Genome-Wide Analysis of Coding and Long Non-Coding RNAs Involved in Cuticular Wax Biosynthesis in Cabbage (Brassica oleracea L. var. capitata)

Abstract
:
1. Introduction
2. Results
2.1. Characterization of the Plant Exhibiting nwgl Phenotype
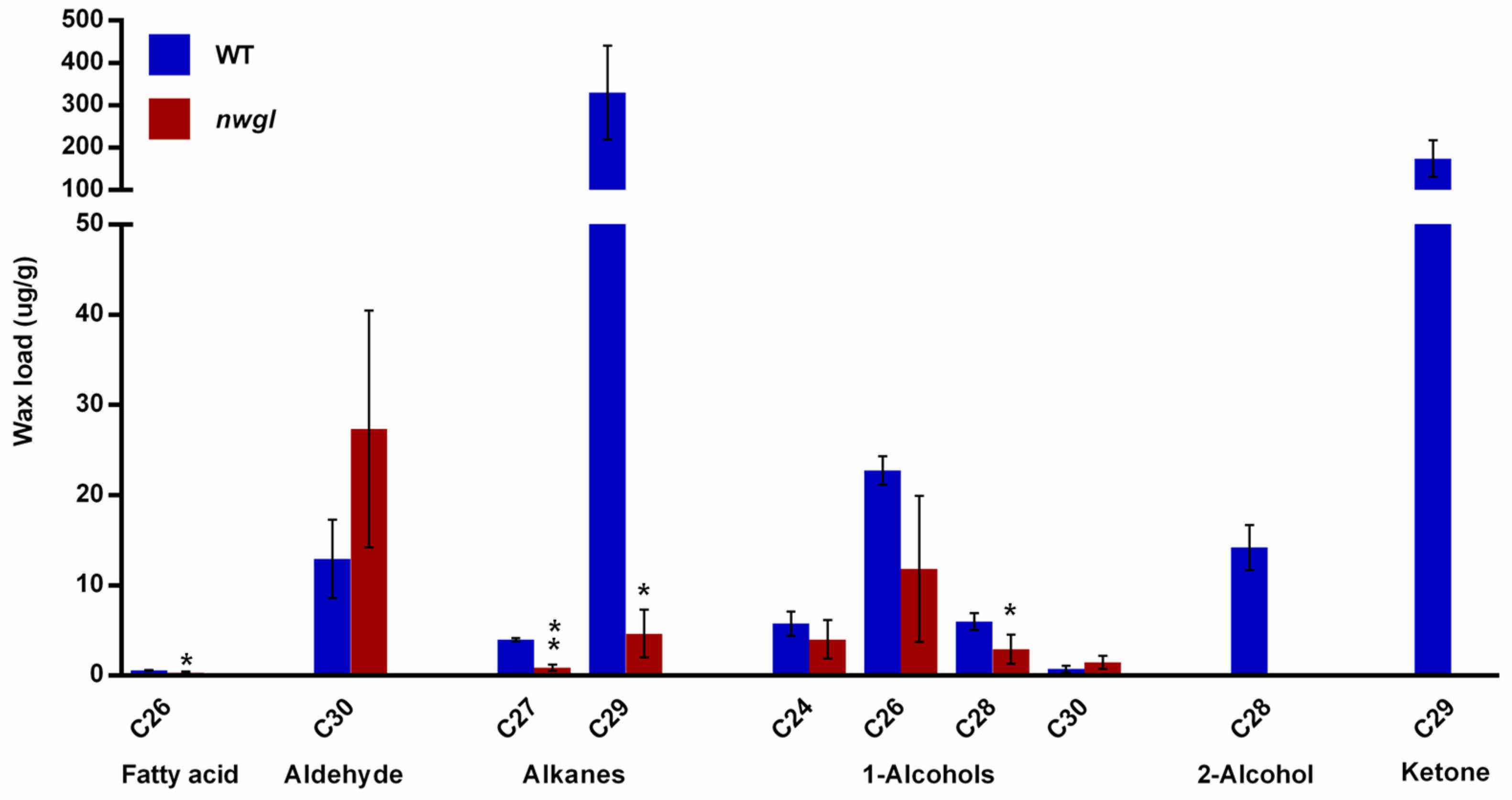
2.2. Cuticular Wax Composition of nwgl Plants
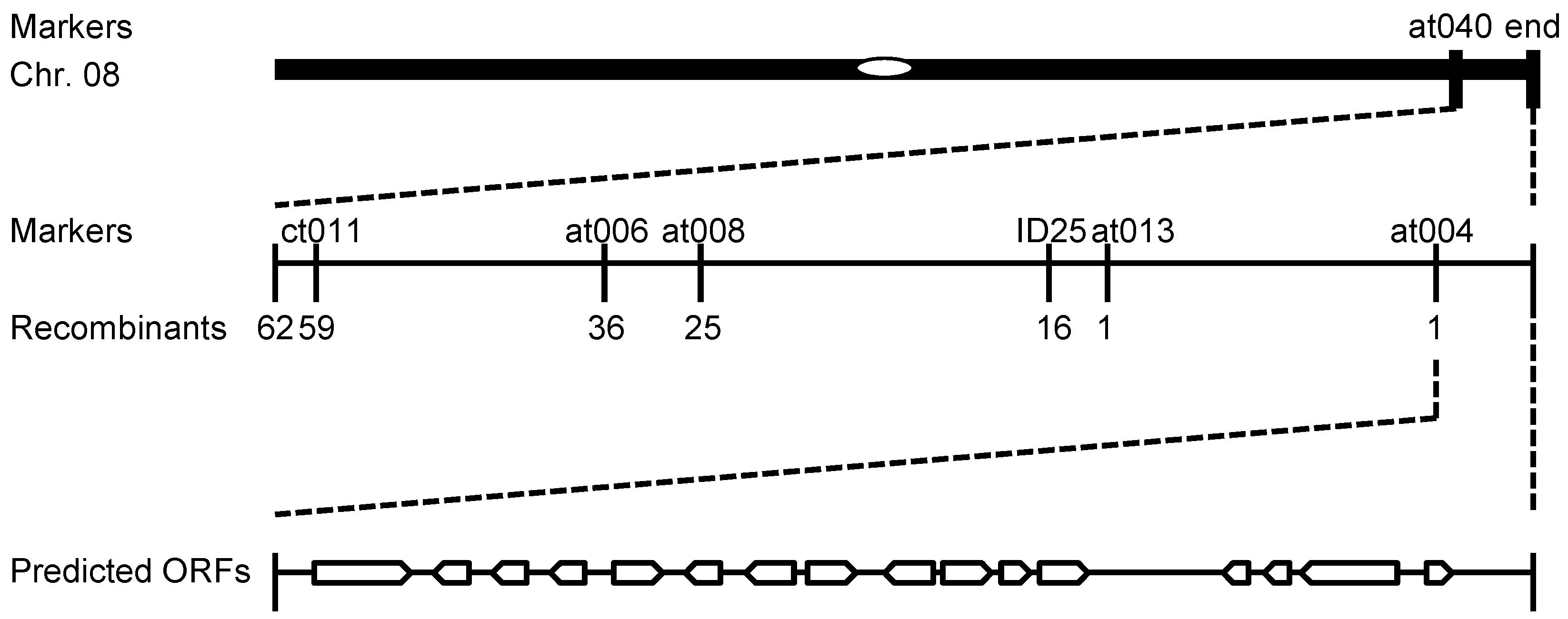
2.3. Fine Mapping of NWGL
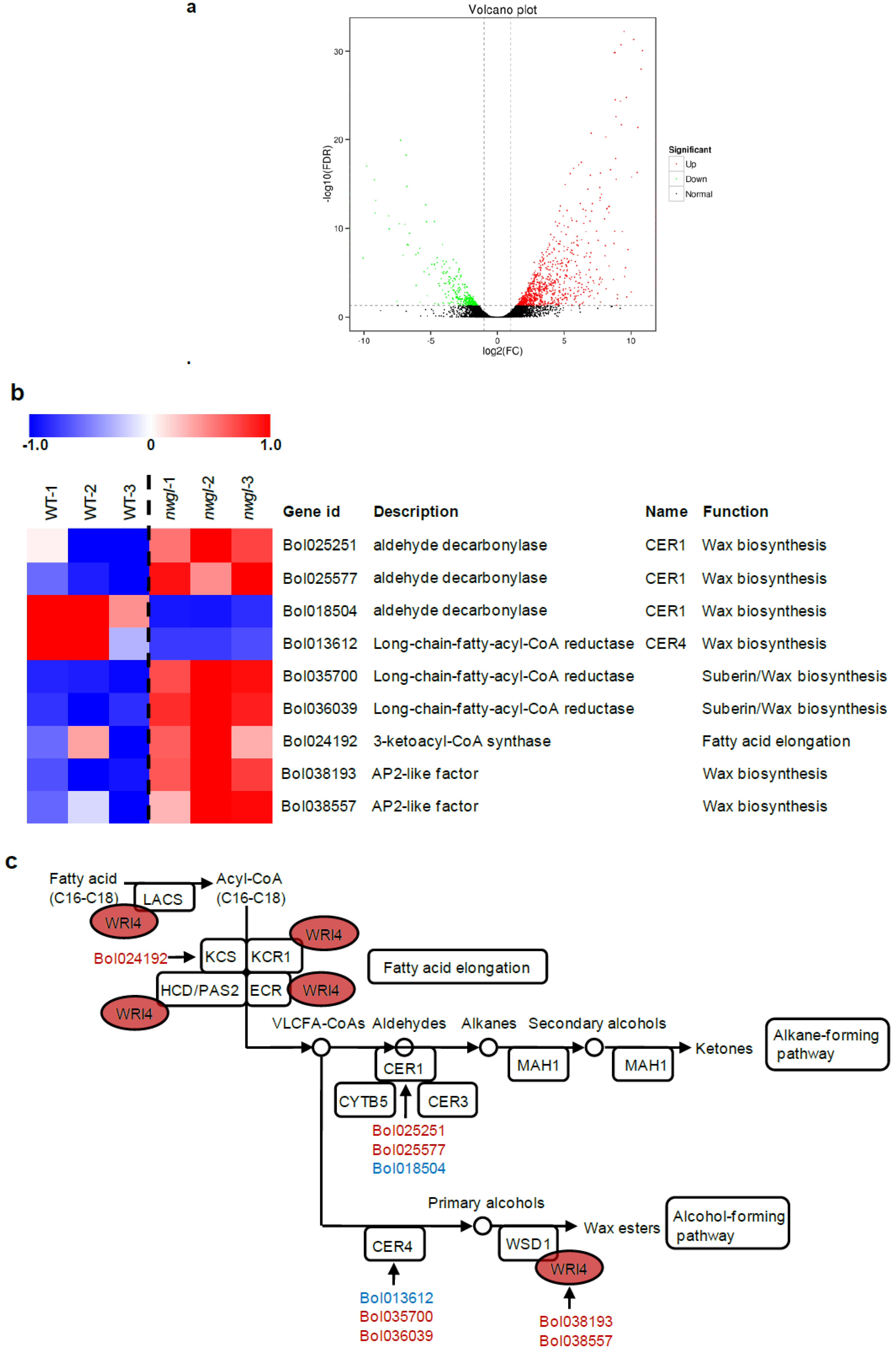
2.4. RNA-seq Analysis of Plants Exhibitingnwgl Phenotype
2.5. Identification and Characterization of lncRNAs in Cabbage
2.6. Identification of Differentially Expressed lncRNAs between nwgl and Wild-Type Plants
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Cryo-scanning Electron Microscopy (Cryo-SEM)
4.3. Cuticular Wax Analysis
4.4. Fine Mapping of NWGL
4.5. RNA Isolation, Library Construction and Illumina Sequencing
4.6. Read Mapping and Transcriptome Assembly
4.7. Identification of lncRNAs
4.8. Analysis of Differential Expression of Genes and lncRNAs
4.9. Target Gene Prediction of lncRNAs
4.10. Functional Enrichment Analysis
4.11. Co-expression Analysis
4.12. McrBC-, HpaⅡ, MspⅠ-PCR
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Samuels, L.; Kunst, L.; Jetter, R. Sealing plant surfaces: Cuticular wax formation by epidermal cells. Annu. Rev. Plant Biol. 2008, 59, 683–707. [Google Scholar] [CrossRef]
- Lee, S.B.; Mi, C.S. Advances in the understanding of cuticular waxes in Arabidopsis thaliana and crop species. Plant Cell Rep. 2015, 34, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Yeats, T.H.; Rose, J.K. The formation and function of plant cuticles. Plant Physiol. 2013, 163, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Joubes, J. Arabidopsis cuticular waxes: Advances in synthesis, export and regulation. Prog. Lipid Res. 2013, 52, 110–129. [Google Scholar] [CrossRef] [PubMed]
- Weidenbach, D.; Jansen, M.; Franke, R.B.; Hensel, G.; Weissgerber, W.; Ulferts, S.; Jansen, I.; Schreiber, L.; Korzun, V.; Pontzen, R.; et al. Evolutionary conserved function of barley and Arabidopsis 3-KETOACYL-CoA SYNTHASES in providing wax signals for germination of powdery mildew fungi. Plant Physiol. 2014, 166, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tian, X. The E3 ligase DROUGHT HYPERSENSITIVE negatively regulates cuticular wax biosynthesis by promoting the degradation of transcription factor ROC4 in rice. Plant Cell 2018, 30, 228–244. [Google Scholar] [CrossRef]
- Aarts, M.G.; Keijzer, C.J.; Stiekema, W.J.; Pereira, A. Molecular characterization of the CER1 gene of Arabidopsis involved in epicuticular wax biosynthesis and pollen fertility. Plant Cell 1995, 7, 2115–2127. [Google Scholar] [CrossRef]
- Brice, B.; Amélie, B.; Frédéric, D.; Stéphanie, P.; Amandine, L.; Dominique, R.; Marjorie, P.; Denis, V.; Haslam, R.P.; Napier, J.A. Overexpression of Arabidopsis ECERIFERUM1 promotes wax very-long-chain alkane biosynthesis and influences plant response to biotic and abiotic stresses. Plant Physiol. 2011, 156, 29–45. [Google Scholar]
- Amélie, B.; Frédéric, D.; Stéphanie, P.; Reinhard, J.; Charlotte, R.; Jean-Denis, F.; Haslam, R.P.; Napier, J.A.; René, L.; Jérome, J. Reconstitution of plant alkane biosynthesis in yeast demonstrates that Arabidopsis ECERIFERUM1 and ECERIFERUM3 are core components of a very-long-chain alkane synthesis complex. Plant Cell 2012, 24, 3106–3118. [Google Scholar]
- Owen, R.; Huanquan, Z.; Hepworth, S.R.; Patricia, L.; Reinhard, J.; Ljerka, K. CER4 encodes an alcohol-forming fatty acyl-coenzyme A reductase involved in cuticular wax production in Arabidopsis. Plant Physiol. 2006, 142, 866–877. [Google Scholar]
- Pierre, B.; Patricia, P.; Erin, O.; CaiZhong, J.; José Luis, R. WIN1, a transcriptional activator of epidermal wax accumulation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 4706–4711. [Google Scholar]
- Zhang, J.; Broeckling, C.; Blancaflor, E.; Sledge, M.; Sumner, L.; Wang, Z. Overexpression of WXP1, a putative Medicago truncatula AP2 domain-containing transcription factor gene, increases cuticular wax accumulation and enhances drought tolerance in transgenic alfalfa (Medicago sativa). Plant J. 2010, 42, 689–707. [Google Scholar] [CrossRef] [PubMed]
- Youhua, W.; Liyun, W.; Lixia, Z.; Zhijin, Z.; Haiwen, Z.; Ruidang, Q.; Shirong, Z.; Rongfeng, H. An ethylene response factor OsWR1 responsive to drought stress transcriptionally activates wax synthesis related genes and increases wax production in rice. Plant Mol. Biol. 2012, 78, 275–288. [Google Scholar]
- Zhou, X.; Jenks, M.A.; Liu, J.; Liu, A.; Zhang, X.; Xiang, J.; Zou, J.; Peng, Y.; Chen, X. Overexpression of transcription factor OsWR2 regulates wax and cutin biosynthesis in rice and enhances its tolerance to water deficit. Plant Mol. Biol. Rep. 2014, 32, 719–731. [Google Scholar] [CrossRef]
- Go, Y.S.; Kim, H.; Kim, H.J.; Suh, M.C. Arabidopsis cuticular wax biosynthesis is negatively regulated by the DEWAX gene encoding an AP2/ERF-type transcription factor. Plant Cell 2014, 26, 1666–1680. [Google Scholar] [CrossRef]
- Park, C.S.; Go, Y.S.; Suh, M.C. Cuticular wax biosynthesis is positively regulated by WRINKLED4, an AP2/ERF-type transcription factorin Arabidopsis stems. Plant J. 2016, 88, 257–270. [Google Scholar] [CrossRef]
- Kim, H.; Go, Y.S.; Suh, M.C. DEWAX2 transcription factor negatively regulates cuticular wax biosynthesis in Arabidopsis leaves. Plant Cell Physiol. 2018, 59, 966–977. [Google Scholar]
- Lam, P.; Zhao, L.; McFarlane, H.E.; Aiga, M.; Lam, V.; Hooker, T.S.; Kunst, L. RDR1 and SGS3, components of RNA-mediated gene silencing, are required for the regulation of cuticular wax biosynthesis in developing inflorescence stems of Arabidopsis. Plant Physiol. 2012, 159, 1385–1395. [Google Scholar] [CrossRef]
- Xia, K.; Ou, X.; Gao, C.; Tang, H.; Jia, Y.; Deng, R.; Xu, X.; Zhang, M. OsWS1 involved in cuticular wax biosynthesis is regulated by osa-miR1848. Plant Cell Environ. 2015, 38, 2662–2673. [Google Scholar] [CrossRef]
- Lam, P.; Zhao, L.; Eveleigh, N.; Yu, Y.; Chen, X.; Kunst, L. The exosome and trans-acting small interfering RNAs regulate cuticular wax biosynthesis during Arabidopsis inflorescence stem development. Plant Physiol. 2015, 167, 323–336. [Google Scholar] [CrossRef]
- Zhao, L.; Kunst, L. SUPERKILLER complex components are required for the RNA exosome-mediated control of cuticular wax biosynthesis in Arabidopsis inflorescence stems. Plant Physiol. 2016, 171, 960–973. [Google Scholar] [PubMed]
- Huang, D.; Feurtado, J.A.; Smith, M.A.; Flatman, L.K.; Koh, C.; Cutler, A.J. Long noncoding miRNA gene represses wheat β-diketone waxes. Proc. Natl. Acad. Sci. USA 2017, 114, E3149–E3158. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y.; Li, Z.; Sun, P.; Tang, J.; Liu, D. Fine-mapping and analysis of Cgl1, a gene conferring glossy trait in cabbage (Brassica oleracea L. var. capitata). Front. Plant Sci. 2017, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Cao, W.; Dong, X.; Liu, Z.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Wang, Y.; Sun, P. A 252-bp insertion in BoCER1 is responsible for the glossy phenotype in cabbage (Brassica oleracea L. var. capitata). Mol. Breed. 2018, 38, 128–135. [Google Scholar] [CrossRef]
- Liu, D.; Tang, J.; Liu, Z.; Dong, X.; Zhuang, M.; Zhang, Y.; Lv, H.; Sun, P.; Liu, Y.; Li, Z.; et al. Fine mapping of BoGL1, a gene controlling the glossy green trait in cabbage (Brassica oleracea L. var. capitata). Mol. Breed. 2017, 37, 69–77. [Google Scholar] [CrossRef]
- Liu, D.; Dong, X.; Liu, Z.; Tang, J.; Zhuang, M.; Zhang, Y.; Lv, H.; Fang, Z.; Yang, L. Fine mapping and candidate gene identification for wax biosynthesis locus, BoWax1 in Brassica oleracea L. var. capitata. Front. Plant Sci. 2018, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Tang, J.; Liu, Z.; Dong, X.; Zhuang, M.; Zhang, Y.; Lv, H.; Sun, P.; Liu, Y.; Li, Z. Cgl2 plays an essential role in cuticular wax biosynthesis in cabbage (Brassica oleracea L. var. capitata). BMC Plant Biol. 2017, 17, 223. [Google Scholar] [CrossRef] [PubMed]
- Ni, E.; Zhou, L.; Li, J.; Jiang, D.; Wang, Z.; Zheng, S.; Qi, H.; Zhou, Y.; Wang, C.; Xiao, S.; et al. OsCER1 plays a pivotal role in very-long-chain alkane biosynthesis and affects plastid development and programmed cell death of tapetum in rice (Oryza sativa L.). Front. Plant Sci. 2018, 9, 1217. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Xu, C.; Ren, J.; Liu, X.; Black, K.; Gai, X.; Wang, Q.; Ren, H. Cucumber ECERIFERUM1 (CsCER1), which influences the cuticle properties and drought tolerance of cucumber, plays a key role in VLC alkanes biosynthesis. Plant Mol. Biol. 2015, 87, 219–233. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, L.; Jiang, J.; Mason, A.S. Genome-wide identification, putative functionality and interactions between lncRNAs and miRNAs in Brassica species. Sci. Rep. 2018, 8, 4960. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hu, J.; Gao, C.; Chen, G.; Wang, B.; Lin, C.; Song, L.; Ding, Y.; Zhou, G. Genome-wide analysis of long non-coding RNAs unveils the regulatory roles in the heat tolerance of Chinese cabbage (Brassica rapa ssp. chinensis). Sci. Rep. 2019, 9, 5002. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Dong, H.; Zhou, D.; Li, M.; Liu, Y.; Zhang, F.; Feng, Y.; Yu, D.; Lin, S.; Cao, J. Systematic identification of long non-coding RNAs during pollen development and fertilization in Brassica rapa. Plant J. 2018, 96, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Kornienko, A.E.; Guenzl, P.M.; Barlow, D.P.; Pauler, F.M. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Hannoufa, A.; Mcnevin, J.; Lemieux, B. Epicuticular waxes of eceriferum mutants of Arabidopsis thaliana. Phytochemistry 1993, 33, 851–855. [Google Scholar] [CrossRef]
- McNevin, J.P.; Woodward, W.; Hannoufa, A.; Feldmann, K.A.; Lemieux, B. Isolation and characterization of eceriferum (cer) mutants induced by T-DNA insertions in Arabidopsis thaliana. Genome 1993, 36, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Jenks, M.A.; Tuttle, H.A.; Eigenbrode, S.D.; Feldmann, K.A. Leaf epicuticular waxes of the eceriferum mutants in Arabidopsis. Plant Physiol. 1995, 108, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Vishwanath, S.J.; Kosma, D.K.; Pulsifer, I.P.; Sabine, S.; Stéphanie, P.; Jérome, J.; Franziska, D.D.; René, L.; Owen, R.; Frédéric, D. Suberin-associated fatty alcohols in Arabidopsis: Distributions in roots and contributions to seed coat barrier properties. Plant Physiol. 2013, 163, 1118–1132. [Google Scholar] [CrossRef]
- Lee, S.B.; Jung, S.J.; Go, Y.S.; Kim, H.U.; Kim, J.K.; Cho, H.J.; Park, O.K.; Suh, M.C. Two Arabidopsis 3-ketoacyl CoA synthase genes, KCS20 and KCS2/DAISY, are functionally redundant in cuticular wax and root suberin biosynthesis, but differentially controlled by osmotic stress. Plant J. 2009, 60, 462–475. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Liao, J.Y.; Li, Z.Y.; Yu, Y.; Zhang, J.P.; Li, Q.F.; Qu, L.H.; Shu, W.S.; Chen, Y.Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E.; et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Wang, Y.; Yao, Y.; Song, N.; Hu, Z.; Qin, D.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tang, G.; Peng, X.; Sun, F.; Liu, S.; Xi, Y. Long non-coding RNAs of switchgrass (Panicum virgatum L.) in multiple dehydration stresses. BMC Plant Biol. 2018, 18, 79. [Google Scholar] [CrossRef]
- Wu, X.; Shi, T.; Iqbal, S.; Zhang, Y.; Liu, L.; Gao, Z. Genome-wide discovery and characterization of flower development related long non-coding RNAs in Prunus mume. BMC Plant Biol. 2019, 19, 64. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucl. Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Using sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucl. Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucl. Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucl. Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucl. Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Wild-type (μg/g) | nwgl (μg/g) |
|---|---|---|
| Fatty acid | 0.59 ± 0.04 | 0.32 ± 0.14 * |
| Aldehyde | 12.93 ± 4.33 | 27.33 ± 13.15 |
| Alkanes | 334.36 ± 110.66 | 5.52 ± 2.97 * |
| 1-Alcohols | 35.19 ± 2.74 | 20.23 ± 12.52 |
| 2-Alcohol | 14.20 ± 2.49 | - |
| Ketone | 174.06 ± 43.09 | - |
| Total | 571.33 ± 152.22 | 53.40 ± 28.31 ** |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, X.; Tai, X.; Ren, Y.; Chen, J.; Bo, T. Genome-Wide Analysis of Coding and Long Non-Coding RNAs Involved in Cuticular Wax Biosynthesis in Cabbage (Brassica oleracea L. var. capitata). Int. J. Mol. Sci. 2019, 20, 2820. https://doi.org/10.3390/ijms20112820
Zhu X, Tai X, Ren Y, Chen J, Bo T. Genome-Wide Analysis of Coding and Long Non-Coding RNAs Involved in Cuticular Wax Biosynthesis in Cabbage (Brassica oleracea L. var. capitata). International Journal of Molecular Sciences. 2019; 20(11):2820. https://doi.org/10.3390/ijms20112820
Chicago/Turabian StyleZhu, Xiaowei, Xiang Tai, Yunying Ren, Jinxiu Chen, and Tianyue Bo. 2019. "Genome-Wide Analysis of Coding and Long Non-Coding RNAs Involved in Cuticular Wax Biosynthesis in Cabbage (Brassica oleracea L. var. capitata)" International Journal of Molecular Sciences 20, no. 11: 2820. https://doi.org/10.3390/ijms20112820