Immortalization of Different Breast Epithelial Cell Types Results in Distinct Mitochondrial Mutagenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Both SV22 and SV1 Cells Exhibit Identical Homoplasmic Mutations, Verifying that Both Cell Types were Derived from the Same Individual
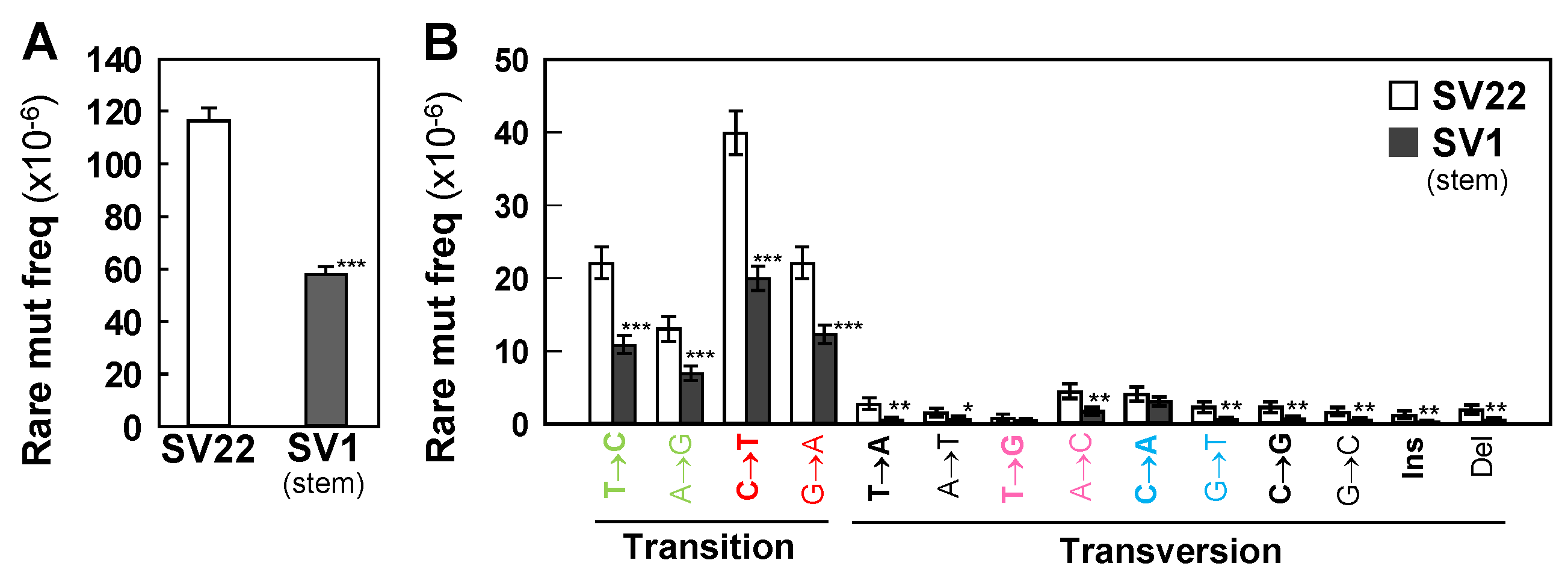
2.2. SV1 Cells Show Significantly Lower Frequencies of Rare Mutations and Subclonal Mutations than do SV22 Cells
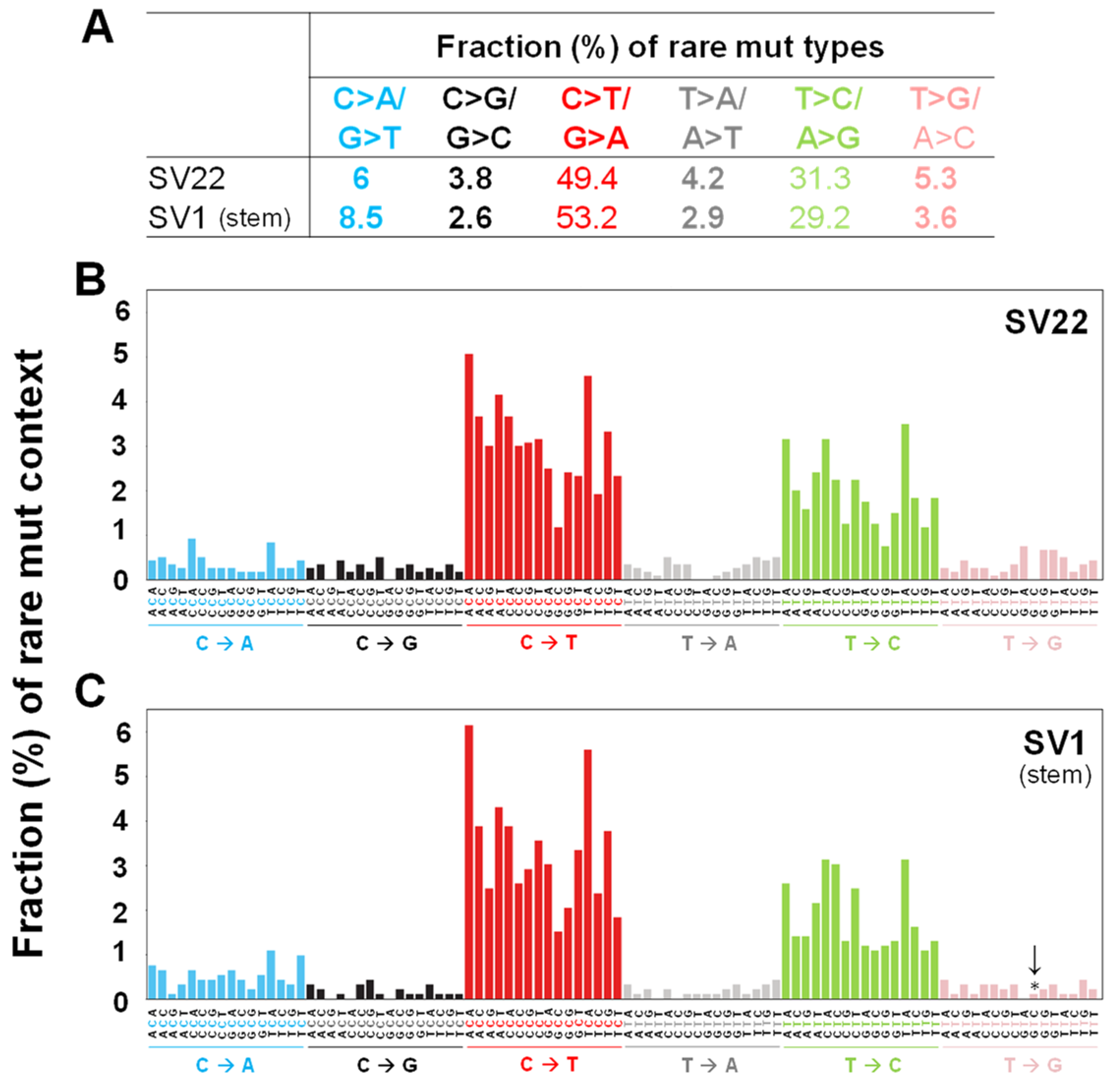
2.3. C>T/G>A Transitions are the Most Prevalent Mutation Types Followed by T>C/A>G in Both Cell Types
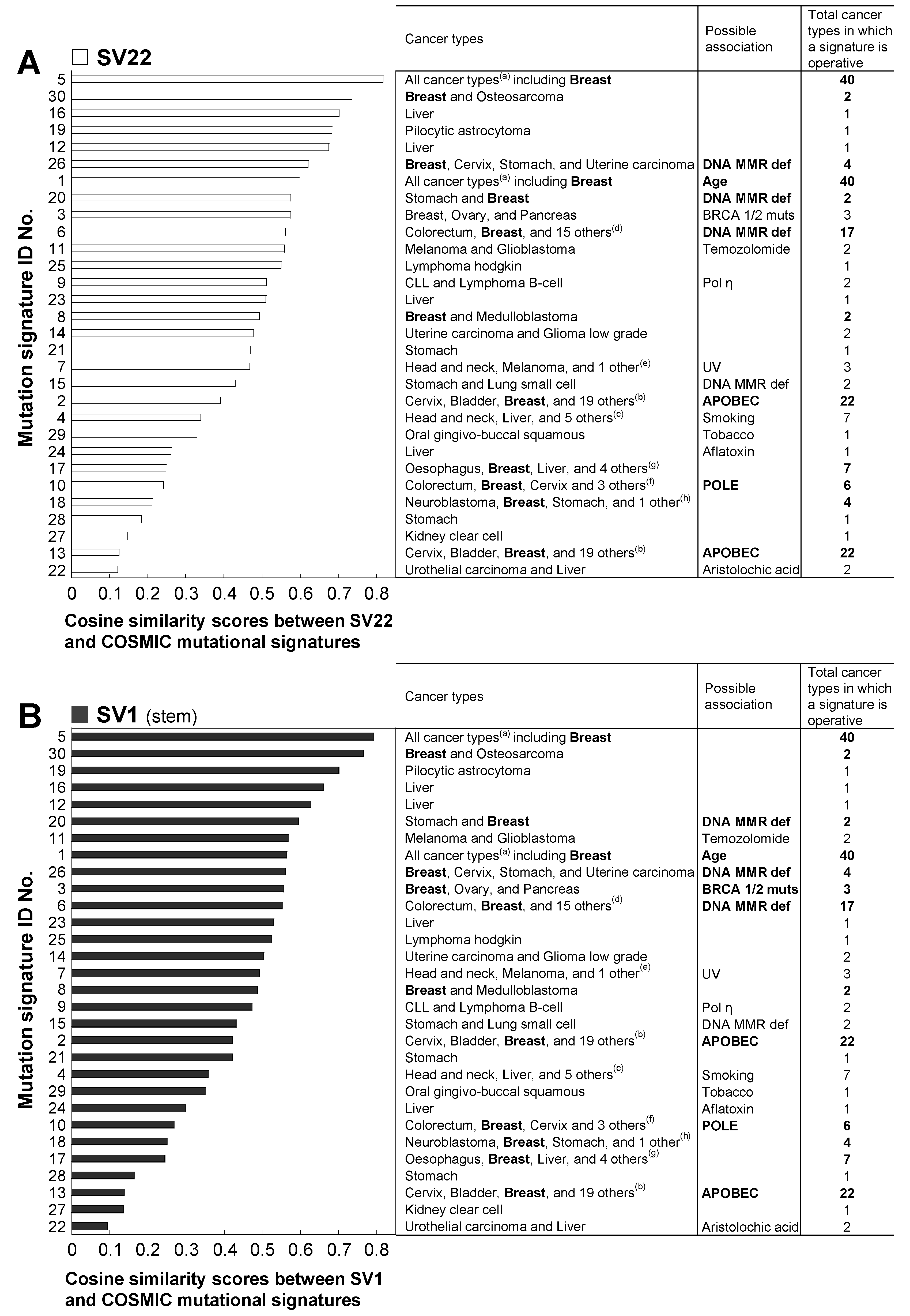
2.4. The Mutation Context Spectra of the Whole mtDNA Rare Mutations in SV22 and SV1 Cells are Compared with the Mutational Signatures of the Catalogue of Somatic Mutations in Cancer (COSMIC)
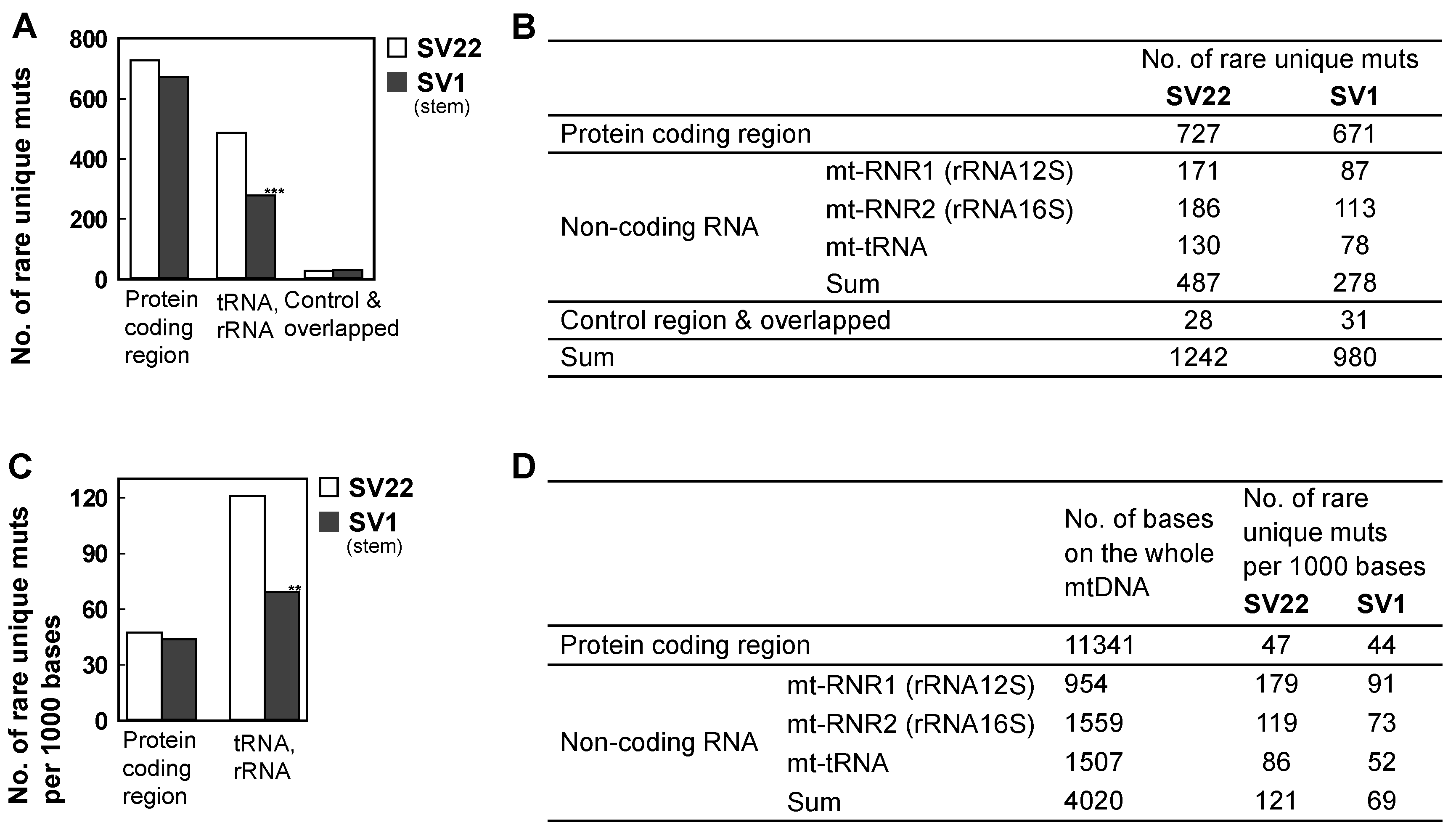
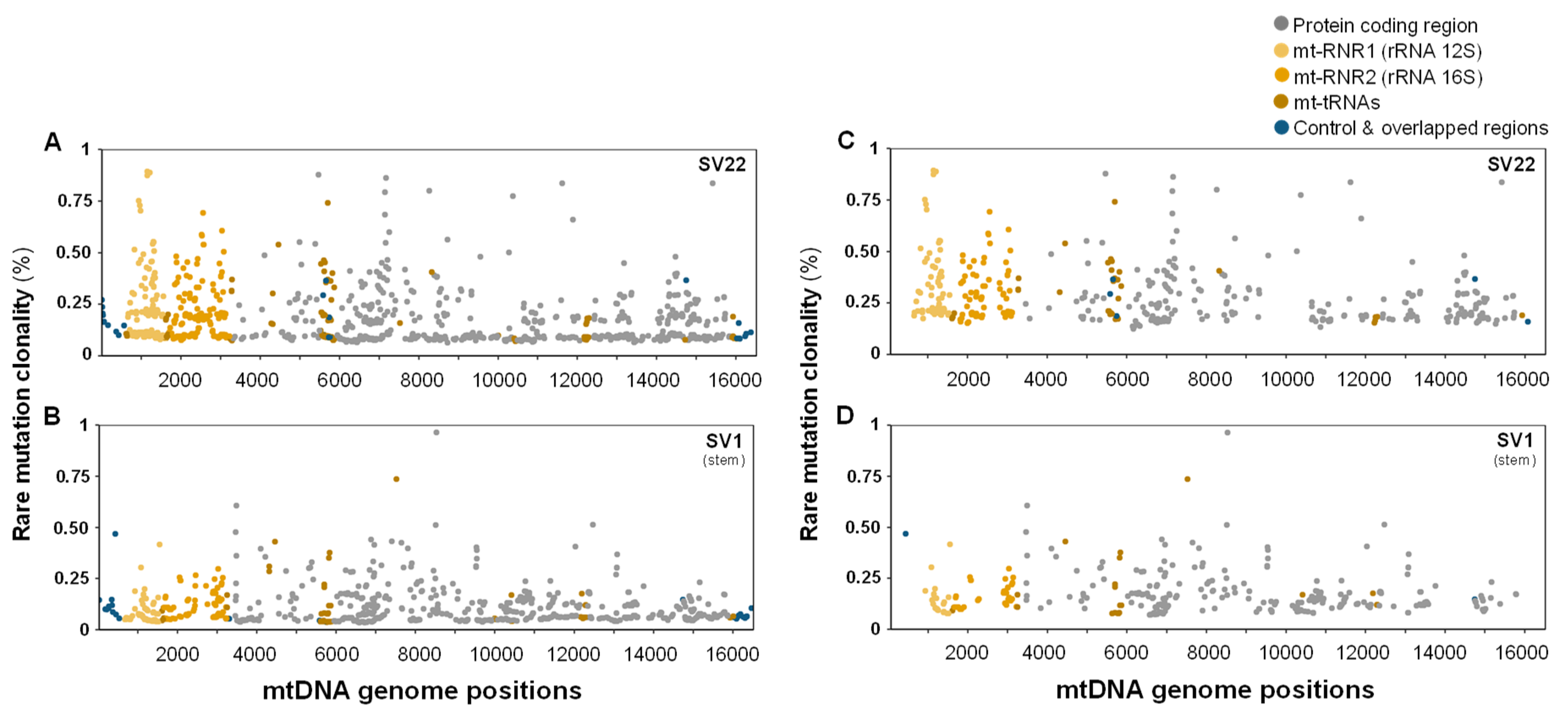
2.5. The Decreased Mitochondrial Mutagenesis of SV1 Cells Occurs Mainly in the Mitochondrial (mt) RNA Gene Region
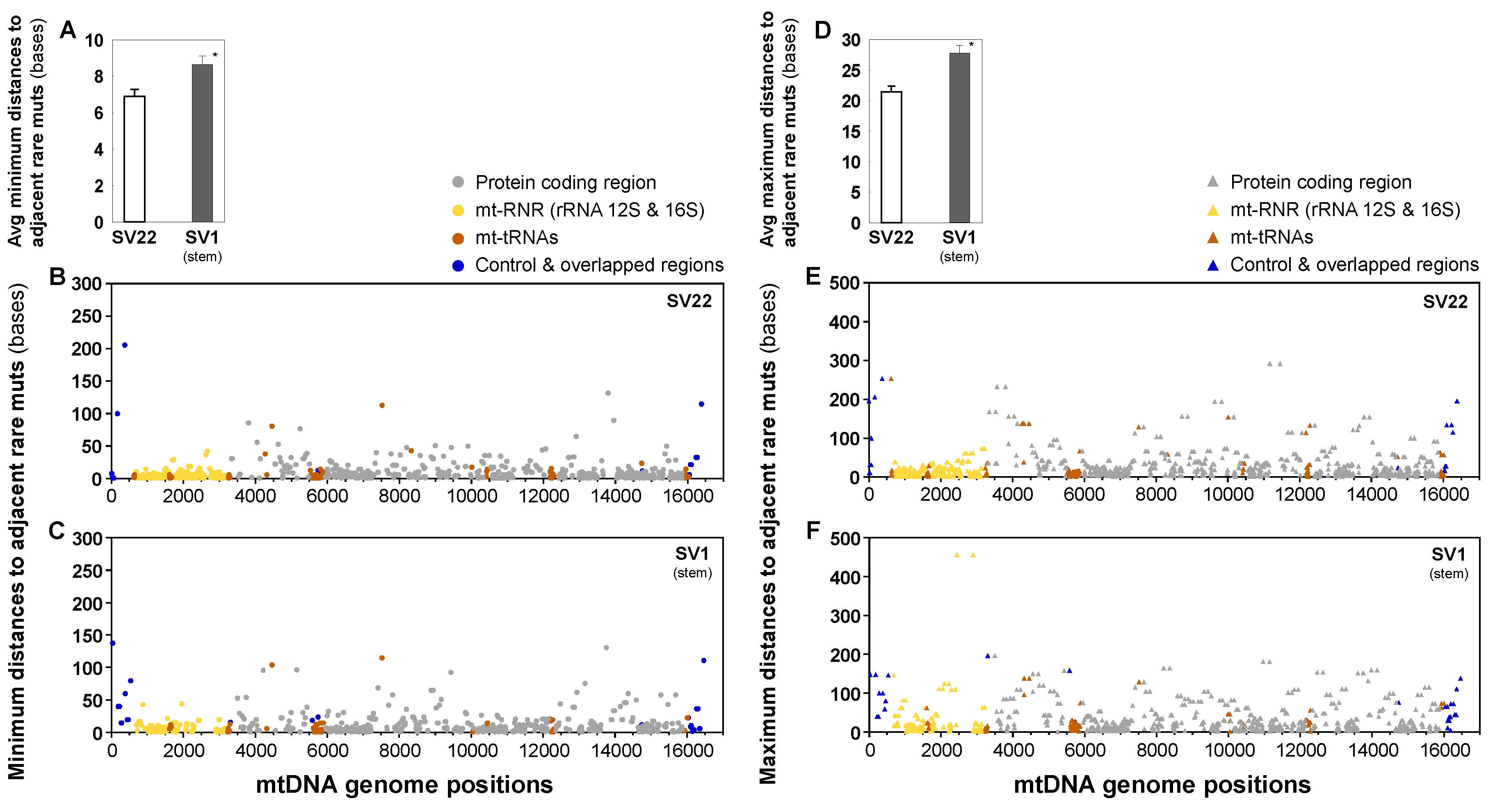
2.6. Distances between Adjacent Mutations are Longer in SV1 Cells than in SV22 Cells
2.7. Proportions (%) of Nonsynonymous Mutations in Mitochondrial Protein Coding Regions and Their Predicted Pathogenicity Scores of Subclonal Mutations are Similar between SV22 and SV1 Cells
2.8. Predicted Pathogenicity Scores of Missense Mutations of Mitochondrial Protein Coding Regions are Similar between SV22 and SV1 Cells
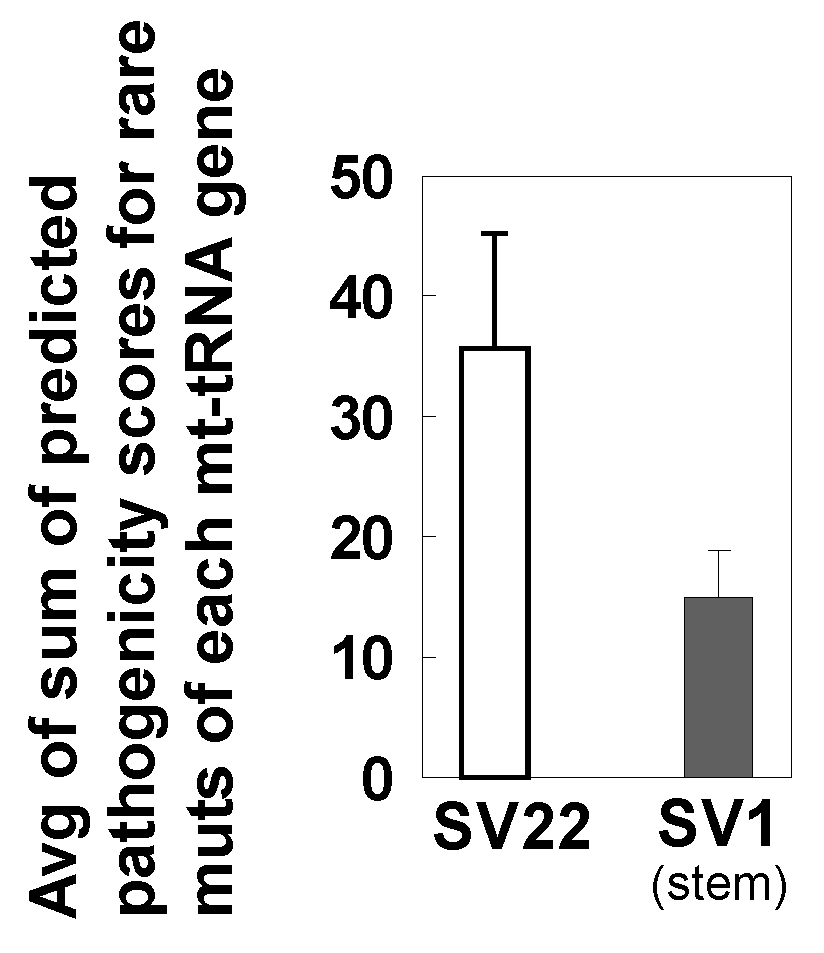
2.9. Predicted Pathogenicity Scores of Rare Mutations of Mitochondrial tRNA Genes Tend to be Lower in SV1 Cells than in SV22 Cells.
2.10. Duplex Sequencing Identifies Many Rare or Subclonal Mutations that are Specific to SV22 and to SV1 Cells
2.11. Independent DNA Library Preparation Experiments of Duplex Sequencing (DS) Generate Reproducible Results
3. Discussion
4. Materials and Methods
4.1. Development and Culture of Immortalized Human Breast Epithelial Cells
4.2. DNA Extraction, Adapter Synthesis, and DNA Library Preparation for Duplex Sequencing (DS)
4.3. Data Processing
4.4. Data Analysis for Positions, Frequencies, Types, and Context Spectra of Mutations and Annotation of Genetic Variants
4.5. Cosine Similarity Analysis and Comparison of Mutation Context Spectra with the COSMIC Mutational Signatures
4.6. Analysis of Distances to Adjacent Mutations
4.7. Predicted Pathogenicity
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DCS | Duplex Consensus Sequence |
| DS | Duplex Sequencing |
| Mt | Mitochondrial |
| HBECs | Human breast epithelial cells |
| SV1 | Normal stem cell (Type-I)-derived immortalized HBECs |
| SV22 | Normal non-stem cell (Type-II)-derived immortalized HBECs |
References
- Böcker, W.; Moll, R.; Poremba, C.; Holland, R.; Van Diest, P.J.; Dervan, P.; Bürger, H.; Wai, D.; Ina Diallo, R.; Brandt, B.; et al. Common adult stem cells in the human breast give rise to glandular and myoepithelial cell lineages: A new cell biological concept. Lab. Investig. 2002, 82, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Al-Hajj, M.; Abdallah, W.M.; Clarke, M.F.; Wicha, M.S. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36 (Suppl. 1), 59–72. [Google Scholar] [CrossRef] [Green Version]
- Welm, B.; Behbod, F.; Goodell, M.A.; Rosen, J.M. Isolation and characterization of functional mammary gland stem cells. Cell Prolif. 2003, 36 (Suppl. 1), 17–32. [Google Scholar] [CrossRef]
- Ince, T.A.; Richardson, A.L.; Bell, G.W.; Saitoh, M.; Godar, S.; Karnoub, A.E.; Iglehart, J.D.; Weinberg, R.A. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 2007, 12, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Keller, P.J.; Arendt, L.M.; Skibinski, A.; Logvinenko, T.; Klebba, I.; Dong, S.; Smith, A.E.; Prat, A.; Perou, C.M.; Gilmore, H.; et al. Defining the cellular precursors to human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Liu, Z.; Jiang, W.; Nagi, C.; Huang, S.; Edwards, D.P.; Jo, E.; Mo, Q.; Creighton, C.J.; Hilsenbeck, S.G.; et al. Mammary Precancerous Stem and Non-Stem Cells Evolve into Cancers of Distinct Subtypes. Cancer Res. 2019, 79, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Tai, M.-H.; Chang, C.-C.; Kiupel, M.; Webster, J.D.; Olson, L.K.; Trosko, J.E. Oct4 expression in adult human stem cells: Evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis 2005, 26, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.S.; Morita, I.; Cruz, A.; Jeon, Y.J.; Trosko, J.E.; Chang, C.C. Expression of estrogen receptors in a normal human breast epithelial cell type with luminal and stem cell characteristics and its neoplastically transformed cell lines. Carcinogenesis 1997, 18, 251–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, C.Y.; Nomata, K.; Oakley, C.S.; Welsch, C.W.; Chang, C.C. Two types of normal human breast epithelial cells derived from reduction mammoplasty: Phenotypic characterization and response to SV40 transfection. Carcinogenesis 1995, 16, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.-Y.; Oakley, C.S.; Welsch, C.W.; Chang, C.-C. Growth requirements and neoplastic transformation of two types of normal human breast epithelial cells derived from reduction mammoplasty. In Vitro Cell. Dev. Biol. Anim. 1997, 33, 282–288. [Google Scholar] [CrossRef]
- Chang, C.C.; Sun, W.; Cruz, A.; Saitoh, M.; Tai, M.H.; Trosko, J.E. A human breast epithelial cell type with stem cell characteristics as target cells for carcinogenesis. Radiat. Res. 2001, 155, 201–207. [Google Scholar] [CrossRef]
- Ahn, E.H.; Chang, C.-C.; Schroeder, J.J. Evaluation of sphinganine and sphingosine as human breast cancer chemotherapeutic and chemopreventive agents. Exp. Biol. Med. 2006, 231, 1664–1672. [Google Scholar] [CrossRef]
- Ahuja, D.; Sáenz-Robles, M.T.; Pipas, J.M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005, 24, 7729–7745. [Google Scholar] [CrossRef] [Green Version]
- Pipas, J.M. SV40: Cell transformation and tumorigenesis. Virology 2009, 384, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Kang, K.S.; Morita, I.; Trosko, J.E.; Chang, C.C. High susceptibility of a human breast epithelial cell type with stem cell characteristics to telomerase activation and immortalization. Cancer Res. 1999, 59, 6118–6123. [Google Scholar] [PubMed]
- Ahn, E.H.; Hirohata, K.; Kohrn, B.F.; Fox, E.J.; Chang, C.-C.; Loeb, L.A. Detection of Ultra-Rare Mitochondrial Mutations in Breast Stem Cells by Duplex Sequencing. PLoS ONE 2015, 10, e0136216. [Google Scholar] [CrossRef]
- Imanishi, H.; Hattori, K.; Wada, R.; Ishikawa, K.; Fukuda, S.; Takenaga, K.; Nakada, K.; Hayashi, J. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS ONE 2011, 6, e23401. [Google Scholar] [CrossRef]
- Sanchez, M.I.; Mercer, T.R.; Davies, S.M.; Shearwood, A.M.; Nygård, K.K.; Richman, T.R.; Mattick, J.S.; Rackham, O.; Filipovska, A. RNA processing in human mitochondria. Cell Cycle 2011, 10, 2904–2916. [Google Scholar] [CrossRef]
- Salgado, J.; Honorato, B.; García-Foncillas, J. Review: Mitochondrial Defects in Breast Cancer. Clin. Med. Oncol. 2008, 2, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Ghaffarpour, M.; Mahdian, R.; Fereidooni, F.; Kamalidehghan, B.; Moazami, N.; Houshmand, M. The mitochondrial ATPase6 gene is more susceptible to mutation than the ATPase8 gene in breast cancer patients. Cancer Cell Int. 2014, 14, 21. [Google Scholar] [CrossRef]
- Hertweck, K.L.; Dasgupta, S. The Landscape of mtDNA Modifications in Cancer: A Tale of Two Cities. Front. Oncol. 2017, 7, 262. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-L.; Meng, H.; Zhang, W.; Qin, Y.-H.; Zhao, N.-M. The role of mitochondrial tRNA variants in female breast cancer. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 3199–3201. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.J.; Kohrn, B.F.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.-C.; Risques, R.-A.; et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 2014, 9, 2586–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, M.W.; Fox, E.J.; Prindle, M.J.; Reid-Bayliss, K.S.; True, L.D.; Radich, J.P.; Loeb, L.A. Sequencing small genomic targets with high efficiency and extreme accuracy. Nat. Methods 2015, 12, 423–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, E.H.; Lee, S.H. Detection of Low-Frequency Mutations and Identification of Heat-Induced Artifactual Mutations Using Duplex Sequencing. Int. J. Mol. Sci. 2019, 20, 199. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.I.; Hussmann, J.A.; McBee, R.M.; Acevedo, A.; Andino, R.; Press, W.H.; Sawyer, S.L. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 19872–19877. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.J.; Reid-Bayliss, K.S.; Emond, M.J.; Loeb, L.A. Accuracy of Next Generation Sequencing Platforms. Next Gener. Seq. Appl. 2014, 1, 1000106. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Govatati, S.; Deenadayal, M.; Shivaji, S.; Bhanoori, M. Mitochondrial displacement loop alterations are associated with endometriosis. Fertil. Steril. 2013, 99, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. MutPred2: Inferring the molecular and phenotypic impact of amino acid variants. bioRxiv 2017. [Google Scholar] [CrossRef]
- Sonney, S.; Leipzig, J.; Lott, M.T.; Zhang, S.; Procaccio, V.; Wallace, D.C.; Sondheimer, N. Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput. Biol. 2017, 13, e1005867. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Gordon, K.; Clouaire, T.; Bao, X.X.; Kemp, S.E.; Xenophontos, M.; de Las Heras, J.I.; Stancheva, I. Immortality, but not oncogenic transformation, of primary human cells leads to epigenetic reprogramming of DNA methylation and gene expression. Nucleic Acids Res. 2014, 42, 3529–3541. [Google Scholar] [CrossRef]
- Saretzki, G.; Walter, T.; Atkinson, S.; Passos, J.F.; Bareth, B.; Keith, W.N.; Stewart, R.; Hoare, S.; Stojkovic, M.; Armstrong, L.; et al. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells 2008, 26, 455–464. [Google Scholar] [CrossRef]
- Frosina, G. The bright and the dark sides of DNA repair in stem cells. J. Biomed. Biotechnol. 2010, 2010, 845396. [Google Scholar] [CrossRef]
- Guan, J.-L.; Simon, A.K.; Prescott, M.; Menendez, J.A.; Liu, F.; Wang, F.; Wang, C.; Wolvetang, E.; Vazquez-Martin, A.; Zhang, J. Autophagy in stem cells. Autophagy 2013, 9, 830–849. [Google Scholar] [CrossRef]
- Joshi, A.; Kundu, M. Mitophagy in hematopoietic stem cells: The case for exploration. Autophagy 2013, 9, 1737–1749. [Google Scholar] [CrossRef] [PubMed]
- McFarland, R.; Swalwell, H.; Blakely, E.L.; He, L.; Groen, E.J.; Turnbull, D.M.; Bushby, K.M.; Taylor, R.W. The m.5650G>A mitochondrial tRNAAla mutation is pathogenic and causes a phenotype of pure myopathy. Neuromuscul. Disord. 2008, 18, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, A.M.; Ghelli, A.; Ceccarelli, C.; Lang, M.; Cenacchi, G.; Capristo, M.; Pennisi, L.F.; Morra, I.; Ciccarelli, E.; Melcarne, A.; et al. The genetic and metabolic signature of oncocytic transformation implicates HIF1alpha destabilization. Hum. Mol. Genet. 2010, 19, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Blakely, E.L.; Yarham, J.W.; Alston, C.L.; Craig, K.; Poulton, J.; Brierley, C.; Park, S.-M.; Dean, A.; Xuereb, J.H.; Anderson, K.N.; et al. Pathogenic mitochondrial tRNA point mutations: Nine novel mutations affirm their importance as a cause of mitochondrial disease. Hum. Mutat. 2013, 34, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Horváth, R.; Bender, A.; Abicht, A.; Holinski-Feder, E.; Czermin, B.; Trips, T.; Schneiderat, P.; Lochmüller, H.; Klopstock, T. Heteroplasmic mutation in the anticodon-stem of mitochondrial tRNA(Val) causing MNGIE-like gastrointestinal dysmotility and cachexia. J. Neurol. 2009, 256, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Glatz, C.; D’Aco, K.; Smith, S.; Sondheimer, N. Mutation in the mitochondrial tRNA(Val) causes mitochondrial encephalopathy, lactic acidosis and stroke-like episodes. Mitochondrion 2011, 11, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-C.; Tseng, L.-M.; Lee, H.-C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, P.-H.; Wu, C.-C.; Lin, J.-C.; Chi, C.-W.; Wei, Y.-H.; Lee, H.-C. Somatic mutations of mitochondrial genome in hepatocellular carcinoma. Mitochondrion 2010, 10, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Levinger, L.; Oestreich, I.; Florentz, C.; Mörl, M. A pathogenesis-associated mutation in human mitochondrial tRNALeu(UUR) leads to reduced 3′-end processing and CCA addition. J. Mol. Biol. 2004, 337, 535–544. [Google Scholar] [CrossRef]
- Kang, K.S.; Sun, W.; Nomata, K.; Morita, I.; Cruz, A.; Liu, C.J.; Trosko, J.E.; Chang, C.C. Involvement of tyrosine phosphorylation of p185(c-erbB2/neu) in tumorigenicity induced by X-rays and the neu oncogene in human breast epithelial cells. Mol. Carcinog. 1998, 21, 225–233. [Google Scholar] [CrossRef]
- Park, J.-S.; Noh, D.-Y.; Kim, S.-H.; Kim, S.-H.; Kong, G.; Chang, C.-C.; Lee, Y.-S.; Trosko, J.E.; Kang, K.-S. Gene expression analysis in SV40-immortalized human breast luminal epithelial cells with stem cell characteristics using a cDNA microarray. Int. J. Oncol. 2004, 24, 1545–1558. [Google Scholar] [PubMed]
- Ahn, E.H.; Chang, C.-C.; Talmage, D.A. Loss of anti-proliferative effect of all-trans retinoic acid in advanced stage of breast carcinogenesis. Anticancer Res. 2009, 29, 2899–2904. [Google Scholar] [PubMed]
- Wang, K.-H.; Kao, A.-P.; Chang, C.-C.; Lee, J.-N.; Hou, M.-F.; Long, C.-Y.; Chen, H.-S.; Tsai, E.-M. Increasing CD44+/CD24(−) tumor stem cells, and upregulation of COX-2 and HDAC6, as major functions of HER2 in breast tumorigenesis. Mol. Cancer 2010, 9, 288. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.H.; Lee, S.H.; Kim, J.Y.; Chang, C.-C.; Loeb, L.A. Decreased Mitochondrial Mutagenesis during Transformation of Human Breast Stem Cells into Tumorigenic Cells. Cancer Res. 2016, 76, 4569–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Huang, C.-H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [Green Version]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, S.; Kim, S.S.; Nebeck, H.E.; Ahn, E.H. Immortalization of Different Breast Epithelial Cell Types Results in Distinct Mitochondrial Mutagenesis. Int. J. Mol. Sci. 2019, 20, 2813. https://doi.org/10.3390/ijms20112813
Kwon S, Kim SS, Nebeck HE, Ahn EH. Immortalization of Different Breast Epithelial Cell Types Results in Distinct Mitochondrial Mutagenesis. International Journal of Molecular Sciences. 2019; 20(11):2813. https://doi.org/10.3390/ijms20112813
Chicago/Turabian StyleKwon, Sujin, Susan S. Kim, Howard E. Nebeck, and Eun Hyun Ahn. 2019. "Immortalization of Different Breast Epithelial Cell Types Results in Distinct Mitochondrial Mutagenesis" International Journal of Molecular Sciences 20, no. 11: 2813. https://doi.org/10.3390/ijms20112813