TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections

 , ,
, ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Marfan Syndrome
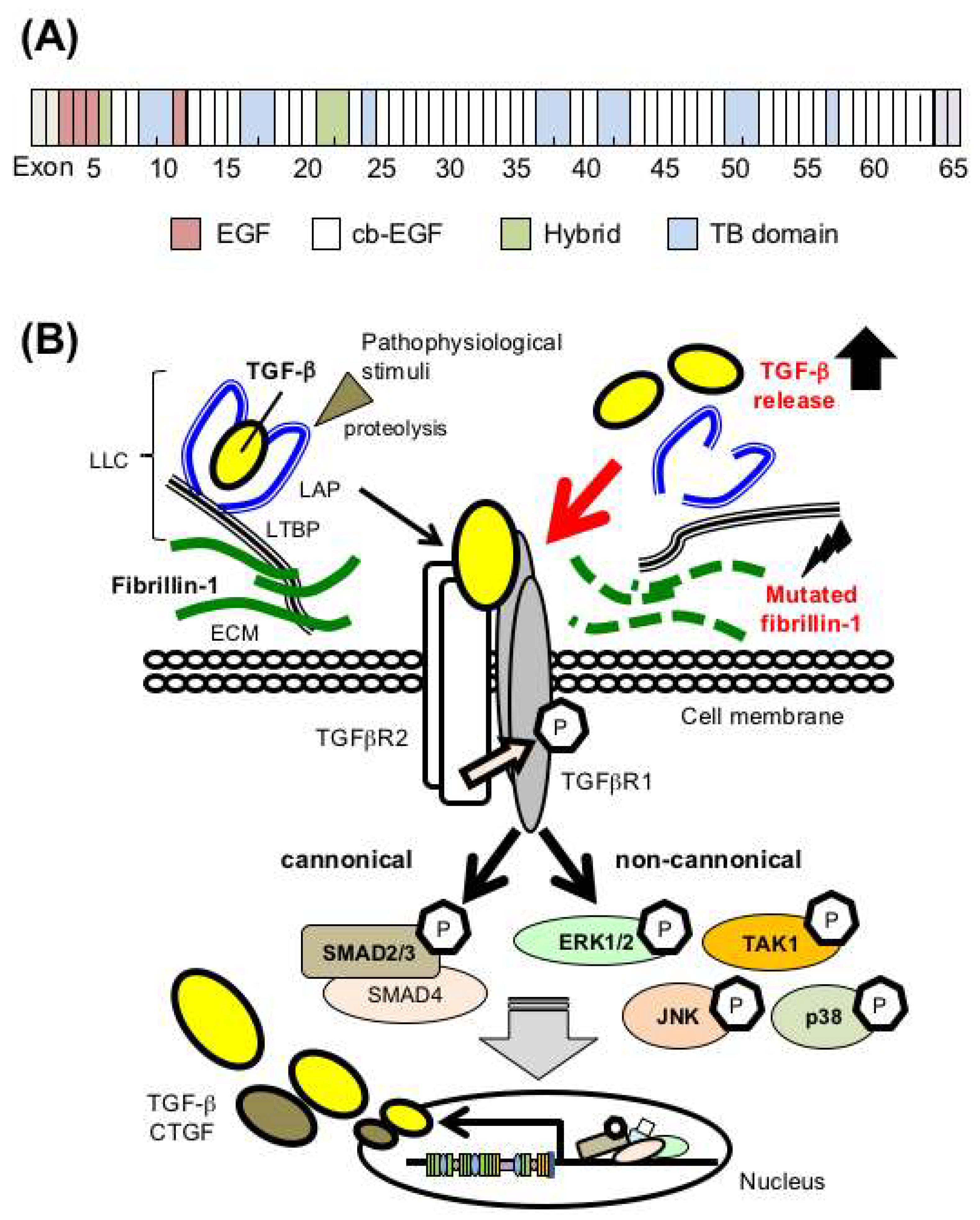
2.1. Fibrillin-1 Regulates TGF-β Bioavailability
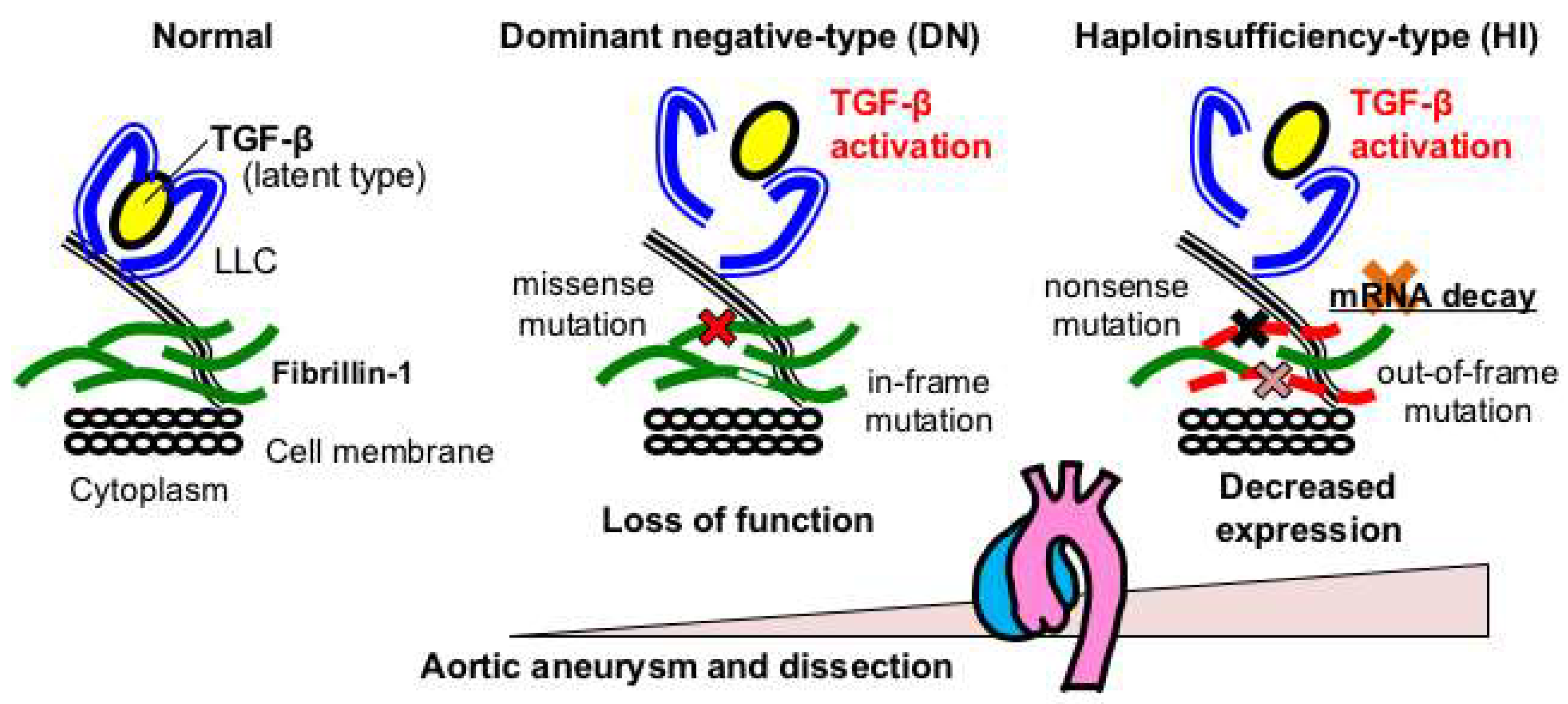
2.2. Genotype-Phenotype Relationships in Marfan Syndrome
2.3. Murine Model of Marfan Syndrome
2.4. Canonical and Noncanonical TGF-β Signaling in Marfan Syndrme
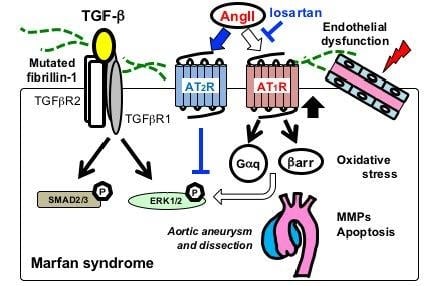
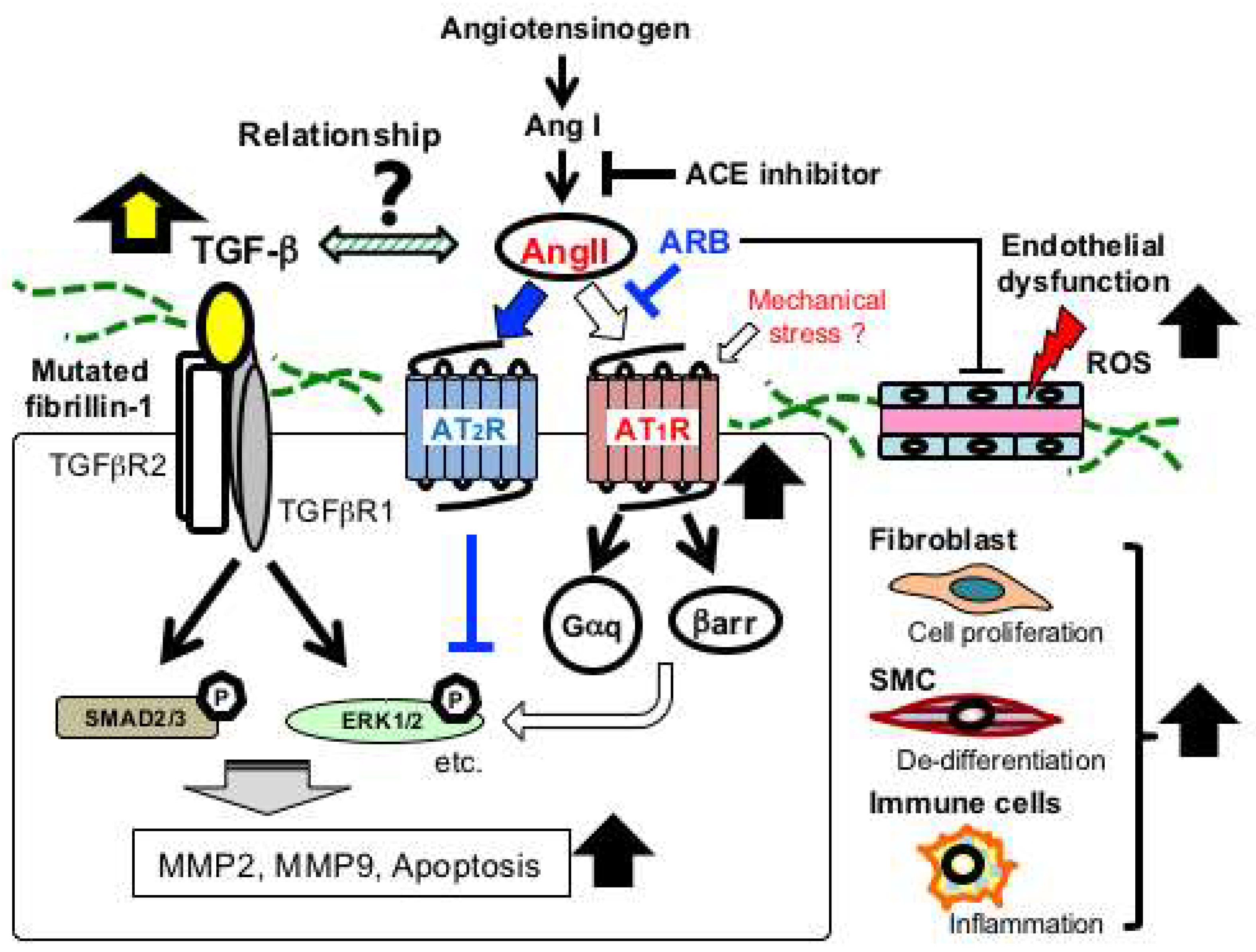
2.5. Angiotensin II Receptor Signaling in MFS
2.6. Endothelial Dysfunction in MFS
2.7. Oxidative Stress in MFS
2.8. Alterations in Other Signal Transduction Pathways and Biomarkers
2.9. Beneficial Roles of TGF-β Signaling during Early Aortic Development
3. Loeys-Dietz Syndrome and Related Genetic Murine Models
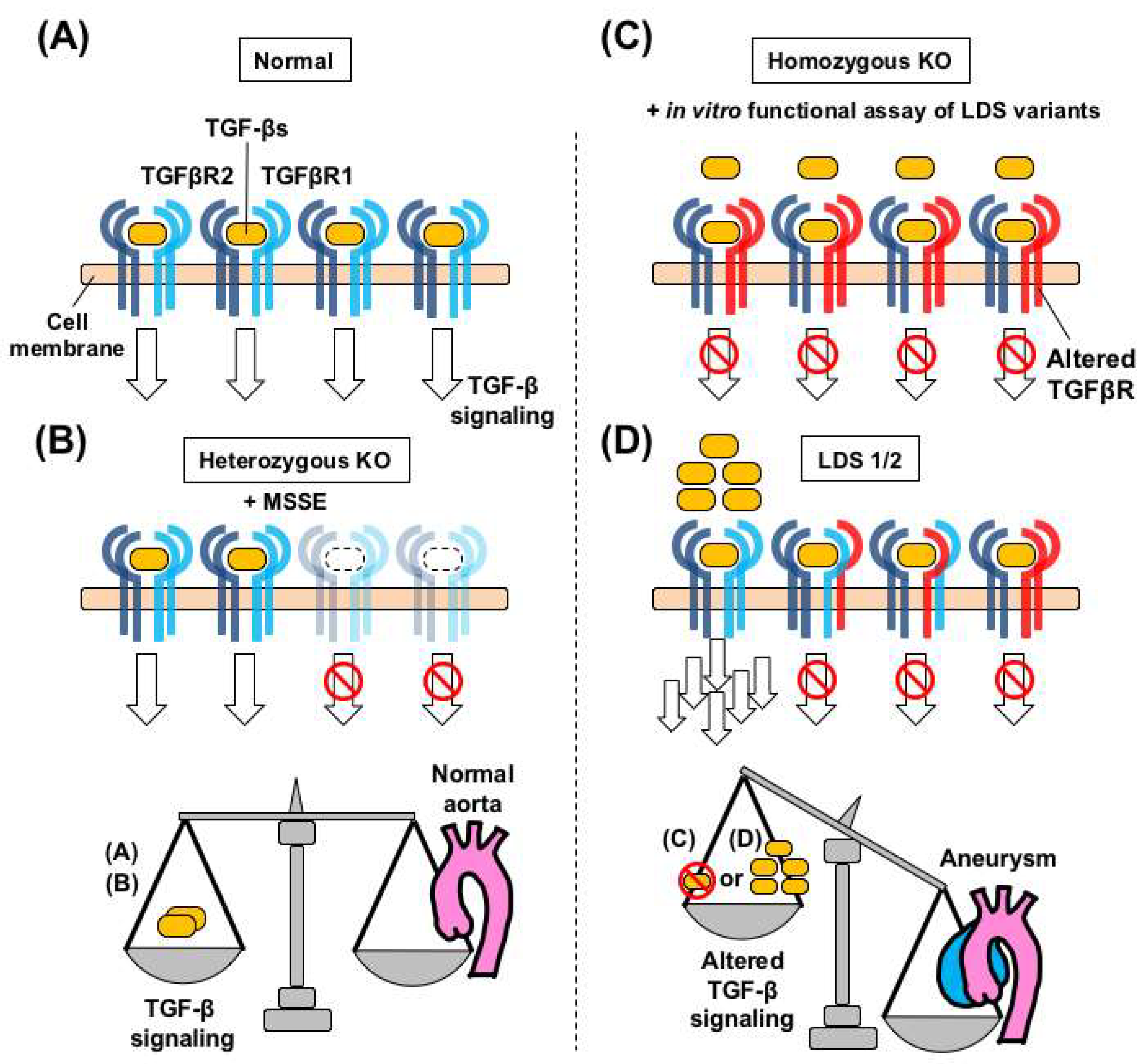
3.1. Heterozygous Loss-of-Function Mutations in TGFBRs Cause Loeys-Dietz Syndrome
3.2. Haploinsufficiency-Type Mutations in TGFBRs do not Cause Loeys-Dietz Syndrome
3.3. Inactivation of Both Tgfbr2 Alleles Causes Aortic Aneurysm in Mice
3.4. A Speculated Mechanism of TGF-β Paradox in Loeys-Dietz Syndrome
3.5. SMAD3 Gene Mutations Cause Loeys-Dietz Syndrome Type 3
3.6. TGFB2 and TGFB3 Mutations Cause Loeys-Dietz Syndrome Types 4 and 5
4. Shprintzen-Goldberg Syndrome
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- El-Hamamsy, I.; Yacoub, M.H. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat. Rev. Cardiol. 2009, 6, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Pomianowski, P.; Elefteriades, J.A. The genetics and genomics of thoracic aortic disease. Ann. Cardiothorac. Surg. 2013, 2, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Yagi, H.; Hara, H.; Fujiwara, T.; Fujita, D.; Nawata, K.; Inuzuka, R.; Taniguchi, Y.; Harada, M.; Toko, H.; et al. Pathophysiology and Management of Cardiovascular Manifestations in Marfan and Loeys-Dietz Syndromes. Int. Heart J. 2016, 57, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Chen, L.; Fan, L.; Gao, D.; Liang, Z.; Wang, R.; Lu, W. The effect of losartan on progressive aortic dilatation in patients with Marfan’s syndrome: A meta-analysis of prospective randomized clinical trials. Int. J. Cardiol. 2016, 217, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Canadas, V.; Vilacosta, I.; Bruna, I.; Fuster, V. Marfan syndrome. Part 2: Treatment and management of patients. Nat. Rev. Cardiol. 2010, 7, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Bunton, T.E.; Biery, N.J.; Myers, L.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Grewal, N.; Gittenberger-de Groot, A.C. Pathogenesis of aortic wall complications in Marfan syndrome. Cardiovasc. Pathol. 2018, 33, 62–69. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Guo, D.C.; Sun, W.; Papke, C.L.; Duraisamy, S.; Estrera, A.L.; Safi, H.J.; Ahn, C.; Buja, L.M.; Arnett, F.C.; et al. Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms, and sporadic aneurysms. J. Thorac. Cardiovasc. Surg. 2008, 136, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Radonic, T.; de Witte, P.; Groenink, M.; de Waard, V.; Lutter, R.; van Eijk, M.; Jansen, M.; Timmermans, J.; Kempers, M.; Scholte, A.J.; et al. Inflammation aggravates disease severity in Marfan syndrome patients. PLoS ONE 2012, 7, e32963. [Google Scholar] [CrossRef] [PubMed]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Fujita, D.; Takeda, N.; Imai, Y.; Inuzuka, R.; Komuro, I.; Hirata, Y. Pathophysiology and Japanese clinical characteristics in Marfan syndrome. Pediatr. Int. 2014, 56, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Isogai, Z.; Ono, R.N.; Ushiro, S.; Keene, D.R.; Chen, Y.; Mazzieri, R.; Charbonneau, N.L.; Reinhardt, D.P.; Rifkin, D.B.; Sakai, L.Y. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 2003, 278, 2750–2757. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.A.; Robertson, I.B.; Handford, P.A. Dissecting the fibrillin microfibril: Structural insights into organization and function. Structure 2012, 20, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Neptune, E.R.; Frischmeyer, P.A.; Arking, D.E.; Myers, L.; Bunton, T.E.; Gayraud, B.; Ramirez, F.; Sakai, L.Y.; Dietz, H.C. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat. Genet. 2003, 33, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Matt, P.; Schoenhoff, F.; Habashi, J.; Holm, T.; Van Erp, C.; Loch, D.; Carlson, O.D.; Griswold, B.F.; Fu, Q.; De Backer, J.; et al. Circulating transforming growth factor-beta in Marfan syndrome. Circulation 2009, 120, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Stheneur, C.; Faivre, L.; Collod-Beroud, G.; Gautier, E.; Binquet, C.; Bonithon-Kopp, C.; Claustres, M.; Child, A.H.; Arbustini, E.; Ades, L.C.; et al. Prognosis factors in probands with an FBN1 mutation diagnosed before the age of 1 year. Pediatr. Res. 2011, 69, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C. Severe infantile Marfan syndrome versus neonatal Marfan syndrome. Am. J. Med. Genet. A 2005, 139, 1. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Collod-Beroud, G.; Loeys, B.L.; Child, A.; Binquet, C.; Gautier, E.; Callewaert, B.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: An international study. Am. J. Hum. Genet. 2007, 81, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Schrijver, I.; Liu, W.; Odom, R.; Brenn, T.; Oefner, P.; Furthmayr, H.; Francke, U. Premature termination mutations in FBN1: Distinct effects on differential allelic expression and on protein and clinical phenotypes. Am. J. Hum. Genet. 2002, 71, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Heesterbeek, T.J.; de Waard, V.; Zwinderman, A.H.; Pals, G.; Mulder, B.J.; Groenink, M. Diagnosis and genetics of Marfan syndrome. Expert Opin. Orphan Drugs 2014, 2, 1049–1062. [Google Scholar] [CrossRef]
- Baudhuin, L.M.; Kotzer, K.E.; Lagerstedt, S.A. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genet. Med. 2015, 17, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Groenink, M.; de Waard, V.; Feenstra, H.M.; Scholte, A.J.; van den Berg, M.P.; Pals, G.; Zwinderman, A.H.; Timmermans, J.; Mulder, B.J. Genotype impacts survival in Marfan syndrome. Eur. Heart J. 2016, 37, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Teixido-Tura, G.; Brion, M.; Forteza, A.; Rodriguez-Palomares, J.; Gutierrez, L.; Garcia Dorado, D.; Pals, G.; Mulder, B.J.; Evangelista, A. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart 2017, 103, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Inuzuka, R.; Maemura, S.; Morita, H.; Nawata, K.; Fujita, D.; Taniguchi, Y.; Yamauchi, H.; Yagi, H.; Kato, M.; et al. Impact of Pathogenic FBN1 Variant Types on the Progression of Aortic Disease in Patients With Marfan Syndrome. Circ. Genom. Precis. Med. 2018, 11, e002058. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.S.; Cain, S.A.; Morgan, A.; Dallas, S.L.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 regulates the bioavailability of TGFbeta1. J. Cell Biol. 2007, 176, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Vollbrandt, T.; Tiedemann, K.; El-Hallous, E.; Lin, G.; Brinckmann, J.; John, H.; Batge, B.; Notbohm, H.; Reinhardt, D.P. Consequences of cysteine mutations in calcium-binding epidermal growth factor modules of fibrillin-1. J. Biol. Chem. 2004, 279, 32924–32931. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, D.P.; Mechling, D.E.; Boswell, B.A.; Keene, D.R.; Sakai, L.Y.; Bachinger, H.P. Calcium determines the shape of fibrillin. J. Biol. Chem. 1997, 272, 7368–7373. [Google Scholar] [CrossRef] [PubMed]
- Hilhorst-Hofstee, Y.; Hamel, B.C.; Verheij, J.B.; Rijlaarsdam, M.E.; Mancini, G.M.; Cobben, J.M.; Giroth, C.; Ruivenkamp, C.A.; Hansson, K.B.; Timmermans, J.; et al. The clinical spectrum of complete FBN1 allele deletions. Eur. J. Hum. Genet. 2011, 19, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Lee, S.Y.; Gayraud, B.; Andrikopoulos, K.; Shapiro, S.D.; Bunton, T.; Biery, N.J.; Dietz, H.C.; Sakai, L.Y.; Ramirez, F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc. Natl. Acad. Sci. USA 1999, 96, 3819–3823. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.M.; Cheng, A.; Myers, L.A.; Martinez-Murillo, F.; Jie, C.; Bedja, D.; Gabrielson, K.L.; Hausladen, J.M.; Mecham, R.P.; Judge, D.P.; et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J. Clin. Investig. 2004, 114, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Biery, N.J.; Keene, D.R.; Geubtner, J.; Myers, L.; Huso, D.L.; Sakai, L.Y.; Dietz, H.C. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Investig. 2004, 114, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Cikach, F.S.; Koch, C.D.; Mead, T.J.; Galatioto, J.; Willard, B.B.; Emerton, K.B.; Eagleton, M.J.; Blackstone, E.H.; Ramirez, F.; Roselli, E.E.; et al. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.; van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011, 332, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Gudey, S.K.; Landstrom, M. The Role of Ubiquitination to Determine Non-Smad Signaling Responses. Methods Mol. Biol. 2016, 1344, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Nataatmadja, M.; West, J.; Prabowo, S.; West, M. Angiotensin II Receptor Antagonism Reduces Transforming Growth Factor Beta and Smad Signaling in Thoracic Aortic Aneurysm. Ochsner J. 2013, 13, 42–48. [Google Scholar] [PubMed]
- Habashi, J.P.; Doyle, J.J.; Holm, T.M.; Aziz, H.; Schoenhoff, F.; Bedja, D.; Chen, Y.; Modiri, A.N.; Judge, D.P.; Dietz, H.C. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 2011, 332, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, N.B.; Qian, L.; Westergaard, N.M.; Kutschke, W.J.; Born, E.J.; Turek, J.W. A Novel Murine Model of Marfan Syndrome Accelerates Aortopathy and Cardiomyopathy. Ann. Thorac. Surg. 2017, 104, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Groenink, M.; den Hartog, A.W.; Franken, R.; Radonic, T.; de Waard, V.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Spijkerboer, A.M.; Marquering, H.A.; et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: A randomized controlled trial. Eur. Heart J. 2013, 34, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Lacro, R.V.; Dietz, H.C.; Sleeper, L.A.; Yetman, A.T.; Bradley, T.J.; Colan, S.D.; Pearson, G.D.; Selamet Tierney, E.S.; Levine, J.C.; Atz, A.M.; et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N. Engl. J. Med. 2014, 371, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Forteza, A.; Evangelista, A.; Sanchez, V.; Teixido-Tura, G.; Sanz, P.; Gutierrez, L.; Gracia, T.; Centeno, J.; Rodriguez-Palomares, J.; Rufilanchas, J.J.; et al. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: A randomized clinical trial. Eur. Heart J. 2016, 37, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Verbrugghe, P.; Verhoeven, J.; Clijsters, M.; Vervoort, D.; Schepens, J.; Meuris, B.; Herijgers, P. The Effect of a Nonpeptide Angiotensin II Type 2 Receptor Agonist, Compound 21, on Aortic Aneurysm Growth in a Mouse Model of Marfan Syndrome. J. Cardiovasc. Pharmacol. 2018, 71, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; Harris, E.M.; Raisch, M.; Mao, L.; Kim, J.; Rockman, H.A.; Lefkowitz, R.J. The role of beta-arrestin2-dependent signaling in thoracic aortic aneurysm formation in a murine model of Marfan syndrome. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1516–H1527. [Google Scholar] [CrossRef] [PubMed]
- Galatioto, J.; Caescu, C.I.; Hansen, J.; Cook, J.R.; Miramontes, I.; Iyengar, R.; Ramirez, F. Cell Type-Specific Contributions of the Angiotensin II Type 1a Receptor to Aorta Homeostasis and Aneurysmal Disease-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Amiya, E.; Watanabe, M.; Omori, K.; Imai, Y.; Fujita, D.; Nishimura, H.; Kato, M.; Morota, T.; Nawata, K.; et al. Impairment of flow-mediated dilation correlates with aortic dilation in patients with Marfan syndrome. Heart Vessel. 2014, 29, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Selamet Tierney, E.S.; Levine, J.C.; Sleeper, L.A.; Roman, M.J.; Bradley, T.J.; Colan, S.D.; Chen, S.; Campbell, M.J.; Cohen, M.S.; De Backer, J.; et al. Influence of Aortic Stiffness on Aortic-Root Growth Rate and Outcome in Patients with the Marfan Syndrome. Am. J. Cardiol. 2018, 121, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Loeper, F.; Oosterhof, J.; van den Dorpel, M.; van der Linde, D.; Lu, Y.; Robertson, E.; Hambly, B.; Jeremy, R. Ventricular-Vascular Coupling in Marfan and Non-Marfan Aortopathies. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Sellers, S.L.; Milad, N.; Chan, R.; Mielnik, M.; Jermilova, U.; Huang, P.L.; de Crom, R.; Hirota, J.A.; Hogg, J.C.; Sandor, G.G.; et al. Inhibition of Marfan Syndrome Aortic Root Dilation by Losartan: Role of Angiotensin II Receptor Type 1-Independent Activation of Endothelial Function. Am. J. Pathol. 2018, 188, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, C.; Becatti, M.; Attanasio, M.; Lucarini, L.; Nassi, N.; Evangelisti, L.; Porciani, M.C.; Nassi, P.; Gensini, G.F.; Abbate, R.; et al. Evidence for oxidative stress in plasma of patients with Marfan syndrome. Int. J. Cardiol. 2010, 145, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.H.; van Breemen, C.; Chung, A.W. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vasc. Pharmacol. 2010, 52, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Altayo, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; Del Blanco, D.G.; Lopez-Luque, J.; Mas-Stachurska, A.; Siegert, A.M.; Bonorino, F.; Barbera, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Oller, J.; Mendez-Barbero, N.; Ruiz, E.J.; Villahoz, S.; Renard, M.; Canelas, L.I.; Briones, A.M.; Alberca, R.; Lozano-Vidal, N.; Hurle, M.A.; et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat. Med. 2017, 23, 200–212. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, D.; McGuinness, J.; Byrne, J.; Terzo, E.; Huuskonen, V.; McAllister, H.; Black, A.; Kearney, S.; Kay, E.; Hill, A.D.; et al. Pravastatin reduces Marfan aortic dilation. Circulation 2011, 124, S168–S173. [Google Scholar] [CrossRef] [PubMed]
- Hibender, S.; Franken, R.; van Roomen, C.; Ter Braake, A.; van der Made, I.; Schermer, E.E.; Gunst, Q.; van den Hoff, M.J.; Lutgens, E.; Pinto, Y.M.; et al. Resveratrol Inhibits Aortic Root Dilatation in the Fbn1C1039G/+ Marfan Mouse Model. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Van der Donckt, C.; Van Herck, J.L.; Schrijvers, D.M.; Vanhoutte, G.; Verhoye, M.; Blockx, I.; Van Der Linden, A.; Bauters, D.; Lijnen, H.R.; Sluimer, J.C.; et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur. Heart J. 2015, 36, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Ijaz, T.; Sun, H.; Lejeune, W.; Vargas, G.; Shilagard, T.; Recinos, A., 3rd; Milewicz, D.M.; Brasier, A.R.; Tilton, R.G. IL-6 regulates extracellular matrix remodeling associated with aortic dilation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J. Am. Heart Assoc. 2014, 3, e000476. [Google Scholar] [CrossRef] [PubMed]
- Ihling, C.; Szombathy, T.; Nampoothiri, K.; Haendeler, J.; Beyersdorf, F.; Uhl, M.; Zeiher, A.M.; Schaefer, H.E. Cystic medial degeneration of the aorta is associated with p53 accumulation, Bax upregulation, apoptotic cell death, and cell proliferation. Heart 1999, 82, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.; Yang, H.H.; Radomski, M.W.; van Breemen, C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ. Res. 2008, 102, e73–e85. [Google Scholar] [CrossRef] [PubMed]
- Emrich, F.C.; Okamura, H.; Dalal, A.R.; Penov, K.; Merk, D.R.; Raaz, U.; Hennigs, J.K.; Chin, J.T.; Miller, M.O.; Pedroza, A.J.; et al. Enhanced caspase activity contributes to aortic wall remodeling and early aneurysm development in a murine model of Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.M.; Zhou, Y.Z.; Cheng, Z.; Liao, X.B.; Zhou, X.M. MicroRNAs: Novel Players in Aortic Aneurysm. BioMed Res. Int. 2015, 2015, 831641. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Rateri, D.L.; Daugherty, A. Abdominal aortic aneurysm: Novel mechanisms and therapies. Curr. Opin. Cardiol. 2015, 30, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Abu-Halima, M.; Ludwig, N.; Radle-Hurst, T.; Keller, A.; Motsch, L.; Marsollek, I.; El Rahman, M.A.; Abdul-Khaliq, H.; Meese, E. Characterization of micro-RNA Profile in the Blood of Patients with Marfan’s Syndrome. Thorac. Cardiovasc. Surg. 2018, 66, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.R.; Chin, J.T.; Dake, B.A.; Maegdefessel, L.; Miller, M.O.; Kimura, N.; Tsao, P.S.; Iosef, C.; Berry, G.J.; Mohr, F.W.; et al. miR-29b participates in early aneurysm development in Marfan syndrome. Circ. Res. 2012, 110, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Emrich, F.; Trojan, J.; Chiu, P.; Dalal, A.R.; Arakawa, M.; Sato, T.; Penov, K.; Koyano, T.; Pedroza, A.; et al. Long-term miR-29b suppression reduces aneurysm formation in a Marfan mouse model. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.R.; Clayton, N.P.; Carta, L.; Galatioto, J.; Chiu, E.; Smaldone, S.; Nelson, C.A.; Cheng, S.H.; Wentworth, B.M.; Ramirez, F. Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Q.; Jiao, Y.; Qin, L.; Ali, R.; Zhou, J.; Ferruzzi, J.; Kim, R.W.; Geirsson, A.; Dietz, H.C.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Investig. 2014, 124, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Wei, H.; Jaffe, M.; Airhart, N.; Du, L.; Angelov, S.N.; Yan, J.; Allen, J.K.; Kang, I.; Wight, T.N.; et al. Postnatal Deletion of the Type II Transforming Growth Factor-beta Receptor in Smooth Muscle Cells Causes Severe Aortopathy in Mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- MacCarrick, G.; Black, J.H., 3rd; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C., 3rd. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, T.; Collod-Beroud, G.; Akiyama, T.; Abifadel, M.; Harada, N.; Morisaki, T.; Allard, D.; Varret, M.; Claustres, M.; Morisaki, H.; et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 2004, 36, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Regalado, E.S.; Guo, D.C.; Villamizar, C.; Avidan, N.; Gilchrist, D.; McGillivray, B.; Clarke, L.; Bernier, F.; Santos-Cortez, R.L.; Leal, S.M.; et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ. Res. 2011, 109, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.; de Graaf, B.M.; van de Beek, G.; Gallo, E.; Kruithof, B.P.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Stenvers, K.L.; Tursky, M.L.; Harder, K.W.; Kountouri, N.; Amatayakul-Chantler, S.; Grail, D.; Small, C.; Weinberg, R.A.; Sizeland, A.M.; Zhu, H.J. Heart and liver defects and reduced transforming growth factor beta2 sensitivity in transforming growth factor beta type III receptor-deficient embryos. Mol. Cell. Biol. 2003, 23, 4371–4385. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Yeo, J.Y.; Chytil, A.; Han, J.; Bringas, P., Jr.; Nakajima, A.; Shuler, C.F.; Moses, H.L.; Chai, Y. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development 2003, 130, 5269–5280. [Google Scholar] [CrossRef] [PubMed]
- Dudas, M.; Kim, J.; Li, W.Y.; Nagy, A.; Larsson, J.; Karlsson, S.; Chai, Y.; Kaartinen, V. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev. Biol. 2006, 296, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Iwata, J.; Hacia, J.G.; Suzuki, A.; Sanchez-Lara, P.A.; Urata, M.; Chai, Y. Modulation of noncanonical TGF-beta signaling prevents cleft palate in Tgfbr2 mutant mice. J. Clin. Investig. 2012, 122, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Horbelt, D.; Guo, G.; Robinson, P.N.; Knaus, P. Quantitative analysis of TGFBR2 mutations in Marfan-syndrome-related disorders suggests a correlation between phenotypic severity and Smad signaling activity. J. Cell Sci. 2010, 123, 4340–4350. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Robertson, S.P.; Daniel, P.B. TGFBR1 mutations associated with Loeys-Dietz syndrome are inactivating. J. Recept. Signal Transduct. Res. 2012, 32, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J. The paradoxical TGF-beta vasculopathies. Nat. Genet. 2012, 44, 838–839. [Google Scholar] [CrossRef] [PubMed]
- Gallo, E.M.; Loch, D.C.; Habashi, J.P.; Calderon, J.F.; Chen, Y.; Bedja, D.; van Erp, C.; Gerber, E.E.; Parker, S.J.; Sauls, K.; et al. Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J. Clin. Investig. 2014, 124, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Smith, M.A.; Wallace, D.C.; James, Z.H.; Renwick, J.H. Multiple self-healing squamous epithelioma. Birth Defects Orig. Artic. Ser. 1971, 7, 157–163. [Google Scholar] [PubMed]
- Goudie, D.R.; D’Alessandro, M.; Merriman, B.; Lee, H.; Szeverenyi, I.; Avery, S.; O’Connor, B.D.; Nelson, S.F.; Coats, S.E.; Stewart, A.; et al. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat. Genet. 2011, 43, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Takeda, N.; Hara, H.; Morita, H.; Kishihara, J.; Inuzuka, R.; Yagi, H.; Maemura, S.; Toko, H.; Harada, M.; et al. Distinct variants affecting differential splicing of TGFBR1 exon 5 cause either Loeys-Dietz syndrome or multiple self-healing squamous epithelioma. Eur. J. Hum. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Van der Pluijm, I.; van Vliet, N.; von der Thusen, J.H.; Robertus, J.L.; Ridwan, Y.; van Heijningen, P.M.; van Thiel, B.S.; Vermeij, M.; Hoeks, S.E.; Buijs-Offerman, R.; et al. Defective Connective Tissue Remodeling in SMAD3 Mice Leads to Accelerated Aneurysmal Growth Through Disturbed Downstream TGF-beta Signaling. EBioMedicine 2016, 12, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Li, C.G.; Liang, Q.Q.; Zhou, Q.; Menga, E.; Cui, X.J.; Shu, B.; Zhou, C.J.; Shi, Q.; Wang, Y.J. A continuous observation of the degenerative process in the intervertebral disc of SMAD3 gene knock-out mice. Spine 2009, 34, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.J.; Doyle, J.J.; Bessling, S.L.; Maragh, S.; Lindsay, M.E.; Schepers, D.; Gillis, E.; Mortier, G.; Homfray, T.; Sauls, K.; et al. Mutations in the TGF-beta repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 2012, 44, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Schepers, D.; Doyle, A.J.; Oswald, G.; Sparks, E.; Myers, L.; Willems, P.J.; Mansour, S.; Simpson, M.A.; Frysira, H.; Maat-Kievit, A.; et al. The SMAD-binding domain of SKI: A hotspot for de novo mutations causing Shprintzen-Goldberg syndrome. Eur. J. Hum. Genet. 2015, 23, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Stroschein, S.L.; Wang, W.; Chen, D.; Martens, E.; Zhou, S.; Zhou, Q. The Ski oncoprotein interacts with the Smad proteins to repress TGFbeta signaling. Genes Dev. 1999, 13, 2196–2206. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Khan, M.M.; Kaul, S.C.; Dong, H.D.; Wadhwa, R.; Colmenares, C.; Kohno, I.; Ishii, S. Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid hormone receptor. Genes Dev. 1999, 13, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.R.; Carta, L.; Benard, L.; Chemaly, E.R.; Chiu, E.; Rao, S.K.; Hampton, T.G.; Yurchenco, P.; Gen, T.A.C.R.C.; Costa, K.D.; et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J. Clin. Investig. 2014, 124, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Tae, H.J.; Petrashevskaya, N.; Marshall, S.; Krawczyk, M.; Talan, M. Cardiac remodeling in the mouse model of Marfan syndrome develops into two distinctive phenotypes. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H290–H299. [Google Scholar] [CrossRef] [PubMed]
- Rouf, R.; MacFarlane, E.G.; Takimoto, E.; Chaudhary, R.; Nagpal, V.; Rainer, P.P.; Bindman, J.G.; Gerber, E.E.; Bedja, D.; Schiefer, C.; et al. Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Goland, S.; Elkayam, U. Cardiovascular problems in pregnant women with marfan syndrome. Circulation 2009, 119, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Houston, L.; Tuuli, M.; Macones, G. Marfan syndrome and aortic dissection in pregnancy. Obstet. Gynecol. 2011, 117, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Fujita, D.; Takeda, N.; Morita, H.; Kato, M.; Nishimura, H.; Inuzuka, R.; Taniguchi, Y.; Nawata, K.; Hyodo, H.; Imai, Y.; et al. A novel mutation of TGFBR2 causing Loeys-Dietz syndrome complicated with pregnancy-related fatal cervical arterial dissections. Int. J. Cardiol. 2015, 201, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Sayama, S.; Takeda, N.; Iriyama, T.; Inuzuka, R.; Maemura, S.; Fujita, D.; Yamauchi, H.; Nawata, K.; Bougaki, M.; Hyodo, H.; et al. Peripartum type B aortic dissection in patients with Marfan syndrome who underwent aortic root replacement: A case series study. BJOG 2018, 125, 487–493. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.; Campens, L.; Muino Mosquera, L. Looking for the Missing Links: Challenges in the Search for Genotype-Phenotype Correlation in Marfan Syndrome. Circ. Genom. Precis. Med. 2018, 11, e002185. [Google Scholar] [CrossRef] [PubMed]
- Quarto, N.; Leonard, B.; Li, S.; Marchand, M.; Anderson, E.; Behr, B.; Francke, U.; Reijo-Pera, R.; Chiao, E.; Longaker, M.T. Skeletogenic phenotype of human Marfan embryonic stem cells faithfully phenocopied by patient-specific induced-pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Granata, A.; Serrano, F.; Bernard, W.G.; McNamara, M.; Low, L.; Sastry, P.; Sinha, S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat. Genet. 2017, 49, 97–109. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeda, N.; Hara, H.; Fujiwara, T.; Kanaya, T.; Maemura, S.; Komuro, I. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. Int. J. Mol. Sci. 2018, 19, 2125. https://doi.org/10.3390/ijms19072125
Takeda N, Hara H, Fujiwara T, Kanaya T, Maemura S, Komuro I. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. International Journal of Molecular Sciences. 2018; 19(7):2125. https://doi.org/10.3390/ijms19072125
Chicago/Turabian StyleTakeda, Norifumi, Hironori Hara, Takayuki Fujiwara, Tsubasa Kanaya, Sonoko Maemura, and Issei Komuro. 2018. "TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections" International Journal of Molecular Sciences 19, no. 7: 2125. https://doi.org/10.3390/ijms19072125
APA StyleTakeda, N., Hara, H., Fujiwara, T., Kanaya, T., Maemura, S., & Komuro, I. (2018). TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. International Journal of Molecular Sciences, 19(7), 2125. https://doi.org/10.3390/ijms19072125