Transcriptome Analysis of Novosphingobium pentaromativorans US6-1 Reveals the Rsh Regulon and Potential Molecular Mechanisms of N-acyl-l-homoserine Lactone Accumulation

Abstract
:1. Introduction
2. Results and Discussion
2.1. Sequence Analysis of RshUS6–1 Protein
2.2. N-Acyl-l-homoserine Lactone (AHL) Accumulation in the Cross Feeding Assay and Extract Assay
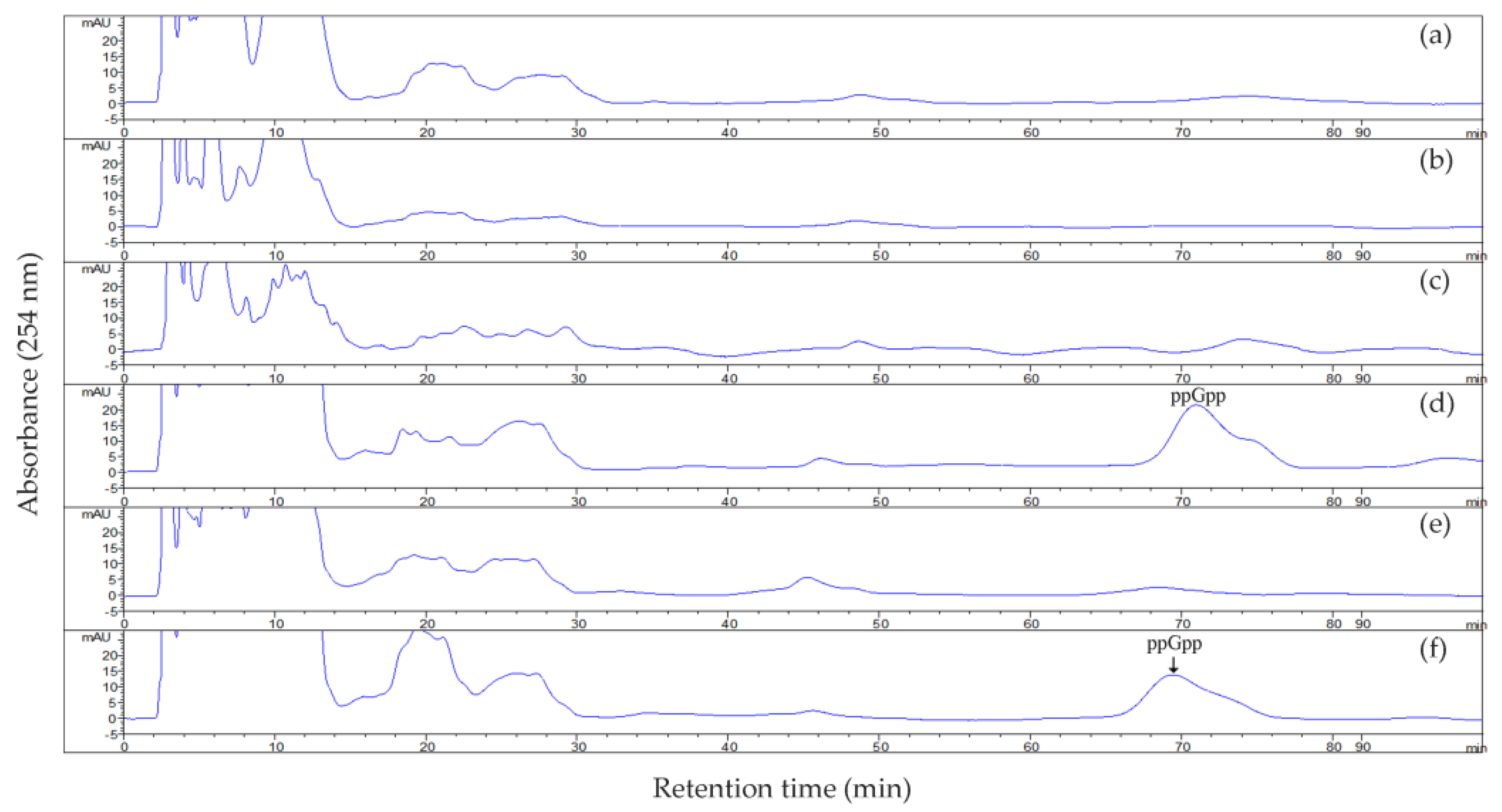
2.3. ppGpp Accumulation in Strain US6-1 and Its Derivatives
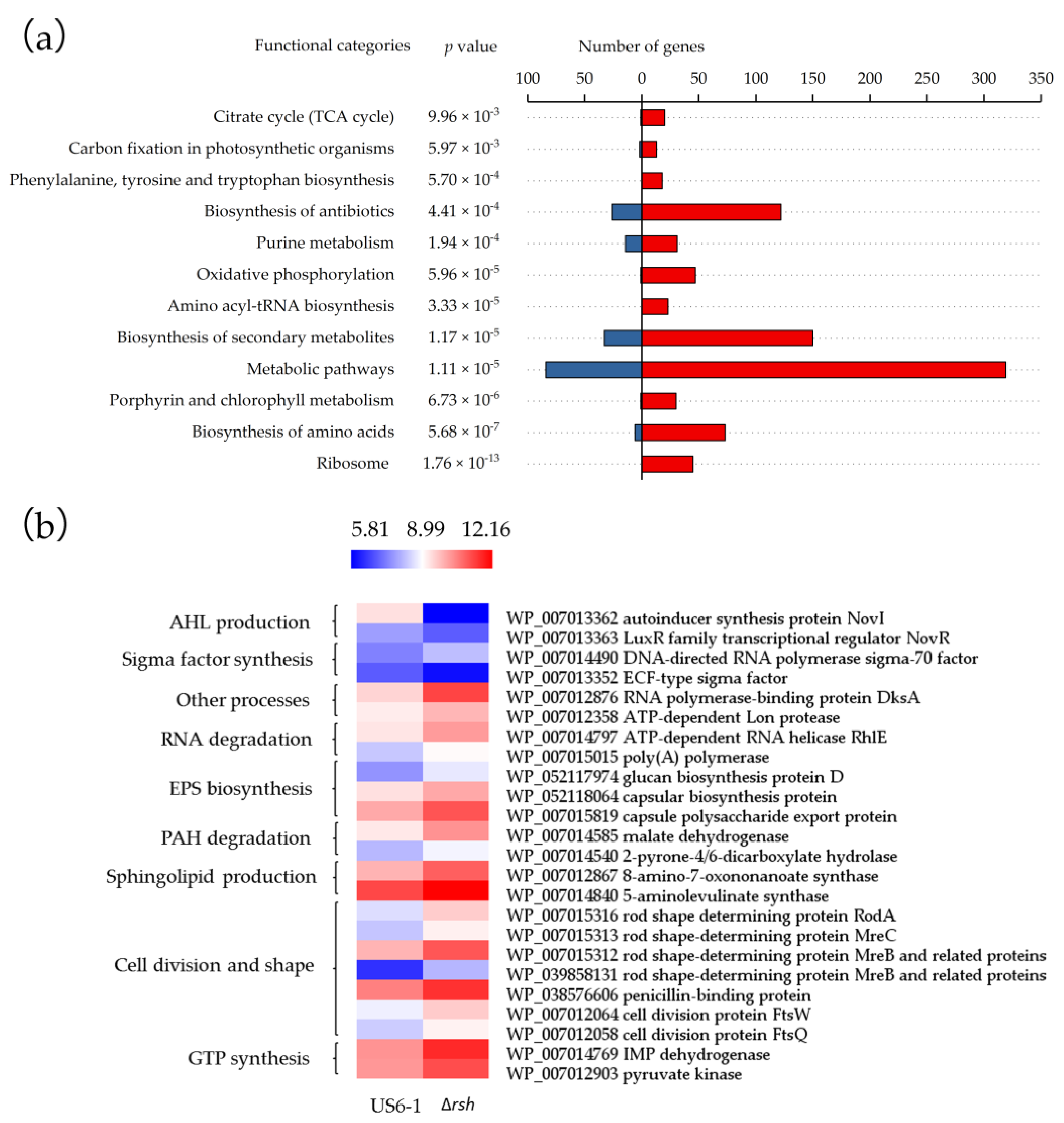
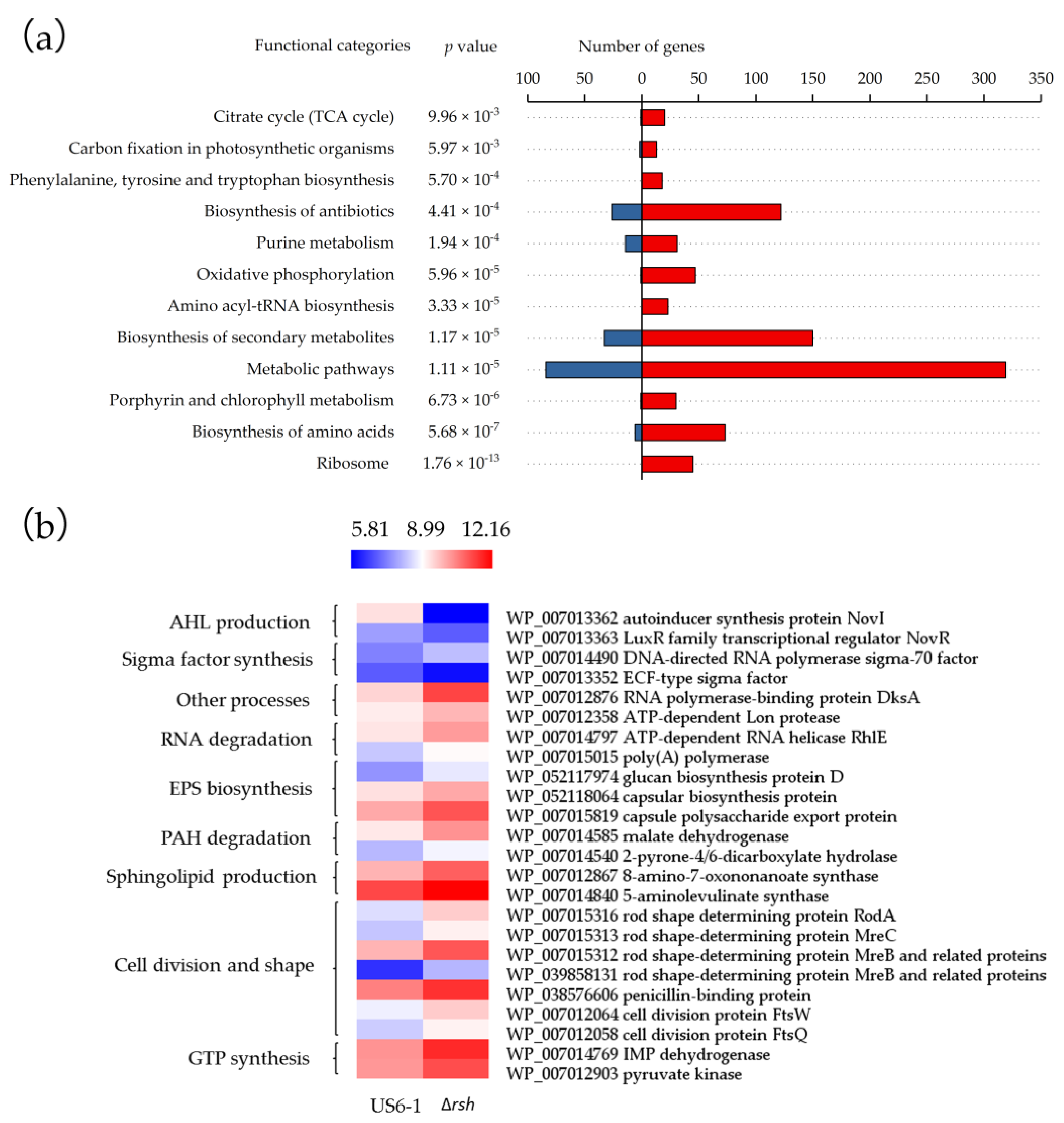
2.4. Global Overview of the RshUS6–1 Regulon
2.5. Essential Genes for AHL Production were Positively Regulated by RshUS6–1 in the Exponential Growth Phase
2.6. Potential Molecular Mechanisms of RshUS6–1-Dependent Expression of novI and novR
2.7. Repressed Exopolysaccharides (EPS) Biosynthesis
2.8. RshUS6–1 Regulon Involved in Other Processes
3. Materials and Methods
3.1. Bacterial Strains, Plasmids and Growth Conditions
3.2. Sequence Analysis of RshUS6–1
3.3. Strain Construction
3.4. Growth Curve
3.5. Detection of AHL Accumulation
3.6. ppGpp Analysis
3.7. Identification of AHL Degradation of ∆rsh in the Exponential Growth Phase
3.8. RNA Extraction and RNA-seq
3.9. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
3.10. Congo Red Binding Assay
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author contribution
Acknowledgments
Conflict of interest
Abbreviations
| AHL | N-acyl-l-homoserine Lactone |
| QS | Quorum Sensing |
| DEGs | Differentially Expressed Genes |
| PAH | Polycyclic Aromatic Hydrocarbon |
| ECF | Extracytoplasmic Function |
| SPT | Serine Palmitoyltransferase |
| EPS | Exopolysaccharide |
References
- Balsalobre, C. Concentration matters!! ppGpp, from a whispering to a strident alarmone. Mol. Microbiol. 2011, 79, 827–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potrykus, K.; Cashel, M. (p)ppGpp: Still magical? Annu. Rev. Microbiol. 2008, 62, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 2015, 13, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durfee, T.; Hansen, A.M.; Zhi, H.; Blattner, F.R.; Jin, D.J. Transcription profiling of the stringent response in Escherichia coli. J. Bacteriol. 2008, 190, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Francois, P.; Liebeke, M.; Fraunholz, M.; Goerke, C.; Krismer, B.; Schrenzel, J.; Lalk, M.; Wolz, C. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Vercruysse, M.; Fauvart, M.; Jans, A.; Beullens, S.; Braeken, K.; Cloots, L.; Engelen, K.; Marchal, K.; Michiels, J. Stress response regulators identified through genome-wide transcriptome analysis of the (p)ppGpp-dependent response in Rhizobium etli. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolz, A. Molecular characteristics of xenobiotic-degrading sphingomonads. Appl. Microbiol. Biotechnol. 2009, 81, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Feng, H.; Lu, H.; Zeng, Y.H. Novel 16S rDNA primers revealed the diversity and habitats-related community structure of sphingomonads in 10 different niches. Anton. Leeuw. 2017, 110, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.; Benimeli, C.S.; Saez, J.M.; Fuentes, M.S.; Cuozzo, S.A.; Polti, M.A.; Amoroso, M.J. Bacterial bio-resources for remediation of hexachlorocyclohexane. Int. J. Mol. Sci. 2012, 13, 15086–15106. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Yano, I.; Masui, M.; Yabuuchi, E. Isolation of a novel sphingoglycolipid containing glucuronic acid and 2-hydroxy fatty acid from Flavobacterium devorans ATCC 10829. J. Biochem. 1978, 83, 1213–1216. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, S.; Moriguchi, R.; Sekiya, K.; Nakai, T.; Ono, E.; Kume, K.; Kawahara, K. The cell envelope structure of the lipopolysaccharide-lacking gram-negative bacterium Sphingomonas paucimobilis. J. Bacteriol. 1994, 176, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Francez-Charlot, A.; Kaczmarczyk, A.; Vorholt, J.A. The branched CcsA/CckA-ChpT-CtrA phosphorelay of Sphingomonas melonis controls motility and biofilm formation. Mol. Microbiol. 2015, 97, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Pollock, T.J.; Armentrout, R.W. Planktonic/sessile dimorphism of polysaccharide-encapsulated sphingomonads. J. Ind. Microbiol. Biotechnol. 1999, 23, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Fida, T.T.; Moreno-Forero, S.K.; Heipieper, H.J.; Springael, D. Physiology and transcriptome of the polycyclic aromatic hydrocarbon-degrading Sphingomonas sp. LH128 after long-term starvation. Microbiology 2013, 159, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.B.; Yew, W.S. Development of quorum-based anti-virulence therapeutics targeting Gram-negative bacterial pathogens. Int. J. Mol. Sci. 2013, 14, 16570–16599. [Google Scholar] [CrossRef] [PubMed]
- Van Delden, C.; Comte, R.; Bally, M. Stringent response activates quorum sensing and modulates cell density-dependent gene expression in Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 5376–5384. [Google Scholar] [CrossRef] [PubMed]
- Erickson, D.L.; Lines, J.L.; Pesci, E.C.; Venturi, V.; Storey, D.G. Pseudomonas aeruginosa relA contributes to virulence in Drosophila melanogaster. Infect. Immun. 2004, 72, 5638–5645. [Google Scholar] [CrossRef] [PubMed]
- Schafhauser, J.; Lepine, F.; McKay, G.; Ahlgren, H.G.; Khakimova, M.; Nguyen, D. The stringent response modulates 4-hydroxy-2-alkylquinoline biosynthesis and quorum-sensing hierarchy in Pseudomonas aeruginosa. J. Bacteriol. 2014, 196, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Moris, M.; Braeken, K.; Schoeters, E.; Verreth, C.; Beullens, S.; Vanderleyden, J.; Michiels, J. Effective symbiosis between Rhizobium etli and Phaseolus vulgatis requires the alarmone ppGpp. J. Bacteriol. 2005, 187, 5460–5469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.B.; Wang, C.; Zhang, L.H. The quormone degradation system of Agrobacterium tumefaciens is regulated by starvation signal and stress alarmone (p)ppGpp. Mol. Microbiol. 2004, 52, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, H.B.; Wang, L.H.; Zhang, L.H. Succinic semialdehyde couples stress response to quorum-sensing signal decay in Agrobacterium tumefaciens. Mol. Microbiol. 2006, 62, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Zeng, Y.H.; Yu, Z.L.; Zhang, J.; Feng, H.; Lin, X.C. In silico and experimental methods revealed highly diverse bacteria with quorum sensing and aromatics biodegradation systems—A potential broad application on bioremediation. Bioresour. Technol. 2013, 148, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.M.; Gan, H.Y.; Ahmad, N.H.; Aziz, N.A.; Hudson, A.O.; Savka, M.A. Whole genome sequencing and analysis reveal insights into the genetic structure, diversity and evolutionary relatedness of luxI and luxR hornologs in bacteria belonging to the Sphingomonadaceae family. Front. Cell. Infect. Microbiol. 2015, 4, 188. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.H.; Zheng, W.; Zheng, T.L.; Tian, Y. Biodegradation of polycyclic aromatic hydrocarbons by Novosphingobium pentaromativorans US6-1. PLoS ONE 2014, 9, e101438. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Kwon, Y.M.; Kwon, K.K.; Kim, S.J. Complete genome sequence of Novosphingobium pentaromativorans US6-1(T). Stand. Genomic Sci. 2015, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Steinchen, W.; Bange, G. The magic dance of the alarmones (p)ppGpp. Mol. Microbiol. 2016, 101, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Amend, A.; Borkowski, J.; Demarco, R.; Bailey, W.; Liu, Y.; Xie, G.C.; Blevins, R. Multiclustal: A systematic method for surveying Clustal W alignment parameters. Bioinformatics 1999, 15, 862–863. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method—A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Felsenstein, J. Confidence-limits on phylogenies—An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.R.; Lin, G.M.; Chen, W.L.; Wang, L.; Zhang, C.C. ppGpp metabolism is involved in heterocyst development in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 2013, 195, 4536–4544. [Google Scholar] [CrossRef] [PubMed]
- Cashel, M.; Gallant, J. Two compounds implicated in the function of the RC gene of Escherichia coli. Nature 1969, 221, 838–841. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.M.; Buckley, L.; Szegedi, E.; Hudson, A.O.; Savka, M.A. Identification of an rsh Gene from a Novosphingobium sp. necessary for quorum-sensing signal accumulation. J. Bacteriol. 2009, 191, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Primm, T.P.; Andersen, S.J.; Mizrahi, V.; Avarbock, D.; Rubin, H.; Barry, C.E. The stringent response of Mycobacterium tuberculosis is required for long-term survival. J. Bacteriol. 2000, 182, 4889–4898. [Google Scholar] [CrossRef] [PubMed]
- Mechold, U.; Cashel, M.; Steiner, K.; Gentry, D.; Malke, H. Functional analysis of a relA/spoT gene homolog from Streptococcus equisimilis. J. Bacteriol. 1996, 178, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Flores, A.; Du Pont, G.; Huerta-Saquero, A.; Merchant-Larios, H.; Servín-González, L.; Durán, S. The stringent response is required for amino acid and nitrate utilization, nod factor regulation, nodulation, and nitrogen fixation in Rhizobium etli. J. Bacteriol. 2005, 187, 5075–5083. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Ki, J.S.; Case, R.J.; Qian, P.Y. Diversity and acyl-homoserine lactone production among subtidal biofilm-forming bacteria. Aquat. Microb. Ecol. 2008, 52, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.Q.; Bittner, A.N.; Wang, J.D. Diversity in (p)ppGpp metabolism and effectors. Curr. Opin. Microbiol. 2015, 24, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnusson, L.U.; Farewell, A.; Nystrom, T. ppGpp: a global regulator in Escherichia coli. Trends Microbiol. 2005, 13, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Eymann, C.; Homuth, G.; Scharf, C.; Hecker, M. Bacillus subtilis functional genomics: global characterization of the stringent response by proteome and transcriptome analysis. J. Bacteriol. 2002, 184, 2500–2520. [Google Scholar] [CrossRef] [PubMed]
- Bowden, S.D.; Eyres, A.; Chung, J.C.S.; Monson, R.E.; Thompson, A.; Salmond, G.P.C.; Spring, D.R.; Welch, M. Virulence in Pectobacterium atrosepticum is regulated by a coincidence circuit involving quorum sensing and the stress alarmone, (p)ppGpp. Mol. Microbiol. 2013, 90, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.K.P.; Al Seesi, S.; Banday, A.R.; Baumgartner, M.; Olthof, A.; Lemoine, C.; Măndoiu, I.I.; Kanadia, R.N. Network-based bioinformatics analysis of spatio-temporal RNA-Seq data reveals transcriptional programs underpinning normal and aberrant retinal development. BMC Genomics 2016, 17, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.K.; Wang, Y.B.; Liu, Z.X.; Cheng, H.; Xue, Y. HemI: A toolkit for illustrating heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef] [PubMed]
- Galloway, W.R.J.D.; Hodgkinson, J.T.; Bowden, S.D.; Welch, M.; Spring, D.R. Quorum sensing in Gram-negative bacteria: Small-molecule modulation of AHL and AI-2 quorum sensing pathways. Chem. Rev. 2011, 111, 28–67. [Google Scholar] [CrossRef] [PubMed]
- Dalebroux, Z.D.; Swanson, M.S. ppGpp: Magic beyond RNA polymerase. Nat. Rev. Microbiol. 2012, 10, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.X.; Ji, L.; Wang, X.; Li, J.R.; Zhu, J.L.; Sun, A.H. Role of RpoS in stress resistance, quorum sensing and spoilage potential of Pseudomonas fluorescens. Int. J. Food Microbiol. 2018, 270, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Guo, M.; Yang, M.J.; Zhang, Y.X.; Zhou, X.H.; Wang, Q.Y. A sigma(E)-mediated temperature gauge controls a switch from LuxR-mediated virulence gene expression to thermal stress adaptation in Vibrio alginolyticus. PLoS Pathog. 2016, 12, e1005645. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Overall, C.C.; Johnson, R.C.; Jones, M.B.; McDermott, J.E.; Heffron, F.; Adkins, J.N.; Cambronne, E.D. ChIP-Seq Analysis of the sigma(E) regulon of Salmonella enterica serovar Typhimurium reveals new genes implicated in heat shock and oxidative stress response. PLoS ONE 2015, 10, e0138466. [Google Scholar] [CrossRef]
- Devescovi, G.; Venturi, V. The Burkholderia cepacia rpoE gene is not involved in exopolysaccharide production and onion pathogenicity. Can. J. Microbiol. 2006, 52, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, L.U.; Gummesson, B.; Joksimović, P.; Farewell, A.; Nyström, T. Identical, independent, and opposing roles of ppGpp and DksA in Escherichia coli. J. Bacteriol. 2007, 189, 5193–5202. [Google Scholar] [CrossRef] [PubMed]
- Jude, F.; Köhler, T.; Branny, P.; Perron, K.; Mayer, M.P.; Comte, R.; van Delden, C. Posttranscriptional control of quorum-sensing-dependent virulence genes by DksA in Pseudomonas aeruginosa. J. Bacteriol. 2003, 185, 3558–3566. [Google Scholar] [CrossRef] [PubMed]
- Takaya, A.; Tabuchi, F.; Tsuchiya, H.; Isogai, E.; Yamamoto, T. Negative regulation of quorum-sensing systems in Pseudomonas aeruginosa by ATP-dependent Lon protease. J. Bacteriol. 2008, 190, 4181–4188. [Google Scholar] [CrossRef] [PubMed]
- Baumgardt, K.; Charoenpanich, P.; Mcintosh, M.; Schikora, A.; Stein, E.; Thalmann, S.; Kogel, K.H.; Klug, G.; Becker, A.; Evguenieva-Hackenberg, E. RNase E affects the expression of the acyl-homoserine lactone synthase gene sinI in Sinorhizobium meliloti. J. Bacteriol. 2014, 196, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, M.; Stone, B.A.; Stanisich, V.A. Curdlan and other bacterial (1 → 3)-β-d-glucans. Appl. Microbiol. Biotechnol. 2005, 68, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.H.; Long, S.R. The Sinorhizobium meliloti stringent response affects multiple aspects of symbiosis. Mol. Microbiol. 2002, 43, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.H.; Choi, C.W.; Lee, S.Y.; Lee, Y.G.; Kwon, J.; Leem, S.H.; Chung, Y.H.; Kahng, H.Y.; Kim, S.J.; Kwon, K.K.; et al. Proteomic characterization of plasmid pLA1 for biodegradation of polycyclic aromatic hydrocarbons in the marine bacterium, Novosphingobium pentaromativorans US6-1. PLoS ONE 2014, 9, e90812. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.C.C.; Johnson, K.A.; Clarke, D.J.; Naismith, J.H.; Campopiano, D.J. The serine palmitoyltransferase from Sphingomonas wittichii RW1: An interesting link to an unusual acyl carrier protein. Biopolymers 2010, 93, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Ikushiro, H.; Hayashi, H.; Kagamiyama, H. A water-soluble homodimeric serine palmitoyltransferase from Sphingomonas paucimobilis EY2395T Strain. J. Biol. Chem. 2001, 276, 18249–18256. [Google Scholar] [CrossRef] [PubMed]
- An, D.D.; Na, C.Z.; Bielawski, J.; Hannun, Y.A.; Kasper, D.L. Membrane sphingolipids as essential molecular signals for Bacteroides survival in the intestine. Proc. Natl. Acad. Sci. USA 2011, 108, 4666–4671. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, M.; Nishikawa, T.; MacDonald, K.; Cavicchioli, R.; Gottschal, J.C.; Kjelleberg, S. Responses to stress and nutrient availability by the marine ultramicrobacterium Sphingomonas sp. strain RB2256. Appl. Environ. Microbiol. 1996, 62, 1287–1294. [Google Scholar] [PubMed]
- Vanoverbeek, L.S.; Eberl, L.; Givskov, M.; Molin, S.; Vanelsas, J.D. Survival of, and induced stress resistance in, carbon-starved Pseudomonas-fluorescens cells residing in soil. Appl. Environ. Microbiol. 1995, 61, 4202–4208. [Google Scholar]
- Simon, R.; Priefer, U.; Pühler, A. A broad host range mobilization system for in vivo genetic engineering: Transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1983, 1, 784–791. [Google Scholar] [CrossRef]
- Mclean, R.J.C.; Whiteley, M.; Stickler, D.J.; Fuqua, W.C. Evidence of autoinducer activity in naturally occurring biofilms. FEMS Microbiol. Lett. 1997, 154, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarczyk, A.; Vorholt, J.A.; Francez-Charlot, A. Markerless gene deletion system for sphingomonads. Appl. Environ. Microbiol. 2012, 78, 3774–3777. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Lidstrom, M.E. Development of improved versatile broad-host-range vectors for use in methylotrophs and other Gram-negative bacteria. Microbiology 2001, 147, 2065–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST plus: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Washio, K.; Lim, S.P.; Roongsawang, N.; Morikawa, M. Identification and characterization of the genes responsible for the production of the cyclic lipopeptide arthrofactin by Pseudomonas sp. MIS38. Biosci. Biotechnol. Biochem. 2010, 74, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ma, A.Z.; Zhuang, X.L.; Zhuang, G.Q. An N-acyl homoserine lactones synthase in the ammonia-oxidizing bacterium Nitrosospira multiformis. Appl. Environ. Microbiol. 2014, 80, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.H.; Wang, Y.L.; Yu, Z.L.; Huang, Y.L. Hypersensitive response of plasmid-encoded ahl synthase gene to lifestyle and nutrient by Ensifer adhaerens X097. Front. Microbiol. 2017, 8, 1160. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Ouahrani-Bettache, S.; Drake, K.L.; Adams, L.G.; Köhler, S.; Occhialini, A. Global Rsh-dependent transcription profile of Brucella suis during stringent response unravels adaptation to nutrient starvation and cross-talk with other stress responses. BMC Genomics 2013, 14, 459. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Z.; Xiao, Y.J.; Nie, H.L.; Huang, Q.Y.; Chen, W.L. Influence of (p)ppGpp on biofilm regulation in Pseudomonas putida KT2440. Microbiol. Res. 2017, 204, 1–8. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Strains | 24 h | 36 h | 48 h | 96 h |
|---|---|---|---|---|---|
| novI | US6-1 | 1.166 ± 0.773 | 1.060 ± 0.466 | 1.115 ± 0.647 | 1.039 ± 0.356 |
| ∆rsh | 0.261 ± 0.083 | 0.011 ± 0.002 * | 0.020 ± 0.004 * | 0.026 ± 0.004 ** | |
| ∆rsh (pCM62-rsh) | 0.501 ± 0.085 *** | 0.648 ± 0.216 | 0.753 ± 0.097 | 3.975 ± 1.547 * | |
| novR | US6-1 | 1.012 ± 0.185 | 1.006 ± 0.129 | 1.001 ± 0.055 | 1.029 ± 0.314 |
| ∆rsh | 0.119 ± 0.018 ** | 0.068 ± 0.005 *** | 0.547 ± 0.017 *** | 0.881 ± 0.354 | |
| ∆rsh (pCM62-rsh) | 0.601 ± 0.050 ** | 0.463 ± 0.076 | 0.543 ± 0.054 | 2.486 ± 0.972 |
| Strains or Plasmids | Relevant Traits | Source or Reference |
|---|---|---|
| Strains | ||
| N. pentaromativorans | ||
| US6-1 | Wild type (JCM 12182) | Microbe division JCM |
| ∆rsh | US6-1, rshUS6-1 deletion mutant, Rifr | This study |
| ∆rsh (pCM62-rsh) | ∆rsh with the plasmid pCM62-rsh, Tcr | This study |
| ∆novI | US6-1, novI deletion mutant, Rifr | Lab stock |
| E. coli | ||
| DH5α | F- hsdR17 endA1 thi-1 gyrA96 relA1 supE44∆lacU169 (ψ80dlacZ∆M15) | TransGen Biotech |
| S17-1 (λpir) | E. coli K-12 Tpr Smr recA thi hsdRM+ RP4::2-Tc::Mu::Km Tn7, λpir phage lysogen | [62] |
| A. tumefaciens A136 (pCF218) (pCF372) | traI-lacZ fusion; AHL biosensor; Tcr Spr | [63] |
| Plasmids | ||
| pMD19-T | T-vector, Ampr | TaKaRa, Dalian, China |
| pAK405 | Plasmid for allelic exchange and markerless gene deletions; Kmr | [64] |
| pAK405-rsh | pAK405 with fusion of up- and downstream regions of rshUS6-1; Kmr | This study |
| pCM62 | Broad-host-range cloning vector; IncP origin of replication; Tcr | [65] |
| pCM62-rsh | pCM62 with rshUS6-1 ORF and 502 bp of upstream region; Tcr | This study |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Huang, Y. Transcriptome Analysis of Novosphingobium pentaromativorans US6-1 Reveals the Rsh Regulon and Potential Molecular Mechanisms of N-acyl-l-homoserine Lactone Accumulation. Int. J. Mol. Sci. 2018, 19, 2631. https://doi.org/10.3390/ijms19092631
Lu H, Huang Y. Transcriptome Analysis of Novosphingobium pentaromativorans US6-1 Reveals the Rsh Regulon and Potential Molecular Mechanisms of N-acyl-l-homoserine Lactone Accumulation. International Journal of Molecular Sciences. 2018; 19(9):2631. https://doi.org/10.3390/ijms19092631
Chicago/Turabian StyleLu, Hang, and Yili Huang. 2018. "Transcriptome Analysis of Novosphingobium pentaromativorans US6-1 Reveals the Rsh Regulon and Potential Molecular Mechanisms of N-acyl-l-homoserine Lactone Accumulation" International Journal of Molecular Sciences 19, no. 9: 2631. https://doi.org/10.3390/ijms19092631
APA StyleLu, H., & Huang, Y. (2018). Transcriptome Analysis of Novosphingobium pentaromativorans US6-1 Reveals the Rsh Regulon and Potential Molecular Mechanisms of N-acyl-l-homoserine Lactone Accumulation. International Journal of Molecular Sciences, 19(9), 2631. https://doi.org/10.3390/ijms19092631