1. Introduction
The Niemann Pick type C (NPC) proteins, NPC1 and NPC2, have received a great deal of attention in recent years, not only as they are involved in the lethal hereditary NPC disease, but also as the NPC1 protein has been identified as being necessary for Ebola and Marburg virus infection [
1,
2]. Specifically, viral infection occurs when the virus glycoprotein (GP) binds to the NPC1 protein. Cells deficient in the NPC1 protein are protected from infection. On the other hand, mutations in either NPC1 or NPC2 can lead to an accumulation of cholesterol and lipids in the late endosomal (LE)/lysosomal (Lys) compartments, the primary phenotype of the Niemann-Pick type C (NPC) disease. Thus, understanding structural features of the NPC1 and NPC2 binding domains is the first step in developing therapeutic treatments for the NPC disease as well as designing inhibitors against Ebola and Marburg viruses.
The exact role of NPC2 is not known, but a model has been proposed in which NPC2 binds cholesterol after receptor-mediated endocytosis of low-density lipoprotein (LDL) [
3]. After binding, NPC2 may transport cholesterol to the membrane-bound NPC1 protein (1278 amino acids), where cholesterol can be transferred to the N-terminal domain (NTD) of NPC1. From there, cholesterol is likely transferred to NPC1’s membrane domain for subsequent biochemical processing [
3]. This mechanism of cholesterol transfer is supported by X-ray crystallography that shows the cholesterol’s isooctyl side chain buried deep inside the hydrophobic pocket of NPC2 with its 3
-hydroxyl group exposed at the protein’s surface [
4]. On the other hand, NPC1(NTD) binds cholesterol in the opposite orientation: the 3
-hydroxyl group is buried in the binding pocket while the cholesterol’s isooctyl side chain is surface exposed [
5]. These findings led to the proposal of a “hydrophobic hand-off” or sliding model, in which cholesterol is transferred between NPC2 and NPC1(NTD) without exposure to water [
6].
Experimental evidence supports the sliding model of cholesterol transfer between NPC2 and NPC1(NTD). Surface plasmon resonance experiments, together with affinity chromatography, have revealed that the second luminal loop-domain of NPC1 maintains the position of NPC2 at the binding interface to facilitate cholesterol transfer [
7]. Biochemical assays have shown that deletion of the NTD (residues 25–257) of NPC1 abolish more than 90% of cholesterol transfer from NPC2 to NPC1(NTD) [
8]. A patch of primarily three residues (M79, V81, and V83) on the surface of NPC2 have been found to be essential for transfer of cholesterol from NPC2 to NPC1(NTD) [
6]. Similarly, critical mutations (L175Q and L176Q) at the surface of NPC1(NTD) abolish cholesterol transfer activity [
6].
Nonetheless, a direct interaction between NPC1(NTD) and NPC2 has thus far not been demonstrated. The geometry of a putative NPC1(NTD)–NPC2 protein complex was proposed by the group of Brown and Goldstein at UT Southwest Medical Center in Texas [
3,
6]. Their structure was assembled from rigid body alignment using the information from point mutation experiments identifying amino acids involved at the potential interface between the two individual proteins [
3,
6]. Using this structure, which we refer to as the “Texas model”, Estiu et al. performed MD simulations and
in silico mutational analyses [
9]. Using the Texas model, we also examined cholesterol isomerization inside the NPC1 and NPC2 binding pockets, proposing a model for cholesterol transfer between the binding pockets of the NPC1(NTD)–NPC2 protein complex [
10].
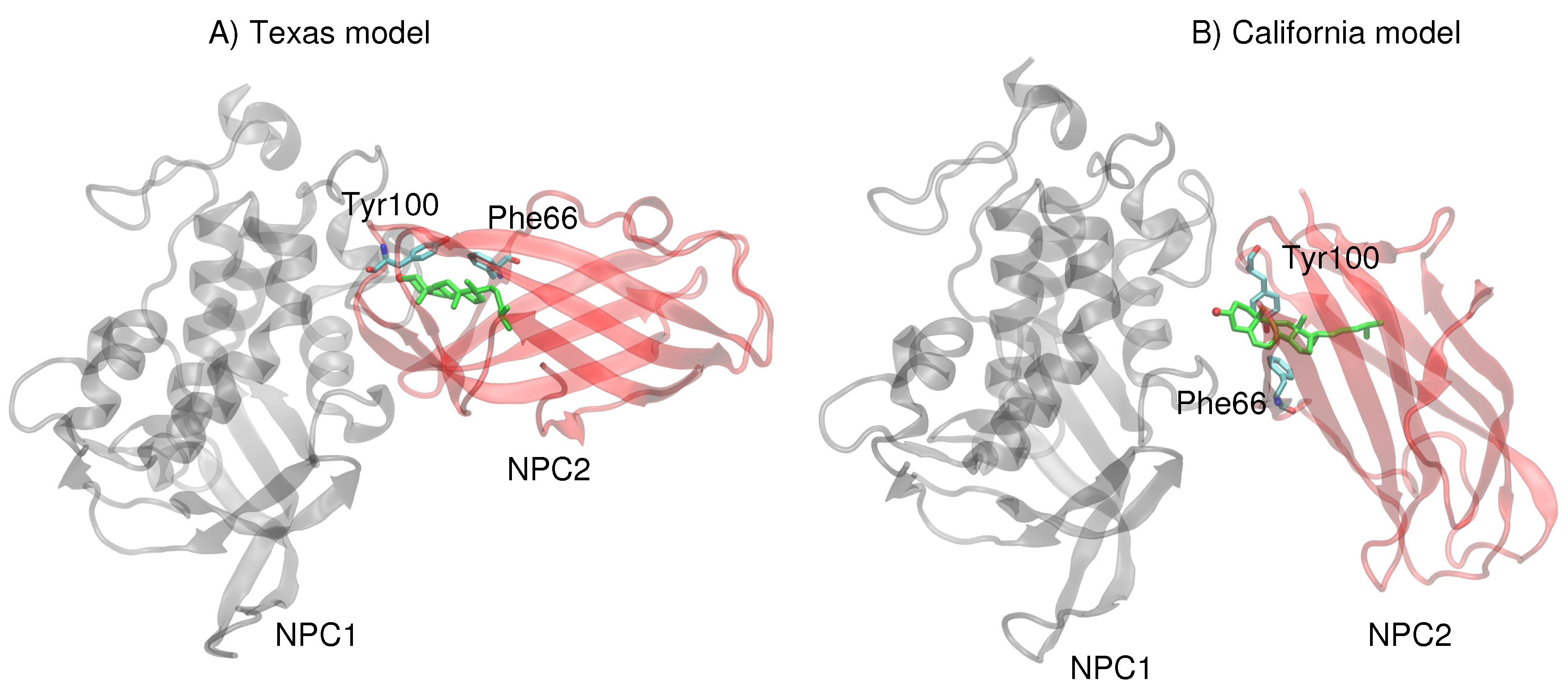
More recently, Li et al. at Stanford University presented a crystal structure (2.4 Å resolution) of the soluble NPC2 protein bound to the NPC1 middle lumenal domain (MLD) (residues 374–620) [
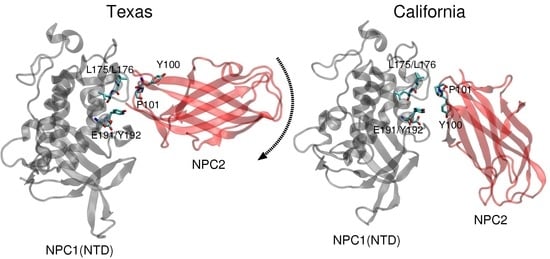
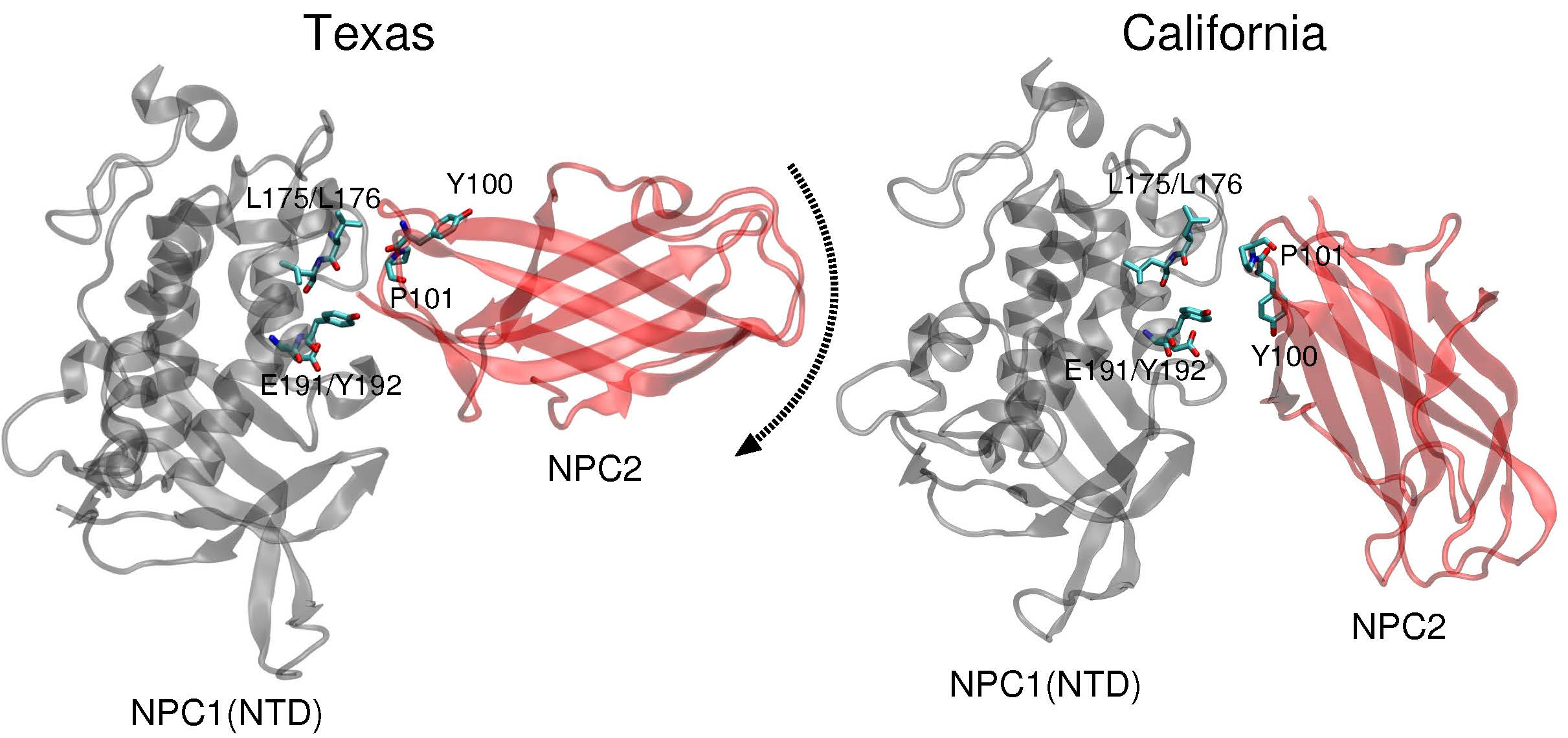
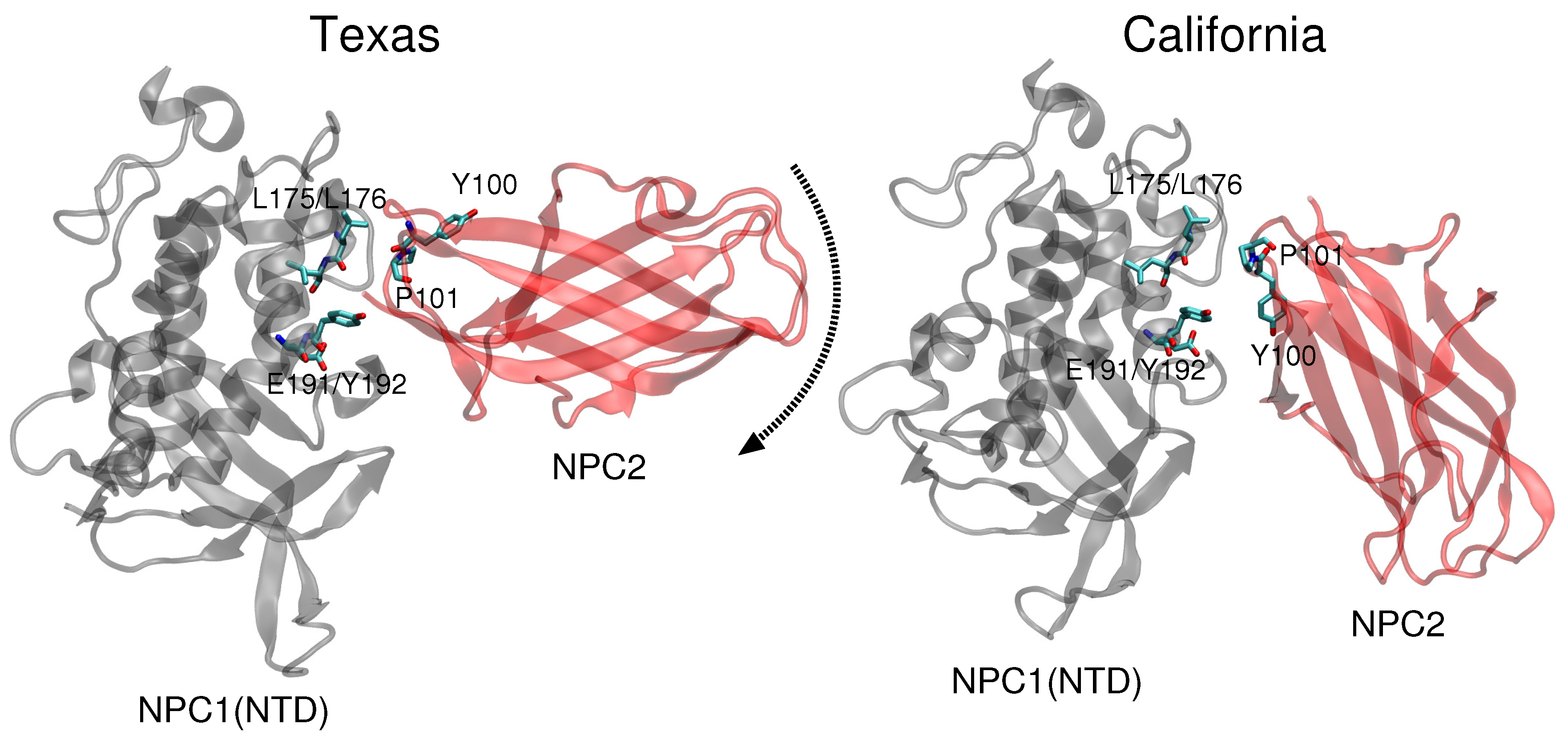
11]. Docking of this complex on a low resolution (4.43 Å) cryo-EM structure of full-length NPC1 protein, Li et al. proposed a new putative NPC1(NTD)–NPC2 interface, which we refer to as the “California model”, that reveals a somewhat different interface but a similar cholesterol transfer tunnel as proposed in the Texas model. In the California model [
11], the angle between NPC2 and NPC1(NTD) is more acute than in the Texas model [
9] (see
Figure 1). In the California model, NPC2’s P120 (corresponding to P101 in PDB 2HKA from Ref. [
4]) is close to NPC1’s NTD K179 (C
-C
separation 4.3 Å) and D180 (C
-C
separation 5.2 Å) [
11]. In comparison, in the Texas model, P120 is farther away from NPC1’s NTD K179 (C
-C
separation 8.1 Å) and D180 (C
-C
separation 10.0 Å) [
9].
We revisited the problem of the NPC1(NTD)–NPC2 interface and analyzed the Texas and California models of the protein–protein complex with respect to cholesterol transfer pathways. We compared the binding energies associated with each of the binding pockets for the two models, and we computed energy barriers corresponding to cholesterol transfer between the NPC binding pockets for each of the two proposed geometries. Based on our analyses, we propose that, depending on the location of the cholesterol ligand, a dynamical interface between the NPC2 and NPC1(NTD) proteins presents structural advantages that can lower the energy barrier and stabilize the passage of the cholesterol substrate. Understanding the atomic details of the interface between the water-soluble NPC2 protein and the membrane-bound NPC1 protein may thus serve as a foundation for therapeutic protein engineering.
2. Results and Discussion
Comparison of the two constructed models (
Figure 1) highlights the difference in relative angle between the NPC1 and NPC2 protein subunits and the alignment of potentially functional amino acids.
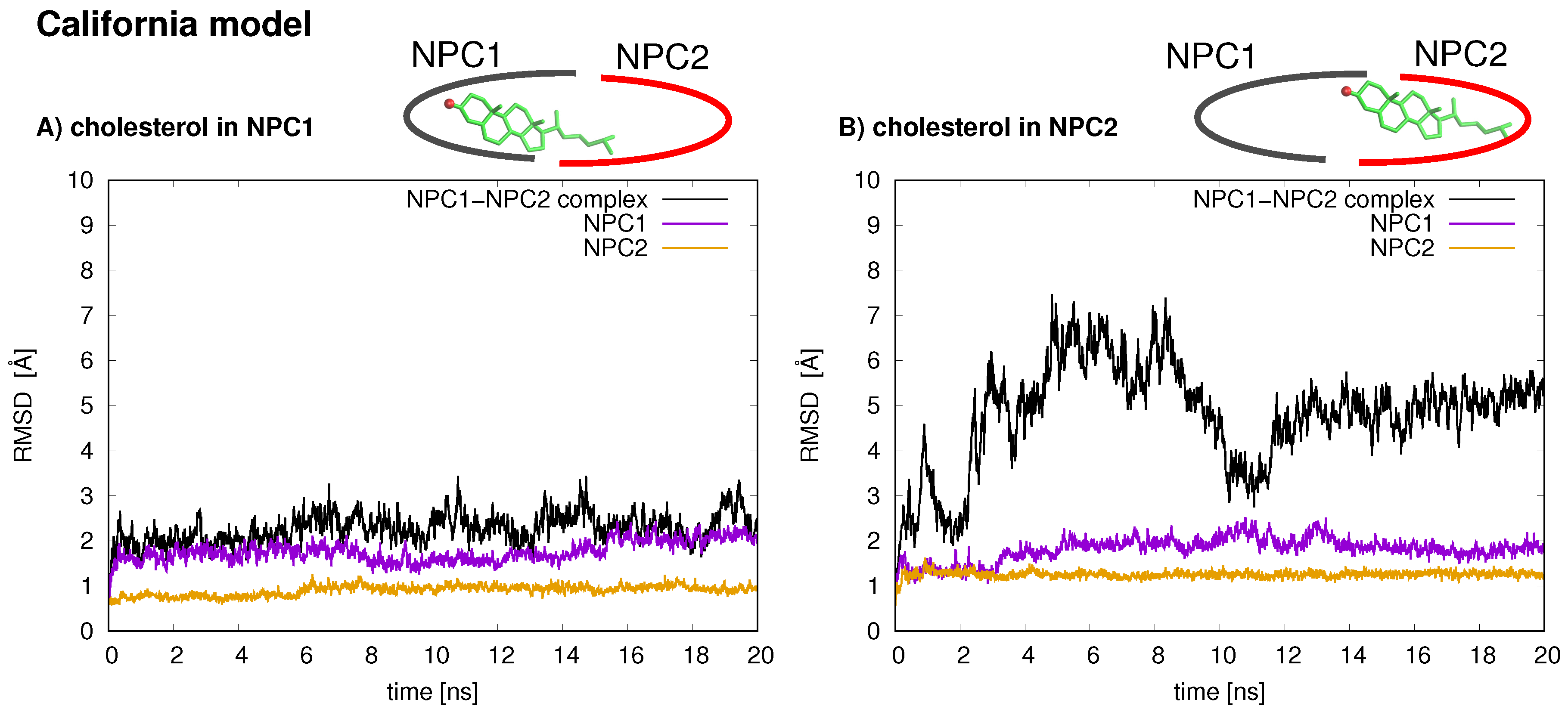
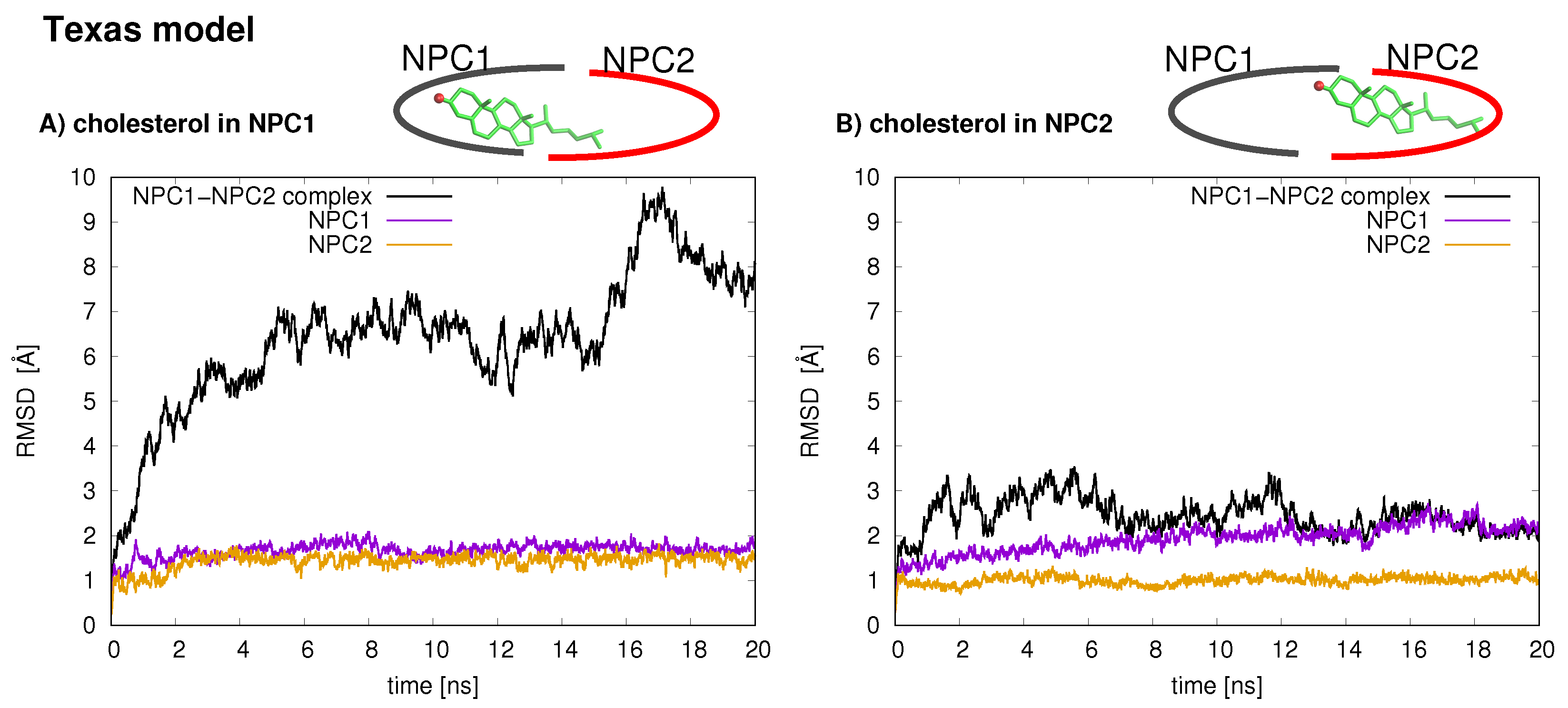
The stabilities of the two protein–protein complexes were checked first by examining the RMSD of protein backbone atoms of the complexes and the individual protein subunits over the 20 ns simulation times. For each model, the cholesterol was simulated in each binding pocket, resulting in four MD trajectories, each 20 ns in length. The RMSD of the Texas model (black line is
Figure 2) when cholesterol is located in the NPC1 binding pocket rapidly increases and peaks around 18 ns at close to 10 Å. Estiu et al. also report large RMSD values for their complex, both in an apo state and when cholesterol is located in the NPC1 binding pocket [
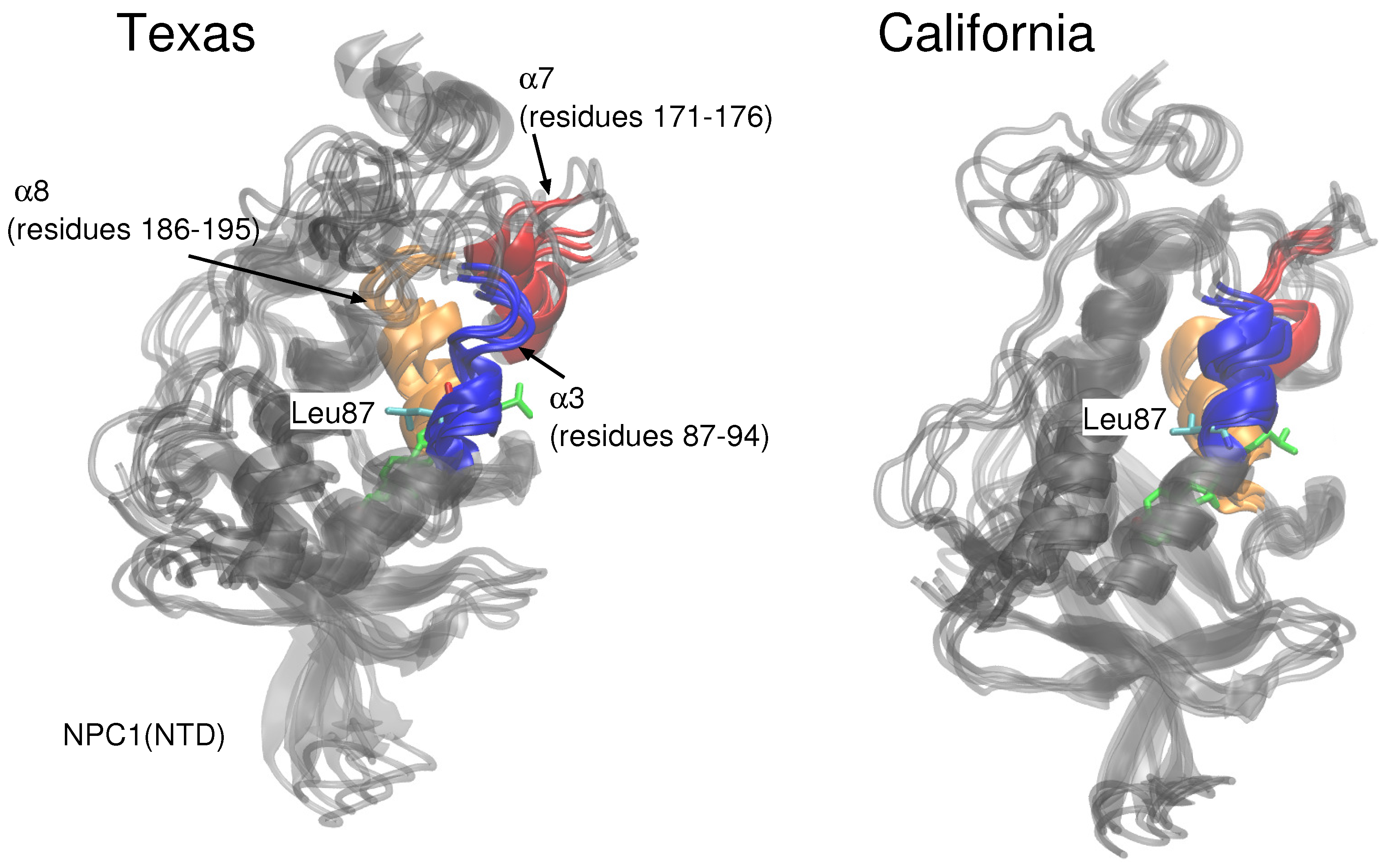
9]. In particular, for the case in which cholesterol is located in the NPC1 binding pocket, they observed significant shifts in the positions of
-helices 3 (residues 87–94), 7 (residues 171–176), and 8 (residues 186–195) [
9] (loop designation according to Ref. [
3]). Estiu et al. attributed the conformational changes to the interaction of Leu87 with the isooctyl chain of the cholesterol. Similar shifts were observed in our simulations with cholesterol in the NPC1 binding pocket, particularly
7 (
Figure 3 Texas model, red helix) demonstrates a large conformational change; Leu87 interacts with the cholesterol ligand. When cholesterol is located in the NPC2 binding pocket, the same model exhibits RMS differences that fluctuate between 2 and 3 Å, also in agreement with the behavior observed by Estiu et al. [
9]. For each binding scenario, the RMSD values of the individual protein subunits were also checked; both NPC1 (purple line) and NPC2 (orange line) demonstrate reasonable structural integrity with RMSD values between 1 and 2 Å.
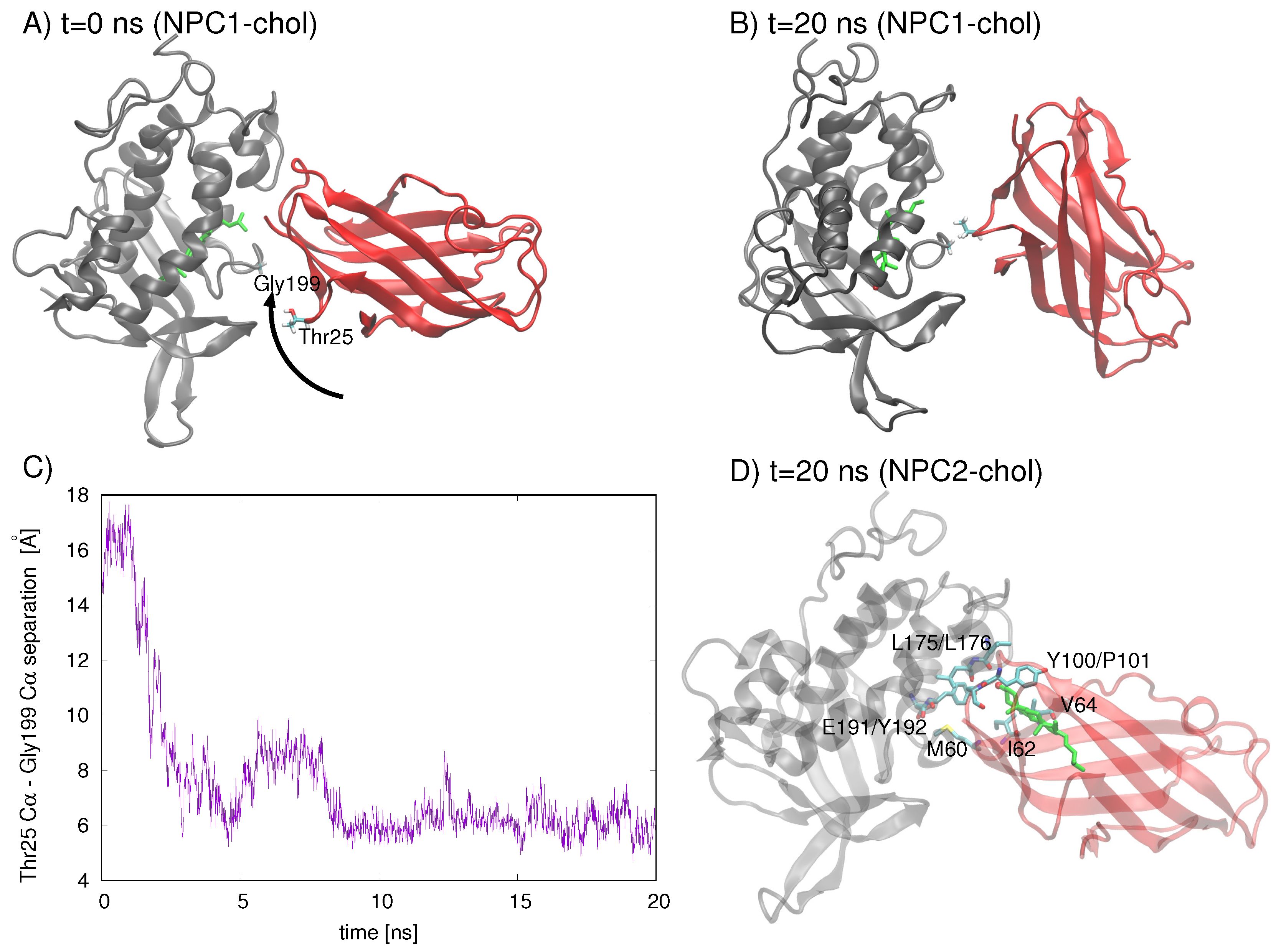
An analysis of residues involved in the Texas model’s interface at
t = 0 ns and
t = 20 ns indicates that the NPC2 protein rotates with respect to NPC1, increasing the separation between the initial interface separation. The rotation brings Gyl199 of NPC1 into close contact (∼5 Å) with Thr25 of NPC2 (
Figure 4C). On the other hand, when cholesterol is in the NPC2 binding pocket of the Texas model, the interface is still intact at
t= 20 ns, with the cholesterol hydroxy group protruding into the interface space (
Figure 4D). Particularly M60, I62, and V64 of NPC2 (corresponding to M79, V81, and V83, respectively, in Ref. [
6]), which have been identified in alanine mutagenesis studies as being critical for preserving transfer function [
6], maintain their position in the interface.
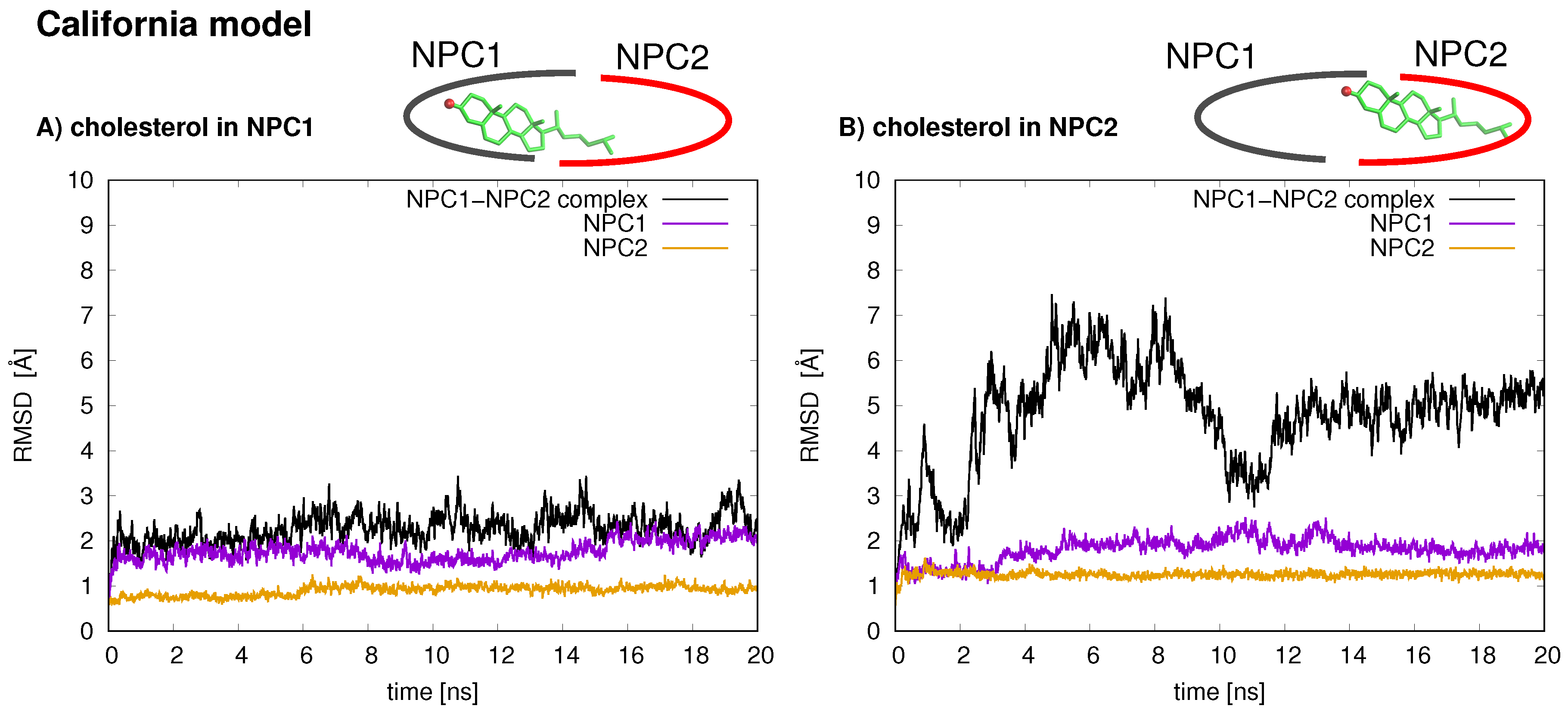
In the California model, the protein–protein complex exhibits relatively small (2–3 Å) RMSD values when the cholesterol ligand is in the NPC1 binding pocket (black line
Figure 5A), as compared to the situation with cholesterol in the NPC2 binding pocket (black line
Figure 5B), which shows fluctuations around 6–7 Å. In the California model, the relative structural integrity of the NPC1 protein with cholesterol in its binding pocket, compared to the Texas model, can be observed in
Figure 3. All three helices,
3 (blue),
7 (red), and
8 (orange), show minimal changes throughout the 20 ns MD simulation. These differences suggest that the relative position of and the angle between the two protein subunits may influence the interface stability, depending on the location of cholesterol. In the Texas model, the complex is more stable with cholesterol located in NPC2 while, in the California model, the reverse is true. Based on these results, one could speculate that the two proteins undergo dynamical rearrangement depending on the location of the cholesterol. One possible interpretation is that the Texas model represents better a stable binding interface before the transfer of cholesterol when it is still in the binding pocket of NPC2. The California model represents a more stable conformation after ligand transfer when cholesterol is located in NPC1(NTD).
To check whether the geometry of the protein–protein complex affects binding energy, we performed solvation energy analyses of the ligand binding strengths using the GBSW approach. Computations indicate that in the Texas model, the NPC2/chol complex is energetically stabilized relative to the complex with cholesterol in the NPC1 binding pocket by approximately −1.48 kcal/mol. In the California model, the corresponding binding energy stabilization is −2.91 kcal/mol. The cholesterol binding behavior of NPC2 differs from that of NPC1(NTD) in in vitro studies as well [
12]. NPC2 binds cholesterol rapidly and reversibly at 4
C as well as at 37
C, behaving—according to the authors—as a “typical” receptor [
12]. NPC1(NTD), on the other hand, binds differently, slowly binding cholesterol at 4
C and not releasing the lipid at this temperature; these on-off rates are accelerated at 37
C [
12]. The authors state that this binding behavior is indicative of a closed conformation of NPC1(NTD) that is opened in the presence of NPC2. It should be noted that experimental binding and transfer rates are obtained via assays that are conducted over a period of several minutes (including incubation and elution); the experimentally measured values are likely an underestimate of the true transfer rates [
12]. In the context of this computational study, the experimentally measured values serve only as a qualitative comparison for the behavior of the two proteins that have been simulated here for hundreds of nanoseconds.
Next, more rigorous free energy perturbation calculations were carried out, using MD simulations with explicit water, to compare the relative free energies of binding of cholesterol in each of the binding pockets for both models. In the Texas model, the free energy of binding in NPC1 was calculated to be −60.0 kcal/mol (±3.1 kcal/mol), and in NPC2 −60.9 kcal/mol (±3.1 kcal/mol). The results are qualitatively similar to the GBSW results, suggesting that the binding pockets in the Texas model indicate no significant free energy gain for cholesterol binding in the NPC1 binding pocket. In the California model, the free energy of binding in NPC1 is somewhat less favorable (−52.1 ± 2.9 kcal/mol) compared to the binding in the NPC2 pocket (−54.4 ± 2.9 kcal/mol). Overall, the California model appears to give weaker cholesterol binding interactions than does the Texas model. Reasons for this tighter binding in the Texas model may be found in the interactions particularly of the 3
-hydroxy end that is involved in
–
interactions with Phe66 and Tyr100 of NPC2 [
13]. In the Texas model, the cholesterol is engaged with Tyr100 and Phe66 which are positioned above the ligand (
Figure 6A). In the California model, on the other hand, both Tyr100 and Phe66 are pointing downward and away from the cholesterol ligand (
Figure 6B). Therefore, the interface of the Texas model may result in more favorable hydrophobic environment, stabilizing the ligand position in the NPC2, as we observed in the RMSD behavior discussed earlier (see
Figure 2).
In vitro data indicate that cholesterol transfer between NPC1 and lipid bilayers facilitated by NPC2 is bidirectional [
12]. Our computations show similar ligand binding strengths for NPC1 and NPC2, which is particularly true for the Texas model. Hence, a bidirectional transfer of cholesterol would be feasible. On the other hand, the orientation of the California model, in which the long axis of the binding pockets of the two protein subunits are more closely aligned, may yield a lower barrier for ligand transfer. Again, our results support the notion of a dynamical interface in which the alignment of the NPC1(NTD) and NPC2 proteins changes depending on the location of the ligand. The pocket alignment of the Texas model may reflect a recognition pose of protein–protein docking, during which the interface is being established. Bidirectional ligand transfer may be possible in the Texas alignment, whereas the California alignment may favor the unidirectional transfer of cholesterol from NPC2 to NPC1.
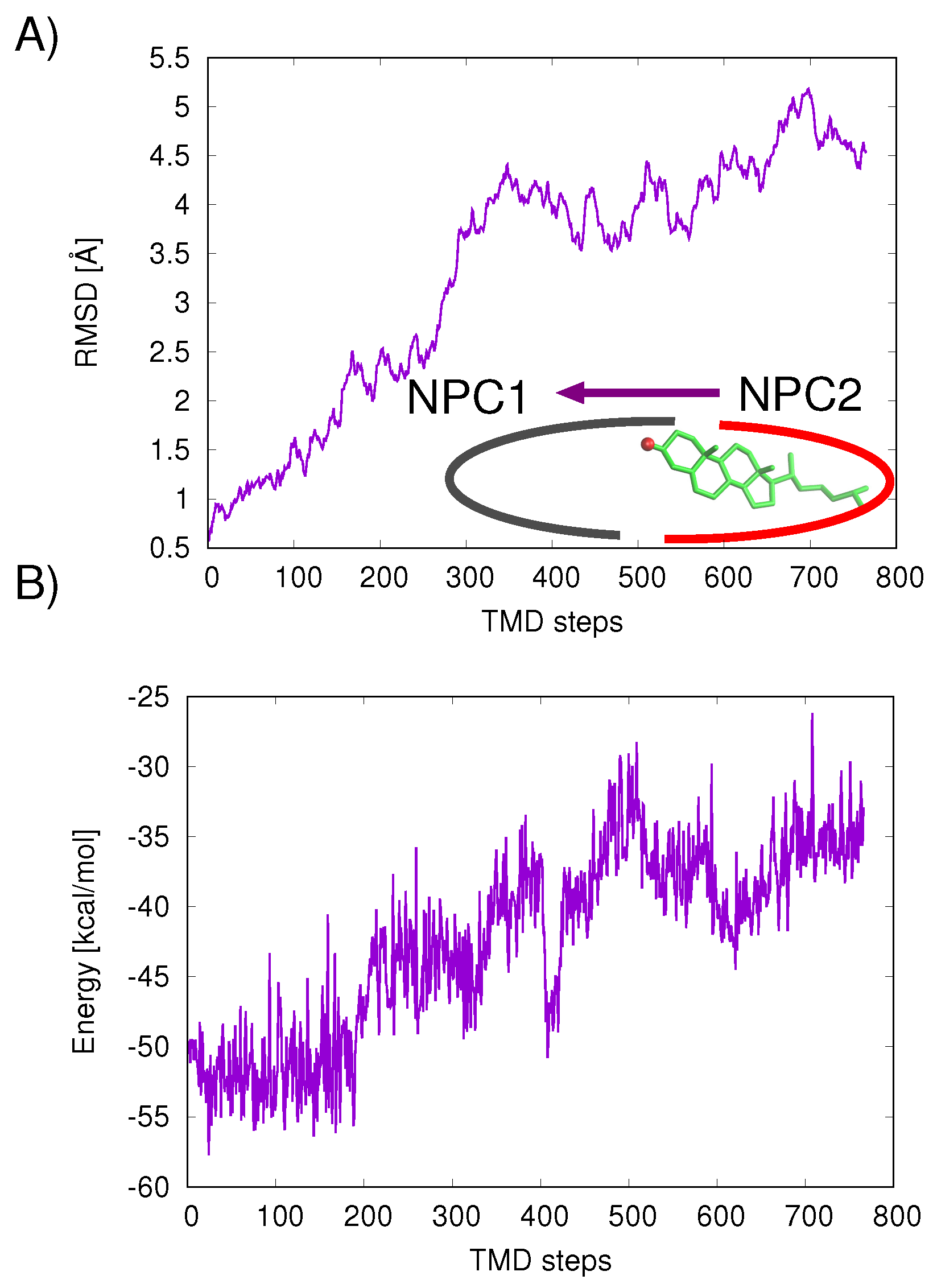
The mechanism of ligand transfer was studied next for both the Texas and California models using TMD simulations. For the Texas model, simulations of cholesterol transfer in the direction NPC2→NPC1 indicated instabilities at the protein–protein interface, evidenced by the increase in protein backbone RMSD values (Å) (
Figure 7A) and increase in cholesterol–protein potential energies (
E = +16.9 kcal/mol) (
Figure 7B). For the California model, relatively lower RMS differences were measured for the cholesterol transfer path NPC2→NPC1 (purple line in
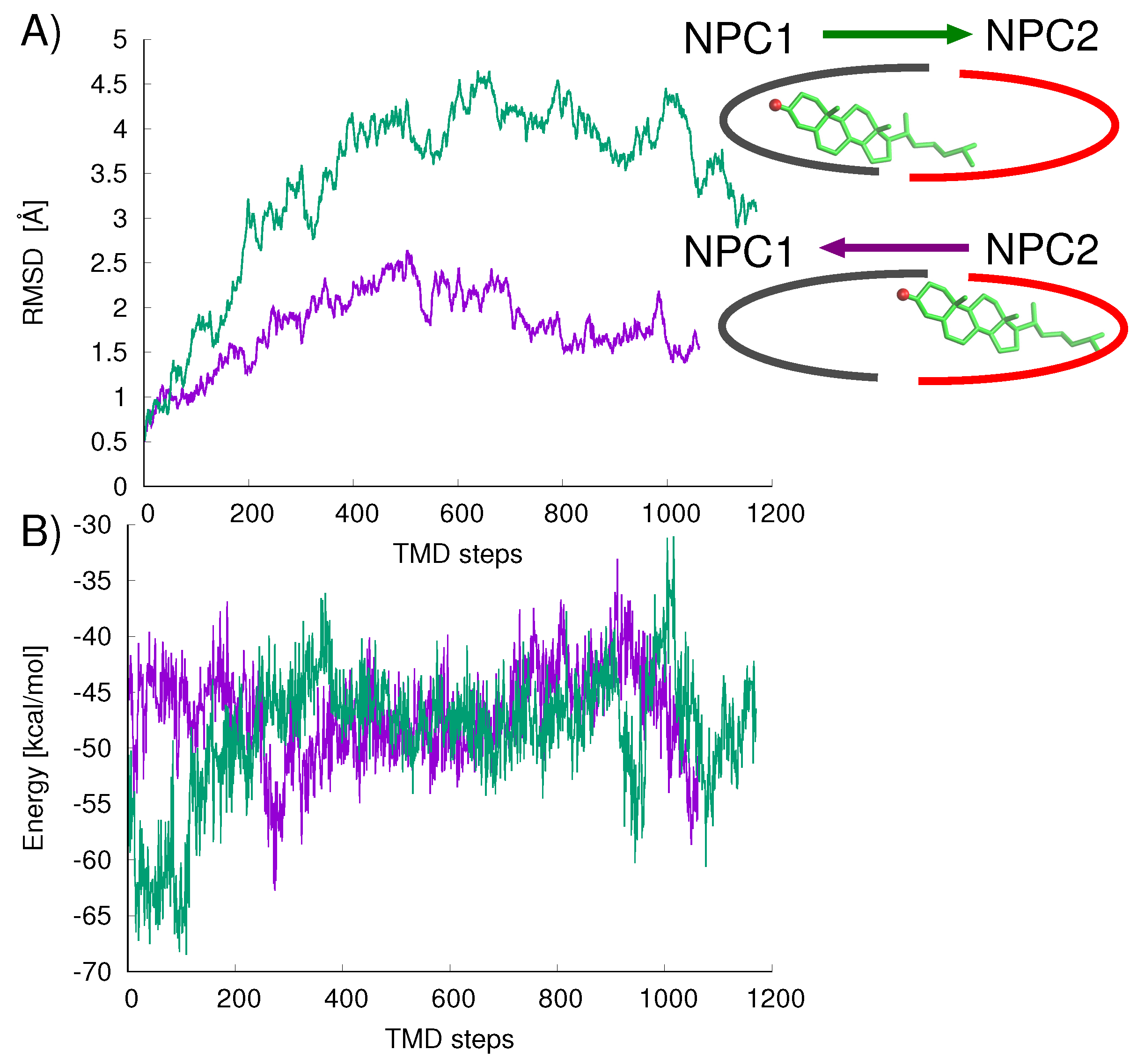
Figure 8A). For the opposition direction, NPC1→NPC2, the California model shows higher protein backbone RMS differences along the path (green line in
Figure 8A). The cholesterol–protein potential energy changes associated with the ligand transfer (
Figure 8B) are overall more favorable for the transfer of cholesterol in the NPC2→NPC1 direction (
E = −12.6 kcal/mol) compared to the transfer in the opposite direction NPC1→NPC2 (
E = +8.3 kcal/mol).
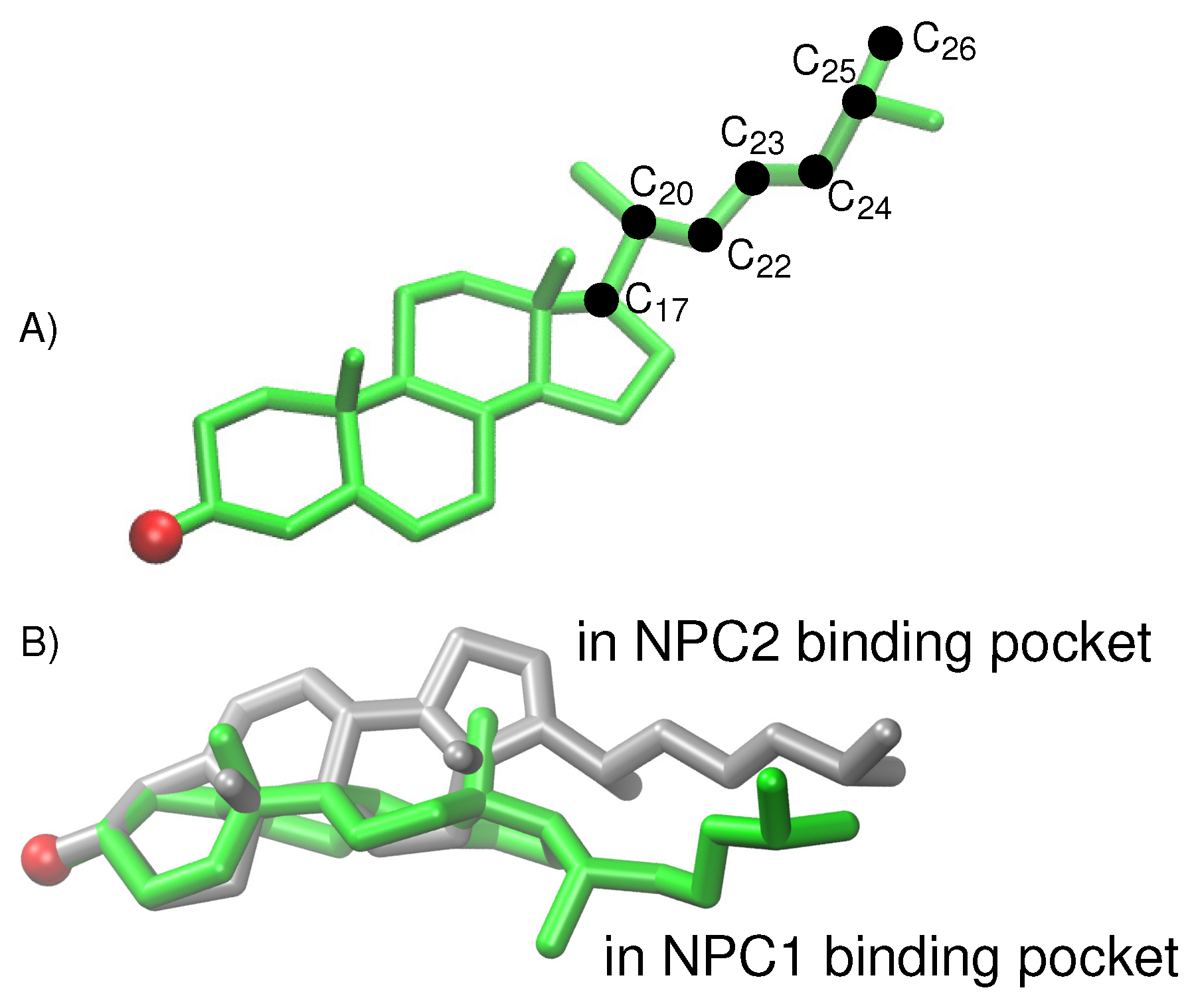
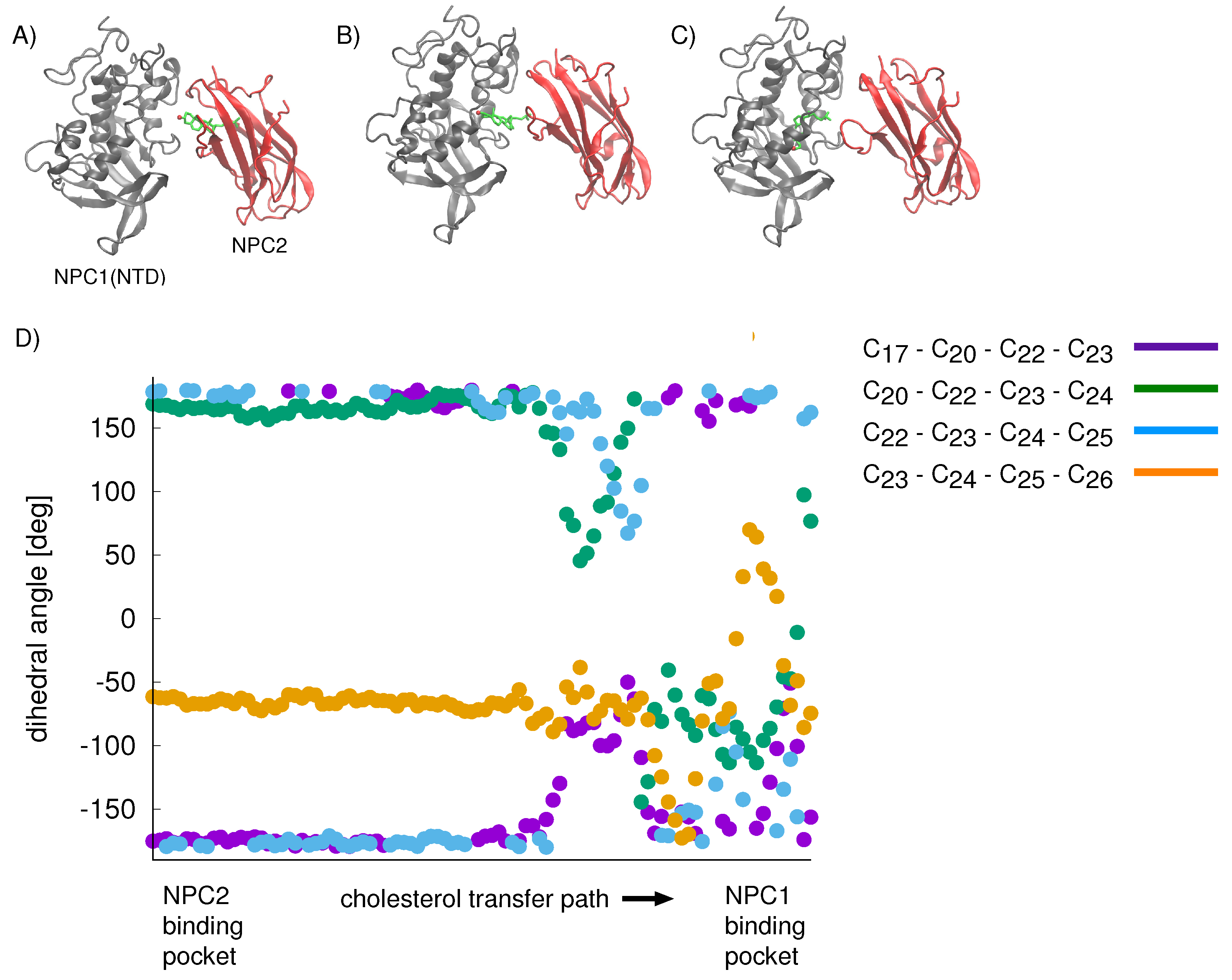
For the cholesterol transfer modeled using the California model, we computed the dihedral angles of the cholesterol tail (
Figure 9A) over the course of the TMD path (
Figure 10D). The coupled twisting of C
-C
-C
-C
and C
-C
-C
-C
dihedral angles is similar to the behavior that was observed previously in our study of cholesterol isomerization inside the binding pockets of NPC1 and NPC2 [
10]. Inside the NPC2 binding pocket (gray model in
Figure 9B), the dihedral angle of cholesterol’s C
-C
-C
-C
atoms is <
(purple dots in
Figure 10D). The C
-C
-C
-C
dihedral angle (blue dots in
Figure 10D) takes on similar values, as observed previously [
10]. During the handover of the ligand at the protein–protein interface (
Figure 10B), the C
-C
-C
-C
dihedral angle approaches −50
, before relaxing to <
inside the NPC1(NTD) binding pocket (green model in
Figure 9B).
4. Conclusions
Here, we have compared two similar binding interfaces of the NPC1(NTD) and NPC2 proteins. MD simulations indicate that the position of the cholesterol in the protein–protein complex affects the stability of the interface, as evidenced by the RMSD values of the models over 20 ns simulation time. In the Texas model, the NPC1(NTD)–NPC2 complex is unstable with cholesterol in the NPC2 binding pocket, whereas, in the California model, the complex is unstable when cholesterol is in the NPC1 binding pocket. As a bidirectional cholesterol transfer has been observed in cholesterol transfer assays, our findings may support the existence of two feasible docking interfaces. According to this hypothesis, the Texas interface may correspond to the situation after cholesterol has been transferred, whereas the California interface is more stable with cholesterol in the NPC2 binding pocket. Free energy analyses of the cholesterol binding strengths in the binding pockets of the two models present arguments that support bidirectional ligand transfer particularly in the Texas model. TMD simulations with the California model have provided us with plausible minimum energy paths connecting the binding pockets and indicate that cholesterol transfer from NPC2 to NPC1 is energetically favorable.
One mechanistic explanation for the difference in proposed protein–protein conformations may be that, in the Texas model, the residues involved in recognition are used to establish the interface. In this conformation, cholesterol transfer may take place. As the protein domains realign according to the California model, forward transfer of cholesterol, e.g., in the direction NPC2→NPC1(NTD), is secured. In this pose, the free energy gain is realized and cholesterol is positioned for subsequent insertion into the lysosomal membrane.
As a direct interaction between NPC2 and NPC1(NTD) has thus far not been detected, one may speculate that the isolated NTD may be insufficient to establish a stable interaction. In fact, domain C of NPC1’s MLD which links the transmembrane domain to the NTD, may be needed to establish the correct binding pose for NPC2 [
8]. Indeed, 28 NPC disease mutations map to NPC1’s domain C [
24]. Furthermore, the NTD also may require specific interactions with the C-terminal domain (CTD) (residues 861-1083), as disruption of this interface has been shown to block cholesterol transfer from the late endosome to ER [
25]. Our ongoing investigations are therefore directed at constructing a more complete NPC1 model that includes several domains (NTD, MLD, and CTD) and simulating this with the NPC2 to analyze additional atomic interactions required for stabilizing the protein–protein interface. These efforts are greatly supported by the emergence of higher resolution X-ray crystallography data as well as cryo-EM data elucidating atomic detail of the NPC1 domains. Our simulations have been carried out with bovine NPC2 which has a highly conserved sequence homology to human NPC2. Nonetheless, repeating our simulations with human NPC2 would be an interesting and worthwhile comparison of binding interactions.
Ultimately, our mechanistic understandings of the interactions between NPC1 and NPC2 may help rationalize the severity of different NPC disease genotypes. This knowledge is also useful for understanding NPC1’s interactions with other proteins found in the lysosomal, including the Ebola GP. Interestingly, at pH = 6, the Ebola GP binds 80× tighter to the NPC1 protein than to NPC2 in vitro [
8]. As our understanding of the molecular interactions involved in protein–protein binding increases, the reasons for this competitive binding may become clearer.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}