Impact of the NO-Sensitive Guanylyl Cyclase 1 and 2 on Renal Blood Flow and Systemic Blood Pressure in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Renal Blood Flow under Basal Conditions
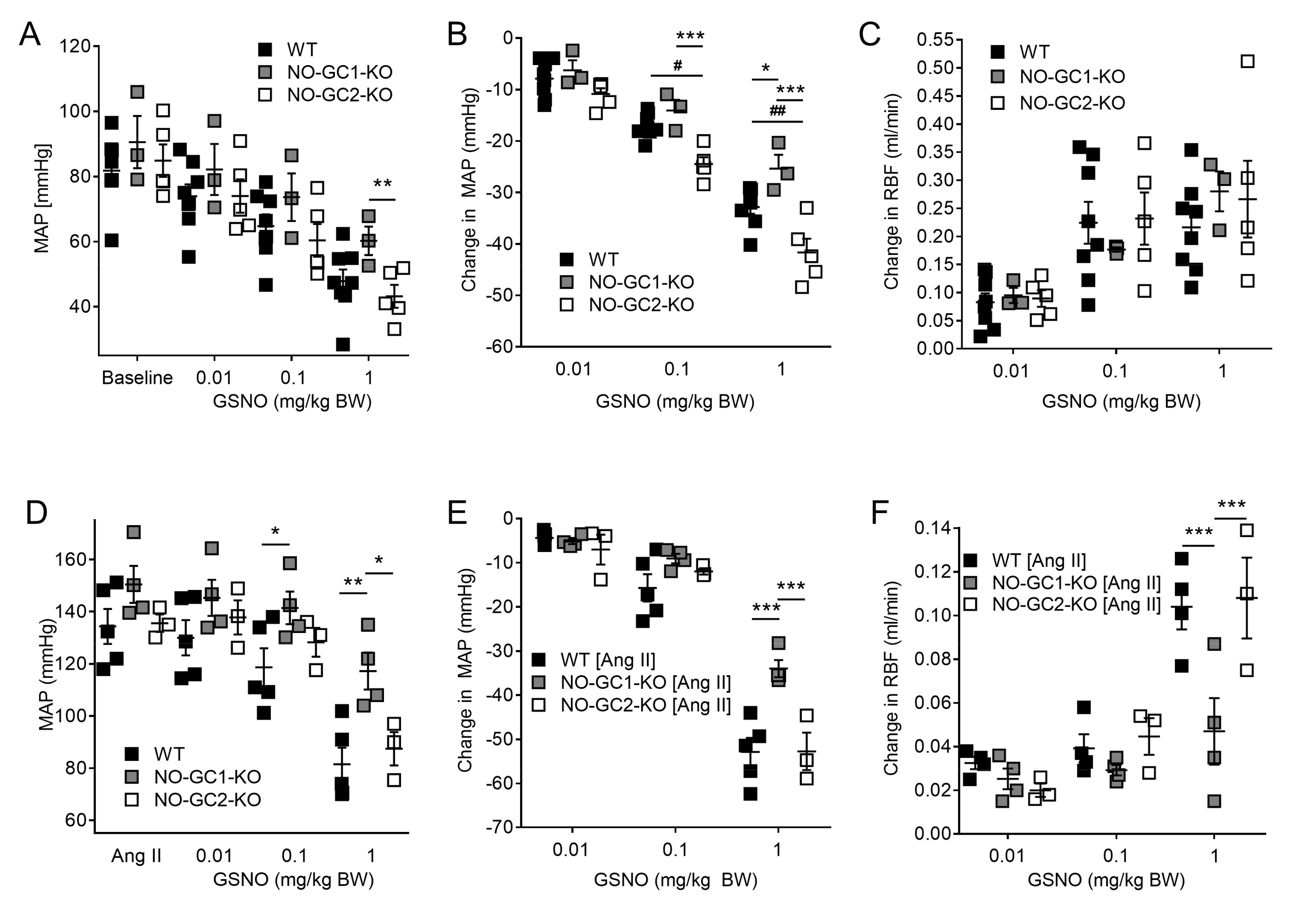
2.2. Effects of NO-sensitive guanylyl cyclase (NO-GC)1 and NO-GC2 on Renal Blood Flow in Response to NO Stimulation
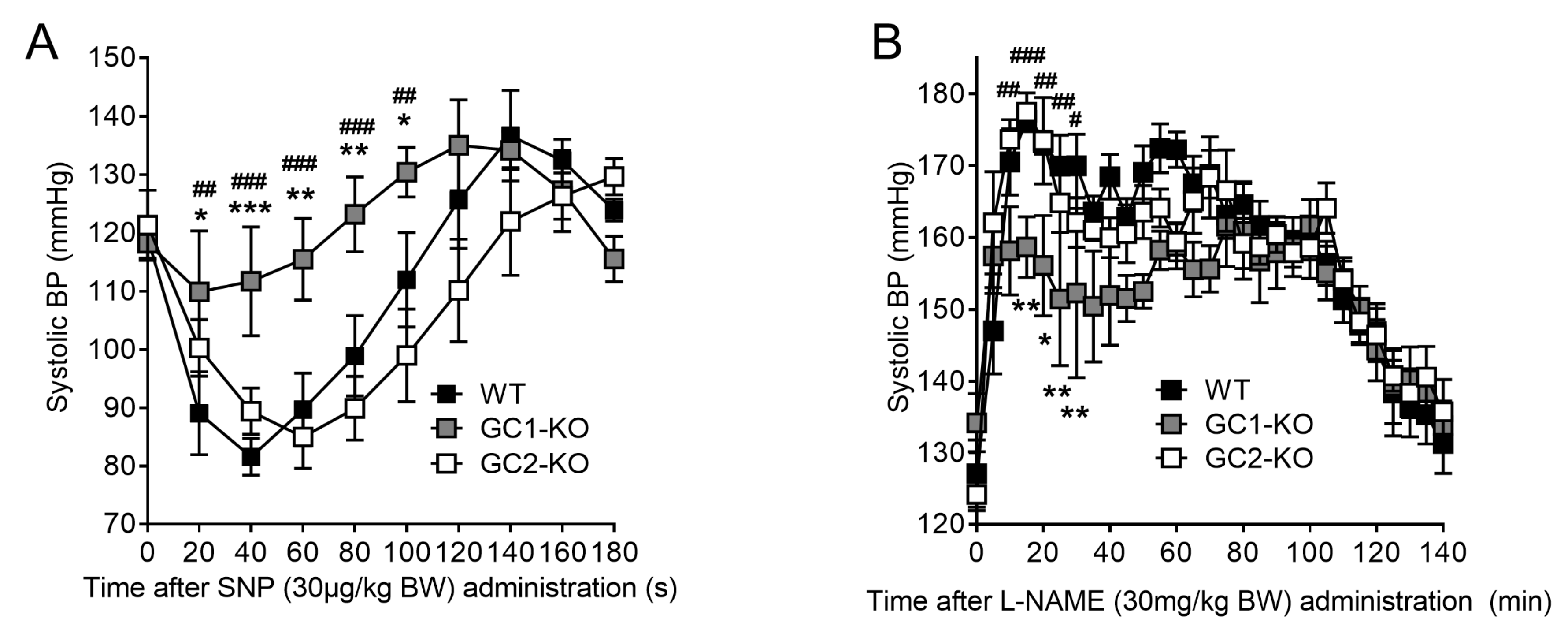
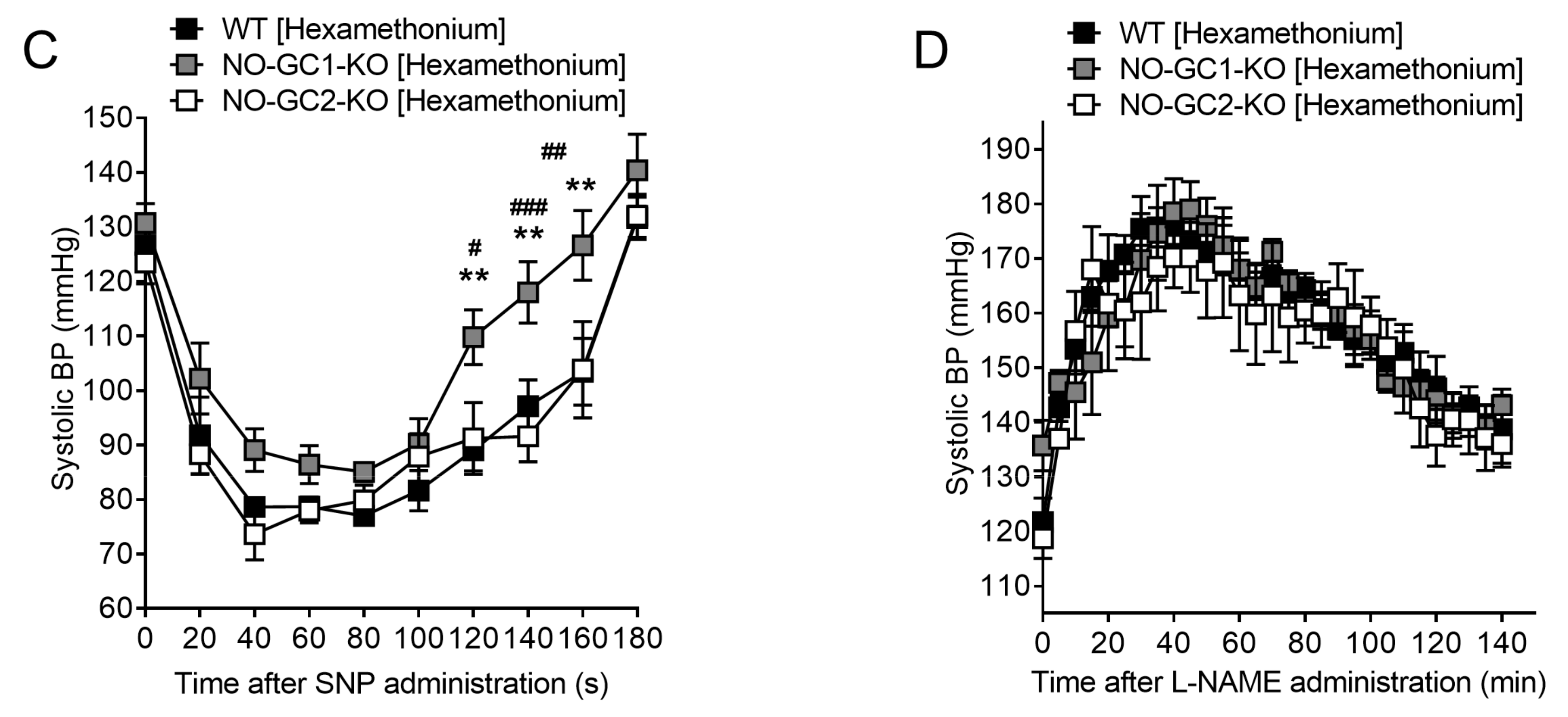
2.3. Inhibition of NO Production by L-NAME Affects Renal Blood Flow in NO-GC1-KO and NO-GC2-KO Mice
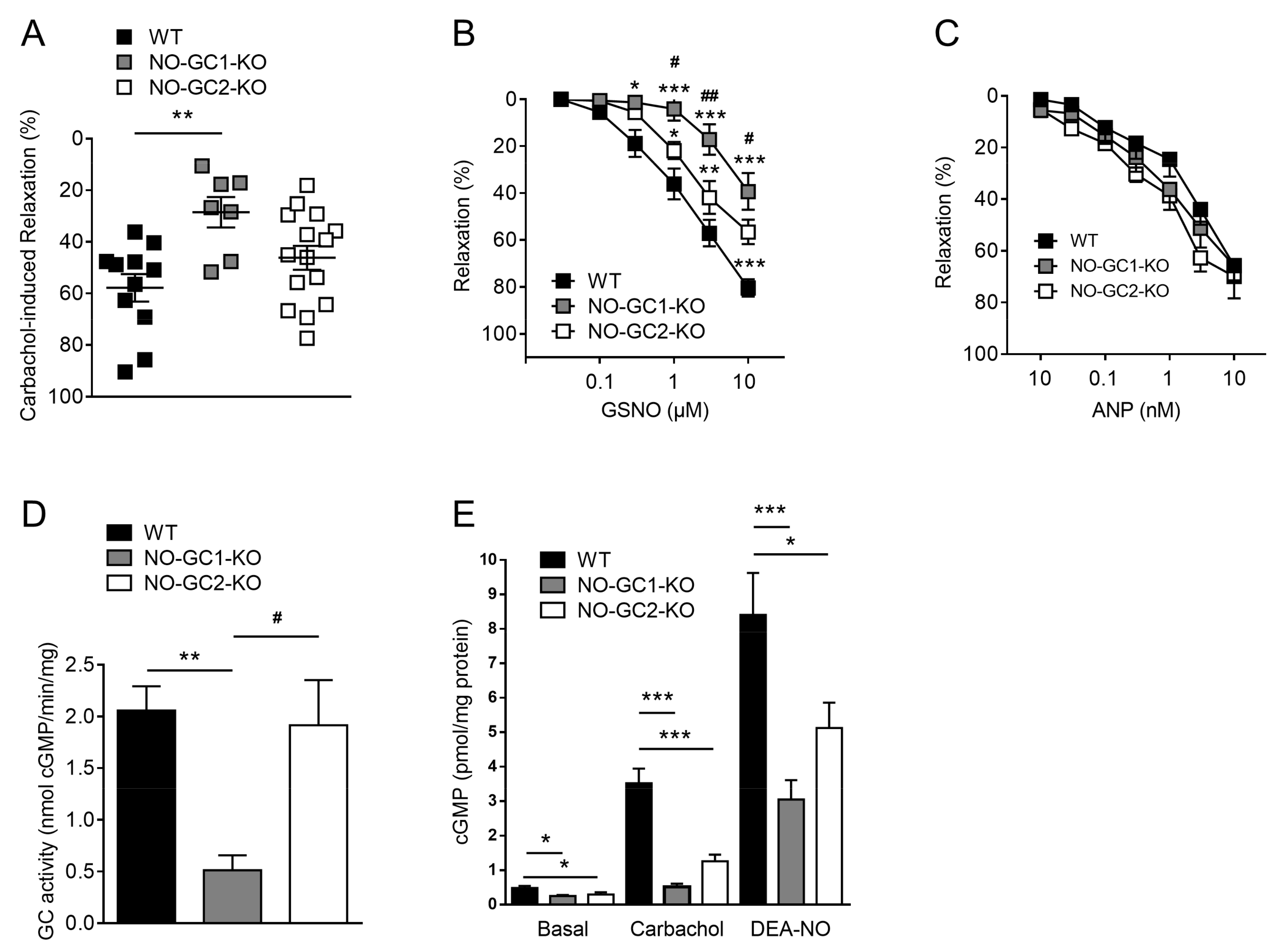
2.4. Contribution of NO-GC1 and NO-GC2 to Renal Vascular Relaxation
2.5. NO-Stimulated cGMP Formation in Kidney Homogenates and Cortical Slices
2.6. Blood Pressure Monitoring in Freely Moving NO-GC1-KO and NO-GC2-KO Mice
3. Discussion
4. Material and Methods
4.1. Animal Models
4.2. Acute Blood Pressure Response and Changes in Renal Blood Flow
4.3. Isolated Perfused Kidneys
4.4. Blood Pressure Measurements in Freely Moving WT, NO-GC1-KO, and NO-GC2-KO Mice
4.5. Measurement of cGMP Content and NO-Stimulated GC-Activity in Renal Cortical Slices
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ANP | atrial natriuretic peptide |
| BP | blood pressure |
| BW | body weight |
| cGMP | cyclic guanosine monophosphate |
| DEA-NO | DEA NONOate |
| GSNO | S-nitrosoglutathione |
| L-NAME | N(G)-Nitro-L-arginine methyl ester |
| MAP | mean arterial blood pressure |
| NO | nitric oxide |
| NO-GC | NO-sensitive guanylyl cyclase (synonym: sGC) |
| RBF | renal blood flow |
| SEM | standard error of the mean |
| SNP | sodium nitroprusside |
References
- Ignarro, L.J. Nitric oxide as a unique signaling molecule in the vascular system: A historical overview. J. Physiol. Pharmacol. 2002, 53 Pt 1, 503–514. [Google Scholar] [PubMed]
- Moncada, S.; Higgs, E.A. Nitric oxide and the vascular endothelium. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2006; Volume 176, pp. 213–254. [Google Scholar]
- Waldman, S.A.; Murad, F. Cyclic GMP synthesis and function. Pharmacol. Rev. 1987, 39, 163–196. [Google Scholar] [PubMed]
- Friebe, A.; Koesling, D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ. Res. 2003, 93, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, M.; Koesling, D. Isoforms of NO-sensitive guanylyl cyclase. Mol. Cell. Biochem. 2002, 230, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Mergia, E.; Friebe, A.; Dangel, O.; Russwurm, M.; Koesling, D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J. Clin. Investig. 2006, 116, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Stegbauer, J.; Friedrich, S.; Potthoff, S.A.; Broekmans, K.; Cortese-Krott, M.M.; Quack, I.; Rump, L.C.; Koesling, D.; Mergia, E. Phosphodiesterase 5 attenuates the vasodilatory response in renovascular hypertension. PLoS ONE 2013, 8, e80674. [Google Scholar] [CrossRef] [PubMed]
- Buys, E.S.; Raher, M.J.; Kirby, A.; Shahid, M.; Baron, D.M.; Hayton, S.R.; Tainsh, L.T.; Sips, P.Y.; Rauwerdink, K.M.; Yan, Q.; et al. Genetic modifiers of hypertension in soluble guanylate cyclase alpha1-deficient mice. J. Clin. Investig. 2012, 122, 2316–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmegeers, S.; Sips, P.; Buys, E.; Brouckaert, P.; van de Voorde, J. Functional role of the soluble guanylyl cyclase α1 subunit in vascular smooth muscle relaxation. Cardiovasc. Res. 2007, 76, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.; Konig, P.; Wirth, A.; Offermanns, S.; Koesling, D.; Friebe, A. Smooth muscle-specific deletion of nitric oxide-sensitive guanylyl cyclase is sufficient to induce hypertension in mice. Circulation 2010, 121, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Cupples, W.A.; Braam, B. Assessment of renal autoregulation. Am. J. Physiol. Renal Physiol. 2007, 292, F1105–F1123. [Google Scholar] [CrossRef] [PubMed]
- Dautzenberg, M.; Just, A. Temporal characteristics of nitric oxide-, prostaglandin-, and EDHF-mediated components of endothelium-dependent vasodilation in the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R987–R998. [Google Scholar] [CrossRef] [PubMed]
- Dautzenberg, M.; Keilhoff, G.; Just, A. Modulation of the myogenic response in renal blood flow autoregulation by NO depends on endothelial nitric oxide synthase (eNOS), but not neuronal or inducible NOS. J. Physiol. 2011, 589 Pt 19, 4731–4744. [Google Scholar] [CrossRef] [PubMed]
- Thieme, M.; Sivritas, S.H.; Mergia, E.; Potthoff, S.A.; Yang, G.; Hering, L.; Grave, K.; Hoch, H.; Rump, L.C.; Stegbauer, J. Phosphodiesterase 5 inhibition ameliorates angiotensin II-dependent hypertension and renal vascular dysfunction. Am. J. Physiol. Renal Physiol. 2017, 312, F474–F481. [Google Scholar] [CrossRef] [PubMed]
- Carlstrom, M.; Wilcox, C.S.; Arendshorst, W.J. Renal autoregulation in health and disease. Physiol. Rev. 2015, 95, 405–511. [Google Scholar] [CrossRef] [PubMed]
- Dautzenberg, M.; Kahnert, A.; Stasch, J.P.; Just, A. Role of soluble guanylate cyclase in renal hemodynamics and autoregulation in the rat. Am. J. Physiol. Renal Physiol. 2014, 307, F1003–F1012. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Schlossmann, J.; Hocher, B. Renal effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Curr. Opin. Pharmacol. 2015, 21, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Mergia, E.; Stegbauer, J. Role of Phosphodiesterase 5 and Cyclic GMP in Hypertension. Curr. Hypertens. Rep. 2016, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, L.S.; Kretschmer, A.; Lawrenz, B.; Hocher, B.; Stasch, J.P. Chronic Activation of Heme Free Guanylate Cyclase Leads to Renal Protection in Dahl Salt-Sensitive Rats. PLoS ONE 2015, 10, e0145048. [Google Scholar] [CrossRef] [PubMed]
- Schinner, E.; Wetzl, V.; Schramm, A.; Kees, F.; Sandner, P.; Stasch, J.P.; Hofmann, F.; Schlossmann, J. Inhibition of the TGFβ signalling pathway by cGMP and cGMP-dependent kinase I. in renal fibrosis. FEBS Open Bio 2017, 7, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Wetzl, V.; Schinner, E.; Kees, F.; Faerber, L.; Schlossmann, J. Differences in the renal antifibrotic cGMP/cGKI-dependent signaling of serelaxin, zaprinast, and their combination. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Mattson, D.L.; Meister, C.J. Renal cortical and medullary blood flow responses to L-NAME and ANG II in wild-type, nNOS null mutant, and eNOS null mutant mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R991–R997. [Google Scholar] [CrossRef] [PubMed]
- Stegbauer, J.; Kuczka, Y.; Vonend, O.; Quack, I.; Sellin, L.; Patzak, A.; Steege, A.; Langnaese, K.; Rump, L.C. Endothelial nitric oxide synthase is predominantly involved in angiotensin II modulation of renal vascular resistance and norepinephrine release. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R421–R428. [Google Scholar] [CrossRef] [PubMed]
- Beierwaltes, W.H.; Potter, D.L.; Shesely, E.G. Renal baroreceptor-stimulated renin in the eNOS knockout mouse. Am. J. Physiol. Renal Physiol. 2002, 282, F59–F64. [Google Scholar] [CrossRef] [PubMed]
- Chester, M.; Seedorf, G.; Tourneux, P.; Gien, J.; Tseng, N.; Grover, T.; Wright, J.; Stasch, J.P.; Abman, S.H. Cinaciguat, a soluble guanylate cyclase activator, augments cGMP after oxidative stress and causes pulmonary vasodilation in neonatal pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L755–L764. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M.A.; Stegbauer, J.; Chen, D.; Gomez, J.A.; Griffiths, R.C.; Azad, H.A.; Herrera, M.; Gurley, S.B.; Coffman, T.M. Vascular Type 1A Angiotensin II Receptors Control BP by Regulating Renal Blood Flow and Urinary Sodium Excretion. J. Am. Soc. Nephrol. 2015, 26, 2953–2962. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, S.A.; Stamer, S.; Grave, K.; Konigshausen, E.; Sivritas, S.H.; Thieme, M.; Mori, Y.; Woznowski, M.; Rump, L.C.; Stegbauer, J. Chronic p38 mitogen-activated protein kinase inhibition improves vascular function and remodeling in angiotensin II-dependent hypertension. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Stegbauer, J.; Chen, D.; Herrera, M.; Sparks, M.A.; Yang, T.; Konigshausen, E.; Gurley, S.B.; Coffman, T.M. Resistance to hypertension mediated by intercalated cells of the collecting duct. JCI Insight 2017, 2, e92720. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mergia, E.; Thieme, M.; Hoch, H.; Daniil, G.; Hering, L.; Yakoub, M.; Scherbaum, C.R.; Rump, L.C.; Koesling, D.; Stegbauer, J. Impact of the NO-Sensitive Guanylyl Cyclase 1 and 2 on Renal Blood Flow and Systemic Blood Pressure in Mice. Int. J. Mol. Sci. 2018, 19, 967. https://doi.org/10.3390/ijms19040967
Mergia E, Thieme M, Hoch H, Daniil G, Hering L, Yakoub M, Scherbaum CR, Rump LC, Koesling D, Stegbauer J. Impact of the NO-Sensitive Guanylyl Cyclase 1 and 2 on Renal Blood Flow and Systemic Blood Pressure in Mice. International Journal of Molecular Sciences. 2018; 19(4):967. https://doi.org/10.3390/ijms19040967
Chicago/Turabian StyleMergia, Evanthia, Manuel Thieme, Henning Hoch, Georgios Daniil, Lydia Hering, Mina Yakoub, Christina Rebecca Scherbaum, Lars Christian Rump, Doris Koesling, and Johannes Stegbauer. 2018. "Impact of the NO-Sensitive Guanylyl Cyclase 1 and 2 on Renal Blood Flow and Systemic Blood Pressure in Mice" International Journal of Molecular Sciences 19, no. 4: 967. https://doi.org/10.3390/ijms19040967
APA StyleMergia, E., Thieme, M., Hoch, H., Daniil, G., Hering, L., Yakoub, M., Scherbaum, C. R., Rump, L. C., Koesling, D., & Stegbauer, J. (2018). Impact of the NO-Sensitive Guanylyl Cyclase 1 and 2 on Renal Blood Flow and Systemic Blood Pressure in Mice. International Journal of Molecular Sciences, 19(4), 967. https://doi.org/10.3390/ijms19040967