap analysis was conducted with 1000 replicates.
4.3. Gene Structural Features and Chromosomal Localization
Exon-intron structures were analyzed using the GSDS server (available online: http://gsds.cbi.pku.edu.cn/) [53]. Meanwhile, the physical positions of all cotton SWEET genes on chromosomes were determined according to the genome annotation files downloaded from the CottonGen website (available online: https://www.cottongen.org). The SWEET genes were mapped using MapInspect software [54] or Circos (version 0.69) software [55]. Tandem duplications were analyzed on the basis of the same or neighboring chromosomal regions [56]. Homologous genes including paralogous and orthologous genes were identified according to phylogenetic trees and sequence alignments [57,58].
4.4. RNA-Sequencing Analysis
To analyze the G. hirsutum SWEET gene expression profiles, the transcriptome data for various G. hirsutum TM-1 tissues were downloaded from the NCBI Sequence Read Archive (accession number PRJNA248163; available online: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA248163/). The gene expression levels were calculated on the basis of the normalized fragments per kilobase of transcript per million mapped fragments (FPKM). Hierarchical clustering was depicted using the Genesis (version 1.7.7) program [59].
4.5. Stress Treatments
Cotton seeds (TM-1) were sown in plastic pots filled with sand for about 10 days at 28 °C. The seedlings were transferred to a liquid culture medium under a 16h light-8h dark photoperiod until the third true leaf appeared. The cotton seedlings were treated with 20% PEG 6000 or heated at 38 °C. The leaves harvested at 0, 1, 3, 6, and 12 h, were immediately frozen in liquid nitrogen and stored at −80 °C for subsequent total RNA extraction.
4.6. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction Analysis
Total RNA was extracted from G. hirsutum leaves using the TRIzol reagent (Tiangen, Beijing, China). The quality and quantity of the purified RNA were assessed by 1.5% gel electrophoresis and with a NanoDrop 2000 spectrophotometer. For each sample, 1 μg RNA was reverse transcribed to cDNA using the PrimeScript™ RT Reagent Kit with gDNA Eraser (TaKaRa, Tokyo, Japan). Gene-specific primers were designed using the Primer-BLAST program (available online: https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome) (Table S1). The G. hirsutum His3 housekeeping gene was used as an internal control. A quantitative real-time polymerase chain reaction (qRT-PCR) was conducted with a Lightcycler480 96 system (Roche, Mannheim, Germany) in a 20 μL reaction mixture containing SYBR Premix Ex Taq (2×) (TaKaRa). The qRT-PCR was completed with three biological replicates, each comprising three technical replicates. The PCR conditions were as follows: 95 °C for 30 s; 40 cycles of 95 °C for 5 s, 60 °C for 1 min, and 72 °C for 10 s; 50 °C for 30 s. The relative gene expression levels were calculated based on the 2−ΔΔCT method [60]. Finally, the gene expression patterns were analyzed using the Origin 8 software (OriginLab Corporation, Northampton, MA, USA).
4.7. Analysis of Genetic Variations and Artificial Selection of G. hirsutum SWEET Genes
The whole genome re-sequencing data for 31 wild and 321 cultivated cotton lines were downloaded from the NCBI Sequence Read Archive (accession number SRP080913) [61]. Single nucleotide polymorphisms (SNPs) in SWEET genes were detected, and Fst values were calculated using Genepop (version 4.0) [62]. The SNP loci with a Fst value > 0.45 were identified as putative sites under selection during domestication.
5. Conclusions
In this study, we comprehensively analyzed G. hirsutum SWEET genes regard to their classification, phylogenetic relationships, structure, cis-acting regulatory elements, chromosomal localization, genetic variations, and expression patterns in diverse tissues and in response to various stresses. Our findings revealed that the G. hirsutum SWEET gene family expanded during evolution, perhaps because of a WGD, some tandem duplications, and a polyploidy event. The GhSWEET genes have significant roles related to cotton development and stress responses. The data presented herein provide a foundation for future functional studies of the most interesting cotton SWEET genes. Our findings may also be relevant for breeding new cotton varieties with enhanced stress tolerance and for accelerating research regarding cotton development.
Supplementary Materials
Supplementary materials can be found at http://www.mdpi.com/1422-0067/19/3/769/s1. Table S1. Sequences of qRT-PCR primers of the selected GhSWEET genes and the GhHis3 internal reference gene. Table S2. Coding sequences of G. hirsutum SWEET genes. Table S3. Details regarding G. raimondii and G. arboreum SWEET genes. Table S4. Analysis of cis-acting regulatory elements in the promoters of putative GhSWEET genes responsive to environmental stresses and phytohormones. Table S5. Fst values of the SNPs in GhSWEET genes between wild and cultivated upland cotton lines. Figure S1. Distribution of orthologous pairs of SWEET genes between the G. hirsutum Dt subgenome and G. raimondii D genome (A) and between the G. hirsutum At subgenome and G. arboreum A genome (B). Different colored lines represent SWEET genes in four clades based on phylogenetic trees.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31401431), and the National Key R&D Program for Crop Breeding (2016YFD0100306). We acknowledge Zhiqiang Zhang and Wei Liu (Agronomy College, Henan Agricultural University, Zhengzhou, China) for revising the original manuscript, and Peng Huo (Zhengzhou Research Center, Institute of Cotton Research of CAAS, Zhengzhou, China) for technical assistance.
Author Contributions
Daigang Yang, Xiongfeng Ma and Wei Li conceived and designed the research. Zhongying Ren, Kuan Sun, Fei Zhang, Chengxiang Song and Xiaojian Zhou performed the experiments. Xiaoyu Pei, Yangai Liu, Kunlun He and Wensheng Zhang prepared the materials. Wei Li, Zhongying Ren and Zhenyu Wang analyzed the data. Wei Li and Zhongying Ren wrote the paper. Daigang Yang, Xiongfeng Ma and Zhenyu Wang revised the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare that they have no competing financial interests.
Abbreviations
| SWEET | Sugars will eventually be exported transporters |
| SUT | Sucrose transporter |
| ABA | Abscisic acid |
| SA | Salicylic acid |
| GA | Gibberellin |
| MeJA | Methyl jasmonate |
| BR | Brassinosteroid |
| MW | Molecular weight |
| pI | Isoelectric point |
| FPKM | Fragments per kilobase of transcript per million mapped fragments |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| SNP | Single nucleotide polymorphism |
| WGD | Whole-genome duplication |
References
- Ruan, Y.L. Sucrose metabolism: Gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef] [PubMed]
- Slewinski, T.L. Diverse functional roles of monosaccharide transporters and their homologs in vascular plants: A physiological perspective. Mol. Plant 2011, 4, 641–662. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Qu, X.Q.; Hou, B.H.; Sosso, D.; Osorio, S.; Fernie, A.R.; Frommer, W.B. Sucrose efflux mediated by sweet proteins as a key step for phloem transport. Science 2012, 335, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Slewinski, T.L.; Meeley, R.; Braun, D.M. Sucrose transporter1 functions in phloem loading in maize leaves. J. Exp. Bot. 2009, 60, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.C.; Ganesan, S.; Ismail, I.O.; Ayre, B.G. Functional characterization of the Arabidopsis AtSUC2 Sucrose/H+ symporter by tissue-specific complementation reveals an essential role in phloem loading but not in long-distance transport. Plant Physiol. 2008, 148, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Sauer, N. Molecular physiology of higher plant sucrose transporters. FEBS Lett. 2007, 581, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U. SWEETs—The missing sugar efflux carriers. Front. Plant Sci. 2011, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Hirose, T.; Scofield, G.N.; Whitfeld, P.R.; Furbank, R.T. The sucrose transporter gene family in rice. Plant Cell Physiol. 2003, 44, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Hamada, M.; Wada, S.; Kobayashi, K.; Satoh, N. Ci-rga, a gene encoding an MtN3/Saliva family transmembrane protein, is essential for tissue differentiation during embryogenesis of the ascidian ciona intestinalis. Differentiation 2005, 73, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liu, F.; Chen, C.; Ma, F.; Li, M. The Malus domestica sugar transporter gene family: Identifications based on genome and expression profiling related to the accumulation of fruit sugars. Front. Plant Sci. 2014, 5, 569. [Google Scholar] [CrossRef] [PubMed]
- Follit, J.A.; Tuft, R.A.; Fogarty, K.E.; Pazour, G.J. The intraflagellar transport protein IFT20 is associated with the golgi complex and is required for cilia assembly. Mol. Biol. Cell 2006, 17, 3781–3792. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Hou, B.H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.Q.; Guo, W.J.; Kim, J.G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Wang, S. Rice MtN3/Saliva/SWEET family genes and their homologs in cellular organisms. Mol. Plant 2013, 6, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.; Valliyodan, B.; Deshmukh, R.; Prince, S.; Nicander, B.; Zhao, M.; Sonah, H.; Song, L.; Lin, L.; Chaudhary, J.; et al. Soybean (Glycine max) SWEET gene family: Insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genom. 2015, 16, 520. [Google Scholar] [CrossRef] [PubMed]
- Manck-Gotzenberger, J.; Requena, N. Arbuscular mycorrhiza symbiosis induces a major transcriptional reprogramming of the potato SWEET sugar transporter family. Front. Plant Sci. 2016, 7, 487. [Google Scholar] [CrossRef] [PubMed]
- Chandran, D. Co-option of developmentally regulated plant SWEET transporters for pathogen nutrition and abiotic stress tolerance. IUBMB Life 2015, 67, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Oikawa, T.; Chiba, Y.; Ishimaru, Y.; Shimizu, T.; Sano, N.; Koshiba, T.; Kamiya, Y.; Ueda, M.; Seo, M. AtSWEET13 and AtSWEET14 regulate gibberellin-mediated physiological processes. Nat. Commun. 2016, 7, 13245. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Osakabe, Y. Sugar compartmentation as an environmental stress adaptation strategy in plants. Semi. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Piron, M.C.; Meyer, S.; Merdinoglu, D.; Bertsch, C.; Mestre, P. The SWEET family of sugar transporters in grapevine: VvSWEET4 is involved in the interaction with botrytis cinerea. J. Exp. Bot. 2014, 65, 6589–6601. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.Y.; Han, J.X.; Han, X.X.; Jiang, J. Genome-wide identification, phylogeny, and expression analysis of the SWEET gene family in tomato. Gene 2015, 573, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, S.; Ma, S.; Sui, X.; Zhang, Z. Spatiotemporal expression and substrate specificity analysis of the cucumber SWEET gene family. Front. Plant Sci. 2017, 8, 1855. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, Z.Y.; Kumar, V.; Xu, X.F.; Yuan, P.; Zhu, X.F.; Li, T.Y.; Jia, B.L.; Xuan, Y.H. Genome-wide identification of the SWEET gene family in wheat. Gene 2018, 642, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Le Hir, R.; Spinner, L.; Klemens, P.A.; Chakraborti, D.; de Marco, F.; Vilaine, F.; Wolff, N.; Lemoine, R.; Porcheron, B.; Gery, C.; et al. Disruption of the sugar transporters AtSWEET11 and AtSWEET12 affects vascular development and freezing tolerance in Arabidopsis. Mol. Plant 2015, 8, 1687–1690. [Google Scholar] [CrossRef] [PubMed]
- Klemens, P.A.; Patzke, K.; Deitmer, J.; Spinner, L.; Le Hir, R.; Bellini, C.; Bedu, M.; Chardon, F.; Krapp, A.; Neuhaus, H.E. Overexpression of the vacuolar sugar carrier AtSWEET16 modifies germination, growth, and stress tolerance in Arabidopsis. Plant Physiol. 2013, 163, 1338–1352. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Lu, K.; Yang, B.; Wang, T.; Zhang, L.; Zhang, A.; Wang, J.; Liu, L.; Qu, C.; Li, J. Genome-wide analysis and expression profiling of the SUC and SWEET gene families of sucrose transporters in oilseed rape (Brassica napus L.). Front. Plant Sci. 2016, 7, 1464. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.W.; Sosso, D.; Chen, L.Q.; Gase, K.; Kim, S.G.; Kessler, D.; Klinkenberg, P.M.; Gorder, M.K.; Hou, B.H.; Qu, X.Q.; et al. Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nature 2014, 508, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Alhassan, Y.; Kumar, N.; Bugaje, I.M.; Pali, H.S.; Kathkar, P. Co-solvents transesterification of cotton seed oil into biodiesel: Effects of reaction conditions on quality of fatty acids methyl esters. Energy Conver. Manag. 2014, 84, 640–648. [Google Scholar] [CrossRef]
- Yang, T.; Zheng, Y. State and trends of oil crops production in China. Oil. Crops Lipids 2016, 23, D603. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.L.; Meng, F.; Wilkins, K.E.; Li, F.; Wang, P.; Booher, N.J.; Carpenter, S.C.D.; Chen, L.Q.; Zheng, H.; Gao, X.; et al. Tal effector driven induction of a SWEET gene confers susceptibility to bacterial blight of cotton. Nat. Commun. 2017, 8, 15588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ruan, Y.L.; Zhou, N.; Wang, F.; Guan, X.; Fang, L.; Shang, X.; Guo, W.; Zhu, S.; Zhang, T. Suppressing a putative sterol carrier gene reduces plasmodesmal permeability and activates sucrose transporter genes during cotton fiber elongation. Plant Cell 2017, 29, 2027–2046. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Bowers, J.E.; Chapman, B.A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. USA 2004, 101, 9903–9908. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Kang, J.Q.; Xin, Z.; Zhen, S.; Qu, L.J.; Gu, H.Y. Variation at the transcriptional level among Chinese natural populations of Arabidopsis thaliana in response to cold stress. Chin. Sci. Bull. 2008, 53, 2989–2999. [Google Scholar]
- Seo, P.J.; Park, J.M.; Kang, S.K.; Kim, S.G.; Park, C.M. An Arabidopsis senescence-associated protein SAG29 regulates cell viability under high salinity. Planta 2011, 233, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, R.; La, C.S.; Atanassova, R.; Dédaldéchamp, F.; Allario, T.; Pourtau, N.; Bonnemain, J.L.; Laloi, M.; Coutos-Thévenot, P.; Maurousset, L.; et al. Source-to-sink transport of sugar and regulation by environmental factors. Front. Plant Sci. 2013, 4, 272. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Peer, Y.V.D.; Rouz, P.; Rombauts, S. Plantcare, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.Z.; Berry, J.C.; Wilson, M.C.; Vijayaraghavan, A.; Burke, J.; Bunn, J.I.; Allen, T.W.; Wheeler, T.; Bart, R.S. Genomics-enabled analysis of the emergent disease cotton bacterial blight. PLoS Genet. 2017, 13, e1007003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. Acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Xu, H.; Xiao, S.; Qin, Y.; Li, Y.; Yan, Y.; Hu, Y. The large soybean (Glycine max) wrky tf family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 2013, 13, 148. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Li, C.; Zhang, Y.; He, Q.; Daud, M.K.; Chen, J.; Zhu, S. Glutathione s-transferase gene family in Gossypium raimondii and g. Arboreum: Comparative genomic study and their expression under salt stress. Front. Plant Sci. 2016, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, Y.; Lu, H.; Cai, X.; Wang, X.; Zhou, Z.; Wang, C.; Wang, Y.; Zhang, Z.; Wang, K.; et al. Genome-wide characterization and expression analysis of the aldehyde dehydrogenase ALDH) gene superfamily under abiotic stresses in cotton. Gene 2017, 628, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Roulin, A.; Auer, P.L.; Libault, M.; Schlueter, J.; Farmer, A.; May, G.; Stacey, G.; Doerge, R.W.; Jackson, S.A. The fate of duplicated genes in a polyploid plant genome. Plant J. 2013, 73, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Haecker, A.; Großhardt, R.; Geiges, B.; Sarkar, A.; Breuninger, H.; Herrmann, M.; Laux, T. Expression dynamics of wox genes mark cell fate decisions duringearly embryonic patterning in Arabidopsis thaliana. Development 2004, 131, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Force, A. The probability of duplicate gene preservation by subfunctionalization. Genetics 2000, 154, 459–473. [Google Scholar] [PubMed]
- Sheehan, M.J.; Kennedy, L.M.; Costich, D.E.; Brutnell, T.P. Subfunctionalization of phyB1 and phyB2 in the control of seedling and mature plant traits in maize. Plant J. 2007, 49, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar]
- Pandey, D.K.; Chaudhary, B. Evolutionary expansion and structural functionalism of the ancient family of profilin proteins. Gene 2017, 626, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006, 7, S10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.Y.; Zhu, Q.H.; Chen, X.; Luo, J.C. GSDS: A gene structure display server. Hereditas 2007, 29, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, W.; He, Q.; Daud, M.K.; Chen, J.; Zhu, S. Characterization of 19 genes encoding membrane-bound fatty acid desaturases and their expression profiles in Gossypium raimondii under low temperature. PLoS ONE 2015, 10, e0123281. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Messer, P.W.; Arndt, P.F. The majority of recent short DNA insertions in the human genome are tandem duplications. Mol. Biol. Evol. 2007, 24, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhao, Y.; Wang, Y.; Liu, Z.; Ijaz, B.; Huang, Y.; Hua, J. Genome-wide identification and expression analysis of the biotin carboxyl carrier subunits of heteromeric acetyl-coa carboxylase in Gossypium. Front. Plant Sci. 2017, 8, 624. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, N.; Deng, T.; Zhang, L.; Zuo, K. Genome-wide cloning, identification, classification and functional analysis of cotton heat shock transcription factors in cotton (Gossypium hirsutum). BMC Genom. 2014, 15, 961. [Google Scholar] [CrossRef] [PubMed]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster analysis of microarray data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-time PCR data by the comparative c(t) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tu, L.; Lin, M.; Lin, Z.; Wang, P.; Yang, Q.; Ye, Z.; Shen, C.; Li, J.; Zhang, L.; et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017, 49, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. Genepop’007: A complete re-implementation of the genepop software for windows and linux. Mol. Ecol. Res. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).