Methylseleninic Acid Induces Lipid Peroxidation and Radiation Sensitivity in Head and Neck Cancer Cells

 , , and
, , and 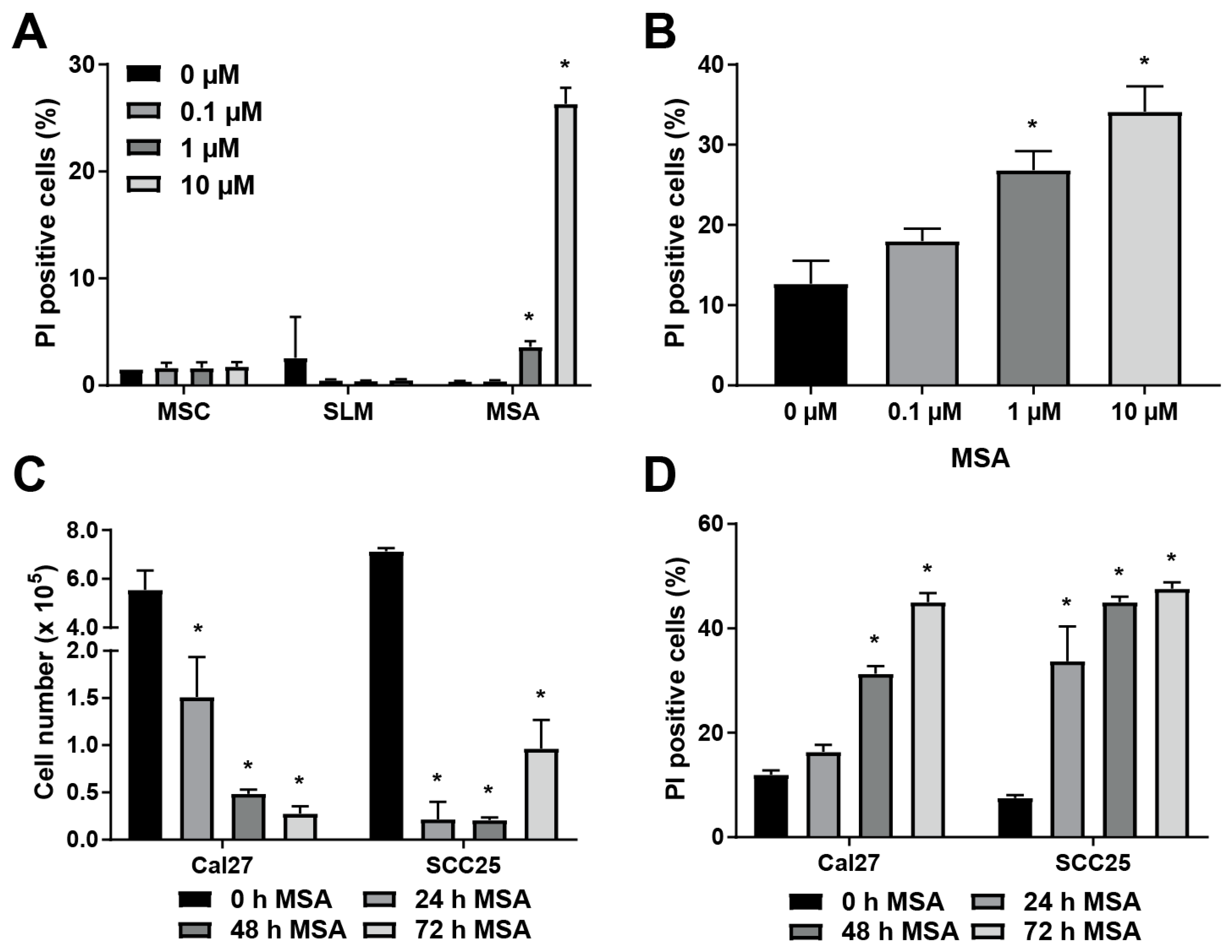{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. MSA is More Toxic to HNSCC Cells than Other Organoselenium Derivatives, and Causes Cell Death in a Dose- and Time-Dependent Manner
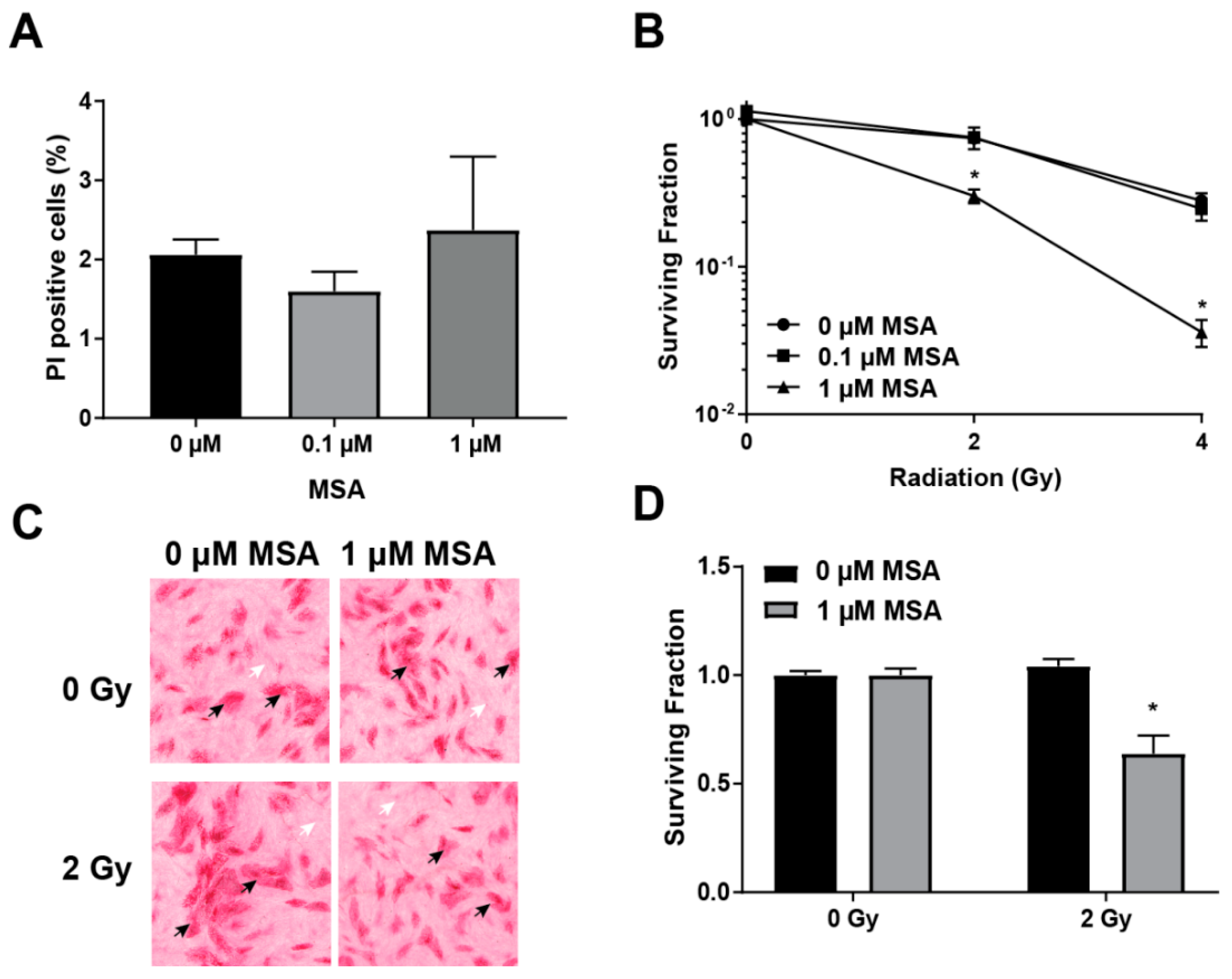
2.2. MSA Treatment Sensitizes HNSCC Cells to Radiation
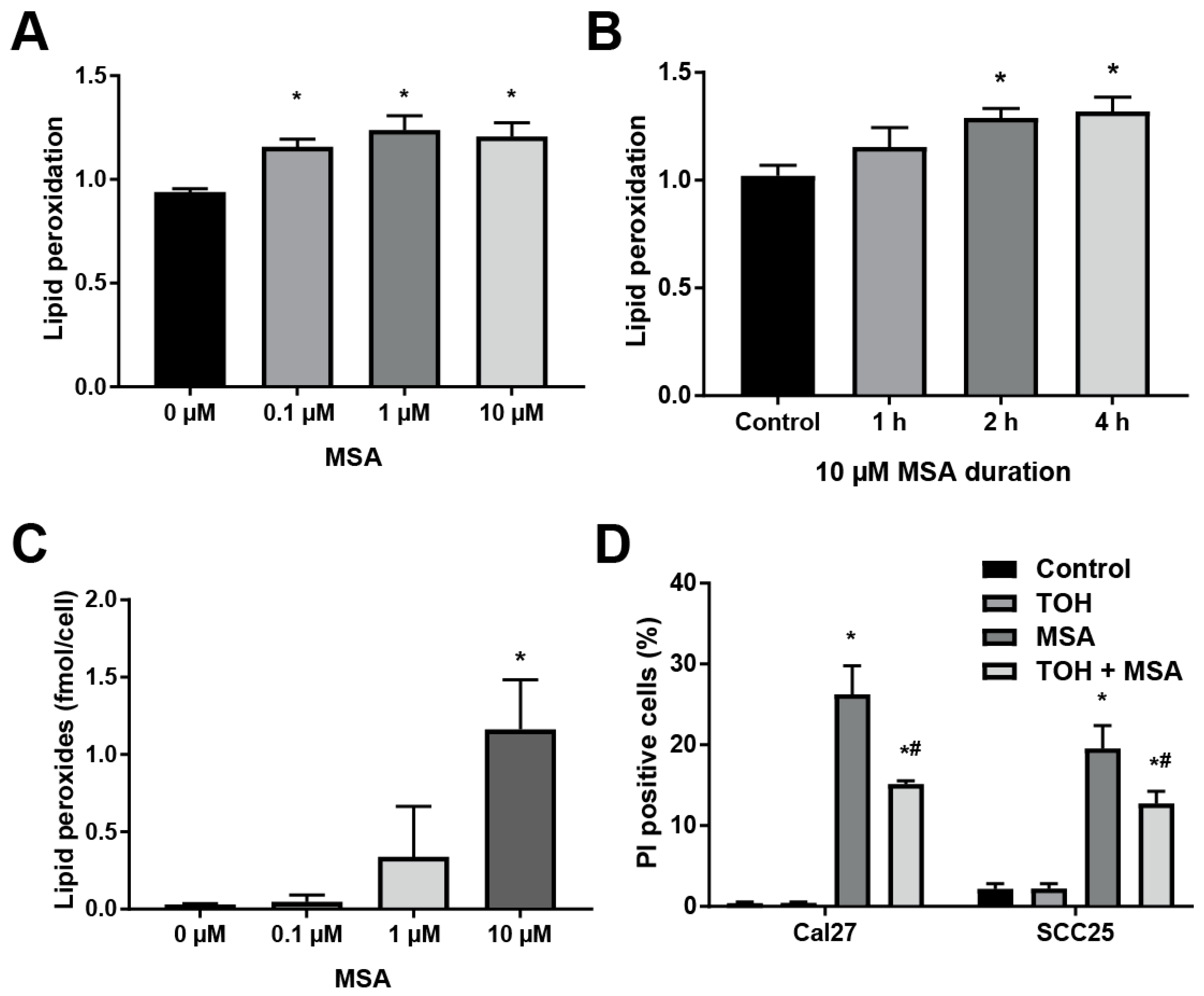
2.3. MSA Treatment Induces Lipid Peroxidation in HNSCC Cells
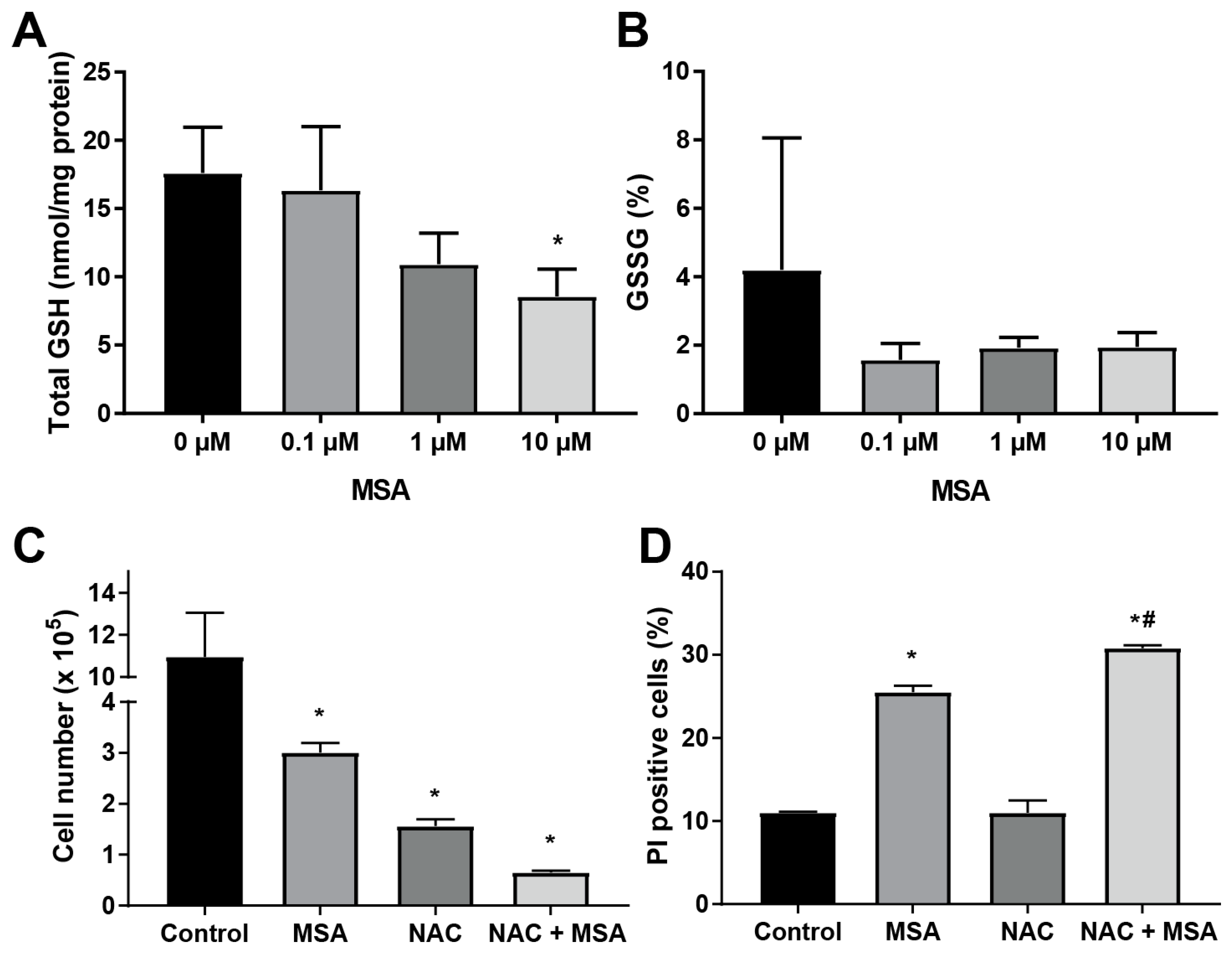
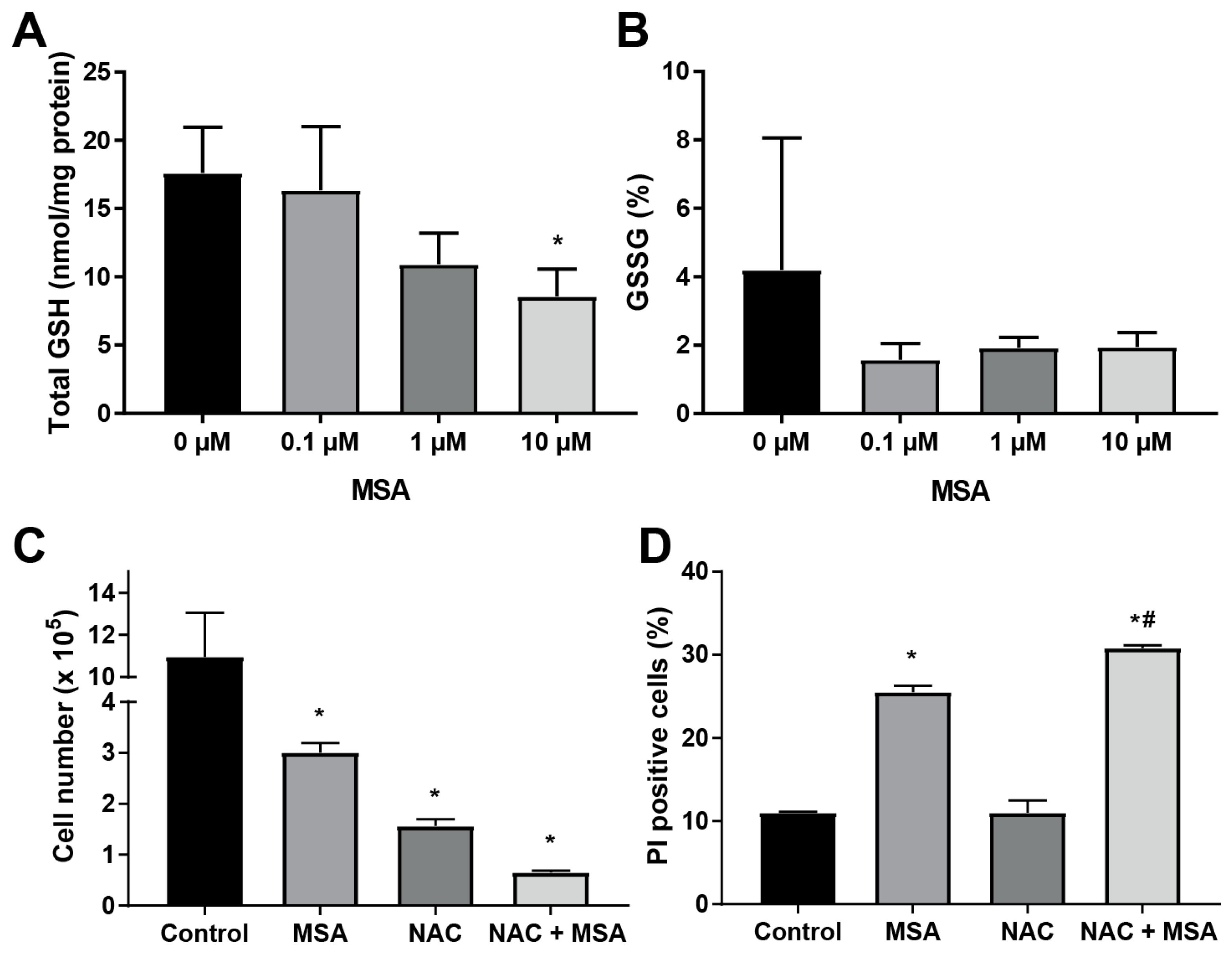
2.4. N-Acetyl-l-Cysteine Exacerbates MSA Toxicity in HNSCC
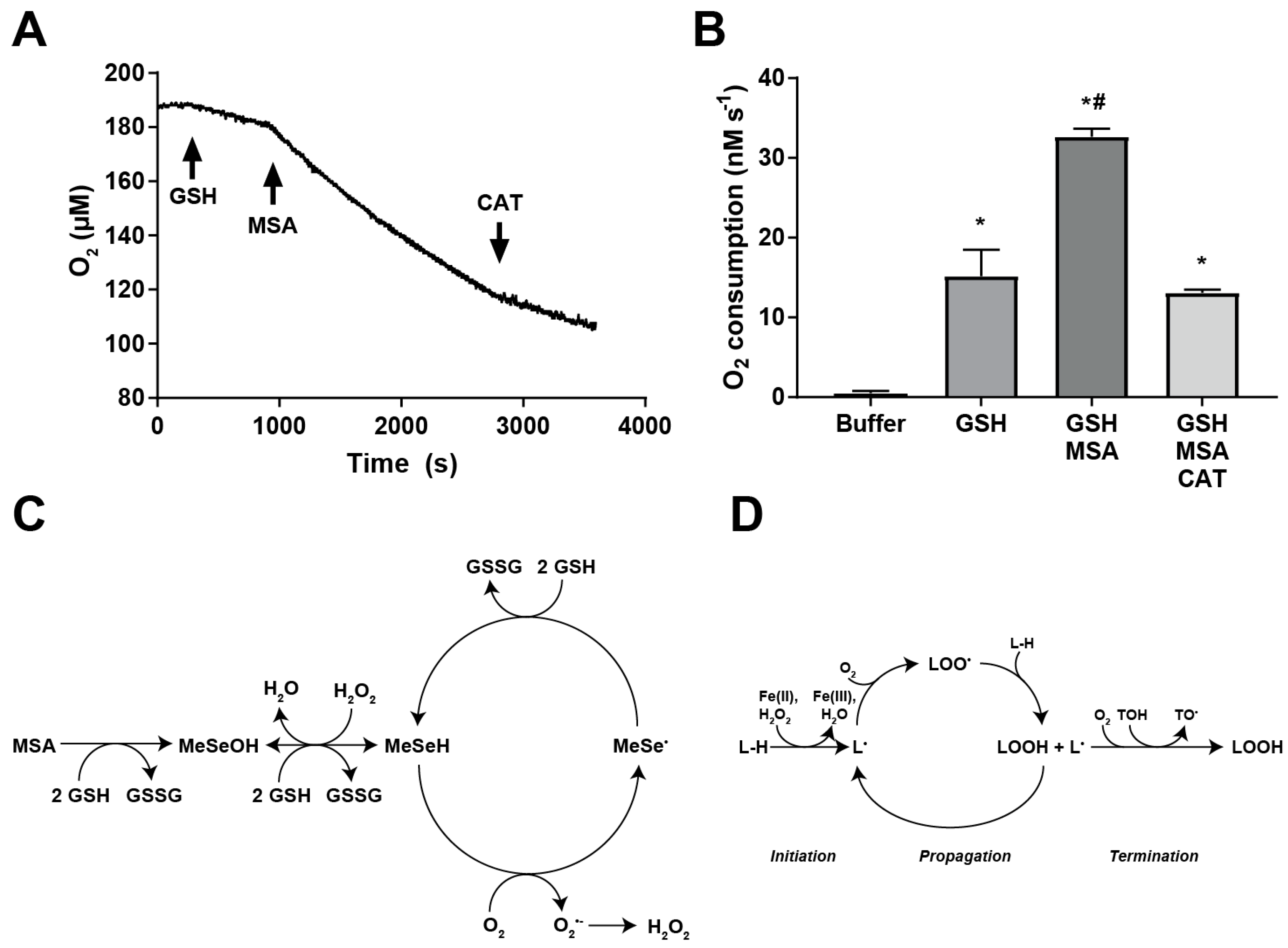
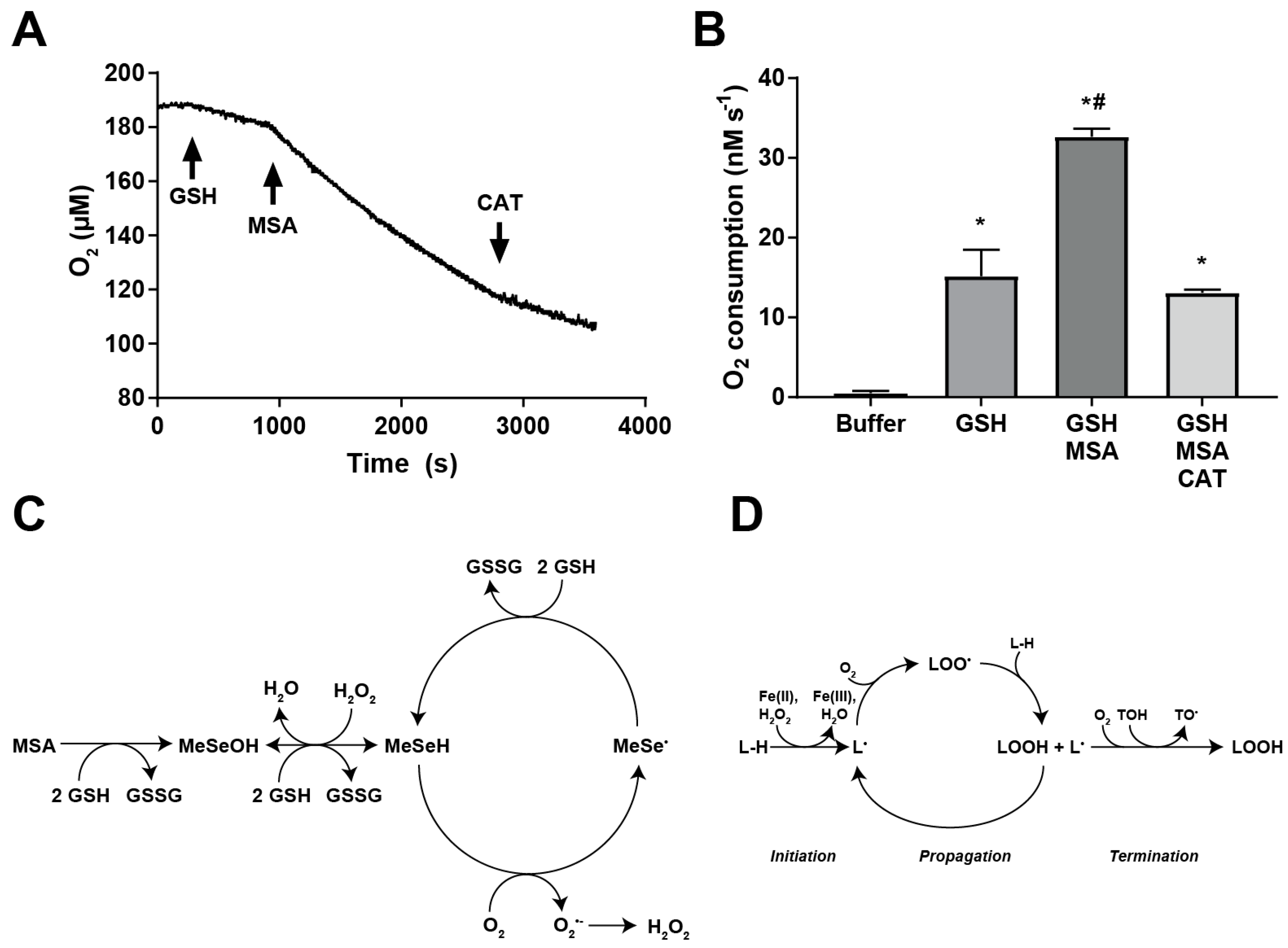
2.5. MSA Treatment Enhances GSH-Dependent O2 Consumption
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Irradiation
4.3. Propidium Iodide Exclusion Assay
4.4. BODIPY C-11 Assay
4.5. Total Lipid Hydroperoxide Determination
4.6. Glutathione Determination
4.7. Oxygen Consumption
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Howlader, N.A.; Krapcho, M.; Miller, D.; Bishop, K.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; et al. SEER Cancer Statistics Review, 1975–2013; National Cancer Institute: Bethesda, MD, USA, 2016.
- Orlandi, E.; Iacovelli, N.A.; Rancati, T.; Cicchetti, A.; Bossi, P.; Pignoli, E.; Bergamini, C.; Licitra, L.; Fallai, C.; Valdagni, R.; et al. Multivariable model for predicting acute oral mucositis during combined IMRT and chemotherapy for locally advanced nasopharyngeal cancer patients. Oral Oncol. 2018, 86, 266–272. [Google Scholar] [CrossRef]
- Pulte, D.; Brenner, H. Changes in survival in head and neck cancers in the late 20th and early 21st century: A period analysis. Oncologist 2010, 15, 994–1001. [Google Scholar] [CrossRef]
- Schueller, P.; Puettmann, S.; Micke, O.; Senner, V.; Schaefer, U.; Willich, N. Selenium influences the radiation sensitivity of C6 rat glioma cells. Anticancer Res. 2004, 24, 2913–2917. [Google Scholar]
- Husbeck, B.; Peehl, D.M.; Knox, S.J. Redox modulation of human prostate carcinoma cells by selenite increases radiation-induced cell killing. Free Radic. Biol. Med. 2005, 38, 50–57. [Google Scholar] [CrossRef]
- Eckers, J.C.; Kalen, A.L.; Xiao, W.; Sarsour, E.H.; Goswami, P.C. Selenoprotein P inhibits radiation-induced late reactive oxygen species accumulation and normal cell injury. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 619–625. [Google Scholar] [CrossRef]
- Puspitasari, I.M.; Abdulah, R.; Yamazaki, C.; Kameo, S.; Nakano, T.; Koyama, H. Updates on clinical studies of selenium supplementation in radiotherapy. Radiat. Oncol. 2014, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Brodin, O.; Eksborg, S.; Wallenberg, M.; Asker-Hagelberg, C.; Larsen, E.H.; Mohlkert, D.; Lenneby-Helleday, C.; Jacobsson, H.; Linder, S.; Misra, S.; et al. Pharmacokinetics and Toxicity of Sodium Selenite in the Treatment of Patients with Carcinoma in a Phase I Clinical Trial: The SECAR Study. Nutrients 2015, 7, 4978–4994. [Google Scholar] [CrossRef] [Green Version]
- Ammar, E.M.; Couri, D. Acute toxicity of sodium selenite and selenomethionine in mice after ICV or IV administration. Neurotoxicology 1981, 2, 383–386. [Google Scholar]
- Shin, S.H.; Yoon, M.J.; Kim, M.; Kim, J.I.; Lee, S.J.; Lee, Y.S.; Bae, S. Enhanced lung cancer cell killing by the combination of selenium and ionizing radiation. Oncol. Rep. 2007, 17, 209–216. [Google Scholar] [CrossRef]
- Ip, C. Lessons from basic research in selenium and cancer prevention. J. Nutr. 1998, 128, 1845–1854. [Google Scholar] [CrossRef]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, H.E. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar]
- Spallholz, J.E.; Palace, V.P.; Reid, T.W. Methioninase and selenomethionine but not Se-methylselenocysteine generate methylselenol and superoxide in an in vitro chemiluminescent assay: Implications for the nutritional carcinostatic activity of selenoamino acids. Biochem. Pharmacol. 2004, 67, 547–554. [Google Scholar] [CrossRef]
- Tan, Y.; Sun, X.; Xu, M.; Tan, X.; Sasson, A.; Rashidi, B.; Han, Q.; Tan, X.; Wang, X.; An, Z.; et al. Efficacy of recombinant methioninase in combination with cisplatin on human colon tumors in nude mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 2157–2163. [Google Scholar]
- Yoshioka, T.; Wada, T.; Uchida, N.; Maki, H.; Yoshida, H.; Ide, N.; Kasai, H.; Hojo, K.; Shono, K.; Maekawa, R.; et al. Anticancer efficacy in vivo and in vitro, synergy with 5-fluorouracil, and safety of recombinant methioninase. Cancer Res. 1998, 58, 2583–2587. [Google Scholar]
- Kokkinakis, D.M.; Hoffman, R.M.; Frenkel, E.P.; Wick, J.B.; Han, Q.; Xu, M.; Tan, Y.; Schold, S.C. Synergy between methionine stress and chemotherapy in the treatment of brain tumor xenografts in athymic mice. Cancer Res. 2001, 61, 4017–4023. [Google Scholar]
- Hu, J.; Cheung, N.K. Methionine depletion with recombinant methioninase: In vitro and in vivo efficacy against neuroblastoma and its synergism with chemotherapeutic drugs. Int. J. Cancer 2009, 124, 1700–1706. [Google Scholar] [CrossRef]
- Spallholz, J.E.; Shriver, B.J.; Reid, T.W. Dimethyldiselenide and methylseleninic acid generate superoxide in an in vitro chemiluminescence assay in the presence of glutathione: Implications for the anticarcinogenic activity of L-selenomethionine and L-Se-methylselenocysteine. Nutr. Cancer 2001, 40, 34–41. [Google Scholar] [CrossRef]
- Li, G.-X.; Lee, H.-J.; Wang, Z.; Hu, H.; Liao, J.D.; Watts, J.C.; Combs, G.F.; Lü, J. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis 2008, 29, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Fu, X.; Xiong, Z.; Zhang, H.; Hill, S.M.; Rowan, B.G.; Dong, Y. Methylseleninic acid enhances paclitaxel efficacy for the treatment of triple-negative breast cancer. PLoS ONE 2012, 7, e31539. [Google Scholar] [CrossRef]
- Aikens, J.; Dix, T.A. Perhydroxyl radical (HOO.) initiated lipid peroxidation. The role of fatty acid hydroperoxides. J. Biol. Chem. 1991, 266, 15091–15098. [Google Scholar]
- Bielski, B.H.; Arudi, R.L.; Sutherland, M.W. A study of the reactivity of HO2/O2- with unsaturated fatty acids. J. Biol. Chem. 1983, 258, 4759–4761. [Google Scholar]
- Rungby, J. Silver-induced lipid peroxidation in mice: Interactions with selenium and nickel. Toxicology 1987, 45, 135–142. [Google Scholar] [CrossRef]
- Sieja, K.; Talerczyk, M. Selenium as an element in the treatment of ovarian cancer in women receiving chemotherapy. Gynecol. Oncol. 2004, 93, 320–327. [Google Scholar] [CrossRef]
- Farina, M.; Soares, F.A.; Zeni, G.; Souza, D.O.; Rocha, J.B. Additive pro-oxidative effects of methylmercury and ebselen in liver from suckling rat pups. Toxicol. Lett. 2004, 146, 227–235. [Google Scholar] [CrossRef]
- Rasheed, M.H.; Beevi, S.S.; Geetha, A. Enhanced lipid peroxidation and nitric oxide products with deranged antioxidant status in patients with head and neck squamous cell carcinoma. Oral Oncol. 2007, 43, 333–338. [Google Scholar] [CrossRef]
- Gupta, A.; Bhatt, M.L.; Misra, M.K. Lipid peroxidation and antioxidant status in head and neck squamous cell carcinoma patients. Oxid. Med. Cell. Longev. 2009, 2, 68–72. [Google Scholar] [CrossRef]
- Chintala, S.; Toth, K.; Cao, S.; Durrani, F.A.; Vaughan, M.M.; Jensen, R.L.; Rustum, Y.M. Se-methylselenocysteine sensitizes hypoxic tumor cells to irinotecan by targeting hypoxia-inducible factor 1alpha. Cancer Chemother. Pharmacol. 2010, 66, 899–911. [Google Scholar] [CrossRef]
- Büntzel, J.; Micke, O.; Glatzel, M.; Schafer, U.; Riesenbeck, D.; Kisters, K.; Bruns, F.; Schänekaes, K.G.; Dawczynski, H.; Mücke, R. Selenium substitution during radiotherapy in head and neck cancer. Trace Elem. Electrolytes 2010, 27, 235–239. [Google Scholar] [CrossRef]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef]
- Griffith, O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 1980, 106, 207–212. [Google Scholar] [CrossRef]
- Menon, S.G.; Sarsour, E.H.; Spitz, D.R.; Higashikubo, R.; Sturm, M.; Zhang, H.; Goswami, P.C. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003, 63, 2109–2117. [Google Scholar]
- Menon, S.G.; Sarsour, E.H.; Kalen, A.L.; Venkataraman, S.; Hitchler, M.J.; Domann, F.E.; Oberley, L.W.; Goswami, P.C. Superoxide signaling mediates N-acetyl-l-cysteine-induced G1 arrest: Regulatory role of cyclin D1 and manganese superoxide dismutase. Cancer Res. 2007, 67, 6392–6399. [Google Scholar] [CrossRef]
- Huber, R.E.; Criddle, R.S. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 1967, 122, 164–173. [Google Scholar] [CrossRef]
- Chaudiere, J.; Courtin, O.; Leclaire, J. Glutathione oxidase activity of selenocystamine: A mechanistic study. Arch. Biochem. Biophys. 1992, 296, 328–336. [Google Scholar] [CrossRef]
- Miki, K.; Xu, M.; Gupta, A.; Ba, Y.; Tan, Y.; Al-Refaie, W.; Bouvet, M.; Makuuchi, M.; Moossa, A.R.; Hoffman, R.M. Methioninase cancer gene therapy with selenomethionine as suicide prodrug substrate. Cancer Res. 2001, 61, 6805–6810. [Google Scholar]
- Drummen, G.P.; van Liebergen, L.C.; Op den Kamp, J.A.; Post, J.A. C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (Micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Rotruck, J.T.; Pope, A.L.; Ganther, H.E.; Swanson, A.B.; Hafeman, D.G.; Hoekstra, W.G. Selenium: Biochemical Role as a Component of Glutathione Peroxidase. Science 1973, 179, 588–590. [Google Scholar] [CrossRef]
- Okuno, T.; Honda, E.; Arakawa, T.; Ogino, H.; Ueno, H. Glutathione-dependent cell cycle G1 arrest and apoptosis induction in human lung cancer A549 cells caused by methylseleninic acid: Comparison with sodium selenite. Biol. Pharm. Bull. 2014, 37, 1831–1837. [Google Scholar] [CrossRef]
- Shen, H.M.; Ding, W.X.; Ong, C.N. Intracellular glutathione is a cofactor in methylseleninic acid-induced apoptotic cell death of human hepatoma HEPG(2) cells. Free Radic. Biol. Med. 2002, 33, 552–561. [Google Scholar] [CrossRef]
- Gordillo, G.M.; Biswas, A.; Khanna, S.; Spieldenner, J.M.; Pan, X.; Sen, C.K. Multidrug Resistance-associated Protein-1 (MRP-1)-dependent Glutathione Disulfide (GSSG) Efflux as a Critical Survival Factor for Oxidant-enriched Tumorigenic Endothelial Cells. J. Biol. Chem. 2016, 291, 10089–10103. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Liochev, S.I.; Fridovich, I. The role of O2.− in the production of HO.: In vitro and in vivo. Free Radic. Biol. Med. 1994, 16, 29–33. [Google Scholar] [CrossRef]
- Agil, A.; Fuller, C.J.; Jialal, I. Susceptibility of plasma to ferrous iron/hydrogen peroxide-mediated oxidation: Demonstration of a possible Fenton reaction. Clin. Chem. 1995, 41, 220–225. [Google Scholar]
- Zhong, W.; Oberley, T.D. Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res. 2001, 61, 7071–7078. [Google Scholar]
- Wagner, B.A.; Buettner, G.R.; Burns, C.P. Free radical-mediated lipid peroxidation in cells: Oxidizability is a function of cell lipid bis-allylic hydrogen content. Biochemistry 1994, 33, 4449–4453. [Google Scholar] [CrossRef]
- Accioly, M.T.; Pacheco, P.; Maya-Monteiro, C.M.; Carrossini, N.; Robbs, B.K.; Oliveira, S.S.; Kaufmann, C.; Morgado-Diaz, J.A.; Bozza, P.T.; Viola, J.P. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008, 68, 1732–1740. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Toth, K.; Peter, I.; Kremmer, T.; Sugar, J. Lipid-rich cell thyroid adenoma: Histopathology with comparative lipid analysis. Virchows Arch. APathol. Anat. Histopathol. 1990, 417, 273–276. [Google Scholar] [CrossRef]
- Ericsson, J.L.E.; Seljelid, R.; Orrenius, S. Comparative light and electron microscopic observations of the cytoplasmic matrix in renal carcinomas. Virchows Archiv für Pathologische Anatomie und Physiologie und für Klinische Medizin 1966, 341, 204–223. [Google Scholar] [CrossRef]
- Kalen, A.L.; Sarsour, E.H.; Venkataraman, S.; Goswami, P.C. Mn-superoxide dismutase overexpression enhances G2 accumulation and radioresistance in human oral squamous carcinoma cells. Antioxid. Redox Signal. 2006, 8, 1273–1281. [Google Scholar] [CrossRef]
- Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]
- Wagner, B.A.; Venkataraman, S.; Buettner, G.R. The rate of oxygen utilization by cells. Free Radic. Biol. Med. 2011, 51, 700–712. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lafin, J.T.; Sarsour, E.H.; Kalen, A.L.; Wagner, B.A.; Buettner, G.R.; Goswami, P.C. Methylseleninic Acid Induces Lipid Peroxidation and Radiation Sensitivity in Head and Neck Cancer Cells. Int. J. Mol. Sci. 2019, 20, 225. https://doi.org/10.3390/ijms20010225
Lafin JT, Sarsour EH, Kalen AL, Wagner BA, Buettner GR, Goswami PC. Methylseleninic Acid Induces Lipid Peroxidation and Radiation Sensitivity in Head and Neck Cancer Cells. International Journal of Molecular Sciences. 2019; 20(1):225. https://doi.org/10.3390/ijms20010225
Chicago/Turabian StyleLafin, John T., Ehab H. Sarsour, Amanda L. Kalen, Brett A. Wagner, Garry R. Buettner, and Prabhat C. Goswami. 2019. "Methylseleninic Acid Induces Lipid Peroxidation and Radiation Sensitivity in Head and Neck Cancer Cells" International Journal of Molecular Sciences 20, no. 1: 225. https://doi.org/10.3390/ijms20010225
APA StyleLafin, J. T., Sarsour, E. H., Kalen, A. L., Wagner, B. A., Buettner, G. R., & Goswami, P. C. (2019). Methylseleninic Acid Induces Lipid Peroxidation and Radiation Sensitivity in Head and Neck Cancer Cells. International Journal of Molecular Sciences, 20(1), 225. https://doi.org/10.3390/ijms20010225