Abstract
G protein-coupled receptors (GPCRs) have the propensity to form homo- and heterodimers. Dysfunction of these dimers has been associated with multiple diseases, e.g., pre-eclampsia, schizophrenia, and depression, among others. Over the past two decades, considerable efforts have been made towards the development of screening assays for studying these GPCR dimer complexes in living cells. As a first step, a robust in vitro assay in an overexpression system is essential to identify and characterize specific GPCR–GPCR interactions, followed by methodologies to demonstrate association at endogenous levels and eventually in vivo. This review focuses on protein complementation assays (PCAs) which have been utilized to study GPCR oligomerization. These approaches are typically fluorescence- and luminescence-based, making identification and localization of protein–protein interactions feasible. The GPCRs of interest are fused to complementary fluorescent or luminescent fragments that, upon GPCR di- or oligomerization, may reconstitute to a functional reporter, of which the activity can be measured. Various protein complementation assays have the disadvantage that the interaction between the reconstituted split fragments is irreversible, which can lead to false positive read-outs. Reversible systems offer several advantages, as they do not only allow to follow the kinetics of GPCR–GPCR interactions, but also allow evaluation of receptor complex modulation by ligands (either agonists or antagonists). Protein complementation assays may be used for high throughput screenings as well, which is highly relevant given the growing interest and effort to identify small molecule drugs that could potentially target disease-relevant dimers. In addition to providing an overview on how PCAs have allowed to gain better insights into GPCR–GPCR interactions, this review also aims at providing practical guidance on how to perform PCA-based assays.
1. Introduction
Membrane receptors are the key players in mediating communication between the cell and the extracellular space. The G protein-coupled receptor (GPCR) protein family represents one of the largest group of cell membrane signaling proteins in the human genome [1]. Currently, approximately 800 members of this superfamily are classified according to the International Union of Pharmacology, Committee on Receptor Nomenclature and Classification (NC-IUPHAR). Herein, GPCRs are grouped into three classes, namely Class A (rhodopsin-like), Class B (secretin receptor family), and Class C (metabotropic glutamate-like) [2]. The remaining GPCRs—fungal mating pheromone receptors, cyclic AMP receptors, and frizzled/smoothened—are also included in alternative classification systems [3].
G protein-coupled receptors are activated by a panel of different triggers, including photons, nucleotides, ions, hormones, peptides, chemokines, and others. Notwithstanding the broad collection of ligands to activate specific GPCRs, they all share a common structure, namely, an N-terminal domain, seven transmembrane α-helices, extra- (ECL1–3) and intracellular loops (ICL1–3) and a C-terminal domain. Specific domains within these GPCRs show plasticity, since conformational changes are clearly distinguishable between their activated and inactive states, as observed from the crystal structures [4]. Upon activation, the motile domains, such as the ICL3 and the C-terminus, are crucial for interaction with intracellular signaling partners, i.e., G-proteins, GPCR kinases (GRKs) and β-arrestins. Given their crucial role in cellular signaling, altered activity or deviating expression levels of GPCRs have frequently been correlated with diseases, including neurodegenerative disorders, depression, and cancer [5,6,7,8,9,10].
Overall, their abundance, regulation in pathophysiology of diverse disease areas, accessibility at the cell surface, and availability of druggable binding sites have made GPCRs the largest drug target family. It is, hence, not surprising that 34% of the current FDA-approved drugs on the market target GPCRs [11]. Interestingly, virtually all pharmaceutical companies have primarily focused on GPCR monomers as a target for drug development. However, the recent knowledge that GPCRs form homo-oligomeric or hetero-oligomeric complexes has opened up unexplored avenues in the development of new drug therapies (e.g., small ligands, peptides, and bivalent ligands) [12,13,14,15].
1.1. GPCR Dimerization
There are approximately 150,000 to 300,000 protein–protein interactions in the human interactome [16,17] and constant efforts are ever-expanding this number. This also holds true for the interest in GPCR–GPCR interactions, as evidenced by the remarkable rise in publications in this field of research, with over 300 reports (Figure 1) covering GPCR multimers during the last 5 years.
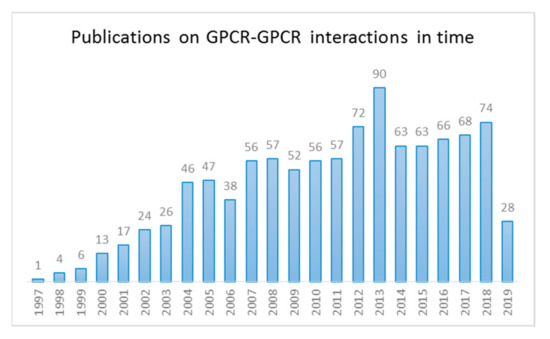
Figure 1.
Number of publications on G protein-coupled receptor (GPCR) dimer and oligomers from 1997–2019 (data incomplete for 2019). (Source: NCBI—a search for the terms ‘‘GPCR* AND DIMER*’ or ‘GPCR* AND OLIGOMER*’’ in the PubMed database).
While the traditional view on GPCRs was a “one ligand-one monomeric GPCR unit-one signaling protein” model, an emerging body of evidence shows that GPCRs can form homo- or hetero-oligomeric complexes. This phenomenon adds an additional layer to the complexity of GPCR signaling. Dysfunction of these dimers has been associated with multiple diseases, e.g., pre-eclampsia (angiotensin II type 1 and bradykinin receptor B2 heterodimer complexes) [18], schizophrenia (dopamine D2 receptor (D2R) heterocomplexes) [19], depression (serotonin 1A receptor 5-HT1A and serotonin 7 receptor 5-HT7 heterocomplexes, galanin receptor type 1 (GalR1), type 2 (GalR2), and 5-HT1A heterocomplexes) [20,21,22,23,24,25,26].
Although the concept of GPCR dimerization has been controversial over the past two decades, several sophisticated studies have led scientists to the validation of the GPCR dimerization theory. An indisputable and widely accepted example is the obligate dimer of Class C GABA receptors [27]. These receptors are obligatory dimers, as GABAB1 is necessary for ligand binding and GABAB2 ensures efficient cell trafficking and downstream signaling. Both protomers are not functional when expressed alone. In addition to obligate GPCR dimers observed in Class C receptors, an increasing amount of convincing evidence suggests there is also -transient dimer formation between Class A GPCRs [28,29,30]. Conflicting observations both in favor of and against oligomerization have been reviewed by Bouvier and Herbert (2014) [31] and by Lambert and Javitch (2014) [32]. Recent insights into Class A GPCR dimerization have shed new light on this concept, i.e., a large-scale comparative study of 60 members of the Rhodopsin family revealed that only a small fraction, i.e., 20%, forms dimers [33]. The dimer formation of Class A GPCRs seems to have a clustered distribution, and furthermore, a striking correlation has been suggested between receptor organization and size of the GPCR family. A restricted family size, as for the small Glutamate family, correlates with predominantly dimeric behavior. This also seems to be true for the receptors exhibiting less diversity like the frizzled GPCRs [33].
Besides the fact that Class A GPCRs seem to dimerize to a lesser extent as compared to Class C receptors, the dimerization event itself is also much more complicated, i.e., Class A GPCRs often seem to interact at multiple interfaces, thus causing a broad impact on functionality [34]. This is noted in the case for the muscarinic acetylcholine M1 or M2 dimerization, wherein reports have suggested that the sites of contact are transient and could involve multiple regions of the receptor [35,36,37]. For the D2R, this transient dimerization phenomenon has been studied in more detail, with a lifetime of 68 ms being assigned to the dimer. This dimer lifetime could be prolonged with a factor of approximately 1.5 by agonist addition (i.e., dopamine and quinpirole) [38,39], whereas incubation with the antagonist spiperone reduced the level of D2R homodimerization by more than 40% [40], indicating a potential role of the dimer in signaling cascades. Dijkman and colleagues introduced the “rolling dimer” interface model for the neurotensin receptor 1 homodimer. This concept unites earlier seemingly conflicting data since multiple dimer conformations could co-exist that interconvert during the dimer lifetime, by rotation of the monomers relative to one another.
In general, the phenomenon of oligomerization can exert a significant impact on receptor–ligand binding, downstream signaling, crosstalk, internalization, and trafficking. For instance, the interaction of D2R with dopamine D3 receptor (D3R) results in a higher potency of certain anti-parkinsonian agents, like pramipexole, compared to their monomers [28]. Furthermore, selective stimulation of D1R or D2R or both by the neurotransmitter dopamine triggered co-internalization of the D1R–D2R heterodimer [29].
1.2. Studying GPCR–GPCR Interactions: Biochemical Methods
In order to target and study GPCR–GPCR interactions, multiple in vitro (and in vivo) biochemical and physical, and functional studies have been reported in the literature, which described approaches such as co-immunoprecipitation (co-IP), proximity ligation assays (PLA), and fluorescence- or bioluminescence-based techniques [41].
The presence of multiple protein–protein interactions (PPIs) has been demonstrated by co-IP or pull-down assays [42,43,44]. On a non-reducing SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis), it has been reported that GPCRs often migrate in a way that corresponds to twice their molecular weight. Co-IP of endogenous GPCRs entirely relies on the selectivity of the antibodies used. Furthermore, this technique necessitates the disruption of biological samples and the solubilization of membrane proteins and generally provides no information about the physical interaction.
On the other hand, fluorescence- or bioluminescence-based energy transfer techniques can be applied in living cells, thereby overcoming some of the limitations of the classical biophysical techniques. For GPCR dimerization, this has been reviewed by Ciruela et al. (2010) [45]. Fluorescence and bioluminescence resonance energy transfer (FRET and BRET) involve the non-radiative energy transfer between a donor and acceptor, which allows the examination of PPIs in their native cell context by monitoring emissions from the acceptor species. Fluorescence resonance energy transfer microscopy offers the advantage that it allows visualization of the interaction inside living cells. Bioluminescence resonance energy transfer avoids some of the troubleshooting involved with FRET such as photobleaching, spectral bleed-through, etc., but does not provide spatial information on the interaction. Below, we will focus primarily on recent advances in yet another biochemical method, namely, bioluminescence and fluorescence-based complementation techniques that have been applied to study GPCR–GPCR interactions.
1.3. Protein Complementation Assays
Protein complementation assays (PCAs), also referred to as split systems, have been implemented over the past 2 decades to study PPIs. In these assays, a reporter protein with enzymatic or fluorescent properties is engineered or “split” into non-active or non-fluorescent fragments. These moieties are fused to potential interacting proteins. Upon interaction, the fragments will be brought into close proximity and re-assemble spontaneously into a functional biosensor. Although these assays do not formally prove direct protein–protein interactions, they do suggest co-localization of the GPCRs of interest since PCAs rely on the fusion partners “interacting”. Complementation-based assays comprise bimolecular fluorescence and luminescence complementation (BiFC/BiLC) as well as β-Lactamase and β-Galactosidase complementation assays. Since 1997, several “split” fluorescent and luminescent proteins have been developed, of which the most commonly used ones are shown in the timeline in Figure 2.
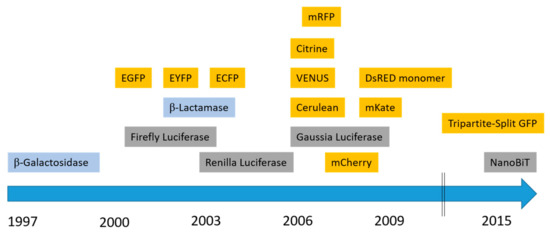
Figure 2.
History of the development of split protein biosensors. Commonly used fluorescent split proteins (yellow), luminescent split reporters (grey), and enzymes with colorigenic substrates (blue) are shown.
These assays have the potential to be implemented as screening assays to identify GPCR–GPCR interactions, which later on should be confirmed by a variety of complementary approaches, for example, Spatial Intensity Distribution Analysis (SpIDA), advanced microscopy techniques, and molecular dynamics, among others [35,39,46,47,48,49]. A benefit of a robust complementation assay is not only its operational simplicity but even more the possibility to test if dimers occur constitutively or whether ligands can alter the oligomeric state of GPCRs. Moreover, these cell-based assays lend themselves for early-stage drug discovery, since molecules that potentially exert an impact on the level of dimerization, whether desired or undesired, can be rapidly identified. In addition, off-target effects can be revealed, such as altered receptor expression, localization or cellular signaling. An overview of the available PCAs as of today and their characteristics are presented in Table 1.

Table 1.
Overview of proteins, already implemented in fluorescence- and luminescence-based complementation assays. N/A = not applicable, nd = not determined.
Many questions remain unanswered in the field of GPCR di- and oligomerization. Therefore, it is a highly warranted first step to fully characterize the in vitro assays featuring robust sensitivity in detecting PPIs of interest. This review aims at offering insight into the progress that has been made in the field of protein complementation assays to study GPCR dimerization. This progress concerns, for example, optimization of complementation assays in stability and light output by a consensus sequence driven strategy. Several mutations applied in fluorescent and luminescent complementation assays in the field of GPCR dimerization will be discussed throughout the following sections of this review, for additional information concerning mutational optimization of split reporter genes in general, we refer to the excellent review by Wehr and Rossner [50].
2. Fluorescence-Based Complementation Assays
One of the well-studied PCAs is BiFC, which is based on the structural and functional complementation of two non-fluorescent protein fragments brought into close proximity by their interacting fusion partners (Figure 3). Upon complementation of the fluorescent protein, the fluorescence can be read via a plate-reader, imaged using a fluorescence microscope or even analyzed by flow cytometry without the need for any treatment of the cells.
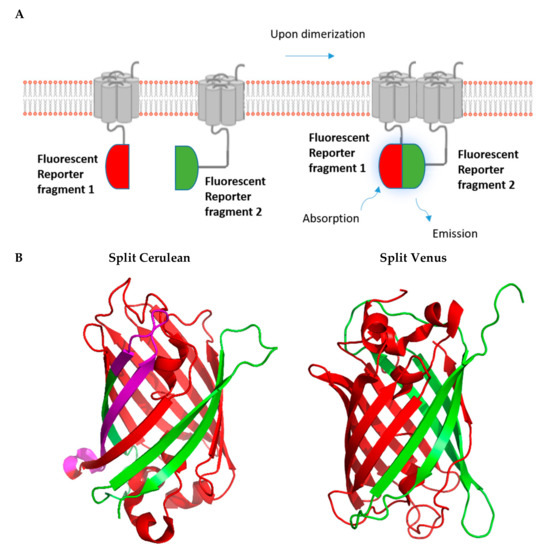
Figure 3.
Fluorescence-based complementation assay. (A). The principle of the fluorescent complementation assay is shown schematically. (B). Split versions of the fluorescent reporters Cerulean and Venus are shown in green and red. Purple refers to overlapping sections. (PDB; Accession no. 3AKO for Venus; PDB: Accession no. 5OXB for Cerulean).
In 1999, Regan and colleagues reported BiFC for the first time in Escherichia coli (E. coli) by using a strategy based on the non-covalent association of the split fragments of green fluorescent protein (GFP), fused to two antiparallel leucine zippers [51]. Subsequently, Hu et al. [52] reported on the development of a BiFC assay based on the split fragments of yellow fluorescent protein (YFP), tagged to two interacting transcription factors in living mammalian cells. Shyu et al. [53] developed Venus, one of the brightest fluorescent proteins, for studying PPIs at physiological conditions using BiFC. Since then, numerous types of fluorescence complementation-based assays have been developed to visualize signaling events involving two or even more interacting proteins, ranging from bacteria to mammalian cells.
2.1. Fluorescent Proteins
2.1.1. Green Fluorescent Protein (GFP)
Regan and colleagues used GFP, the first fluorescent protein, for characterizing BiFC by tagging the split fragments of GFP, i.e., NGFP and CGFP, split at residue 157–158, to strongly interacting antiparallel leucine zippers. The split fragments did not self-assemble in bacteria, whereas upon fusion to antiparallel leucine zippers, the fragments could properly fold to form a functional GFP protein, and hence, gain fluorescence. A recent study performed by Son and colleagues [54] made use of enhanced GFP (EGFP), an improved version of GFP that is brighter and more photostable. The EGFP molecule was split to yield N-EGFP (1–158) and C-EGFP (159–238), respectively. These split parts were fused to α-factor receptor (Ste2p), to study homodimerization in S. cerevisiae. Both full length and C-terminally truncated forms of the receptors led to fluorescence, indicating dimerization.
A recent advance in BiFC based on GFP is Tripartite GFP, introduced by Cabantous et al. [55] in 2013 in an attempt to cope with the problem of poor folding and self-assembly seen with split GFP. Tripartite GFP is based on the tripartite association of two twenty amino-acid residue fragments of GFP, coined as GFP 10 and GFP 11, that can be fused to the proteins under investigation, and the complementary GFP 1–9 detector. Upon interaction between the protein partners, GFP 10 and GFP 11 associate with GFP 1–9 to form the functional GFP molecule. This system was initially characterized in E. coli and in mammalian cells, where the rapamycin induced association of FRB (FKBP-rapamycin binding domain of mTOR) and FKBP12 (FK506- and rapamycin-binding protein) was studied. This system has extensively been exploited for the validation of multiple reporter systems. Although use of this Tripartite GFP system has not been reported for studying GPCR dimerization, it could be an advantageous platform to be used over the conventional split GFP.
2.1.2. Yellow Fluorescent Protein (YFP) Variants
In 2001, Nagai et al. [56] first demonstrated the use of the yellow fluorescent protein (YFP) in BiFC, wherein they used a variant of YFP to study the effect of Ca2+ on PPI in living cells. Shortly after, Griesbeck and colleagues [52,57] tried several permutations and combinations of split regions in EYFP (S65G, S72A, T203Y) to improve the fluorescence signal along with minimal self-assembly of the split fragments. Several variants of YFP have been developed such as Citrine (a variant having a Q69M mutation) and Venus (a variant carrying a F46L mutation), that folds even at a physiological temperature of 37 °C [58]. This led to the usage of these fluorescent protein fragments to study GPCR–GPCR interactions.
Vidi et al. (2008) [59,60] used split fragments of Venus fused to the adenosine A2A receptor (A2A) to address the question about A2A oligomerization in the CAD neuronal cell line. Co-expression of A2A-VN and A2A-VC in these cells, as well as in HEK293 cells, resulted in fluorescence, indicating A2A dimerization, in contrast to negative controls wherein cells transfected with A2A-VN/D1R-VC/M4R-VC did not show any fluorescence, pointing at the specificity of the interaction.
Similarly, Kilpatrick et al. (2014) [61] assessed dimerization of a receptor for NPY, Neuropeptide Y Y1/Y5. This neuropeptide is a widely expressed modulator of the central nervous system, mainly known for its role in the regulation of appetite upon its release from hypothalamic arcuate neurons. The receptors, tagged with split YFP, showed heterodimerization and the irreversible nature of these fragment tags also helped to elucidate the cellular localization of these dimers. Evaluation of the effect of ligands on the internalization of dimers, as opposed to individual protomers, demonstrated that Y1/Y5 receptor dimers displayed altered ligand pharmacology, indicative of allosteric interaction.
Przybyla and Watts [62] in 2010 used Venus-based BiFC to demonstrate heterodimerization between the cannabinoid receptor 1 (CB1) and the dopamine D2Long receptor (D2LR) in CAD cells, wherein CB1 and D2LR receptors were fused at their C-terminus to VN and the C-terminal fragment of Cerulean (CC), respectively (see Section 2.2.1).
Ang et al. [63] in 2018 assessed homo- and heterodimerization of free fatty acid receptors, GPCRs that are expressed on mammalian cells to sense the short-chain fatty acids derived from the microbiota. To study dimerization, HEK293T cells were transfected with VN- or VC-tagged (free fatty acid receptor 2/3) FFAR2/3 receptor. Homodimerization of FFAR3 and FFAR2, as well as heterodimerization between FFAR2-VN and FFAR3-VC were observed, as compared to negative control FFAR2/3 tagged to VN or VC plus P2RY1-VN or P2RY1-VC (for heterodimerization) and P2RY1-VN plus P2RY1-VC (for homodimerization).
Xue et al. [64] in 2018 used a BiFC assay based on split Venus to detect heterodimerization between growth hormone secretagogue receptor 1α (GHSR1a) and Orexin 1 receptor (OX1R). Recently, Navarro et al. [65] (2018) used interfering peptides corresponding to regions in TM1–7 domain for A2A and TM5–7 domain for A1, each fused to TAT peptides, to disrupt the interaction between the A1–A2A heteromer. They therefore expressed receptors tagged with split fragments of YFP (nYFP and cYFP) in HEK293T cells. In the absence of the TM peptides, fluorescence complementation was detected for A2A-nYFP and A2A-cYFP in the presence of untagged A1, suggesting homodimerization of A2A. The interaction sites were mapped to TM4 and 5 of A2A with the help of interfering peptides. In the same way, a fluorescence signal corresponding to the heterodimer (A1-nYFP and A2A-cYFP) was also detected. Upon addition of TM peptides corresponding to regions 5/6, a reduction in fluorescence was observed, suggesting the involvement of these regions in the formation of the heterodimer. In the same year (2018), BiFC using split fragments of YFP was utilized by Hinz et al. [66] for studying homodimerization between A2A and heterodimerization with A2B receptor. For this purpose, CHO cells were transfected with a constant amount of A2A-NYFP and increasing amounts of HA-A2A-CYFP and a strong fluorescence signal was observed, indicative of the A2A-A2A homodimer. For the purpose of studying A2B-A2A heterodimerization, the GPCRs were tagged with NYFP and CYFP, respectively. CHO cells transfected with constant amounts of A2B-NYFP and increasing amounts of HA-A2A-CYFP showed significantly higher fluorescence when compared to the negative control comprising of GABAB2-NYFP and increasing amounts of HA-A2A-CYFP.
An interesting study was performed by Song et al. [67] in 2019, where a homo-molecular fluorescence complementation (homo-FC) probe was designed by splitting a single fluorescent protein (superfolder GFP, sfGFP) into two fragments and linking the C-terminal fragment (strands 8–11) with a short linker N-terminally to the N-terminal fragment (strands 1–7). This “flopped fusion” construct, which does not show intramolecular self-complementation, was used to tag the β2 adrenergic receptor (β2-AR), to study its homodimerization. A fluorescent signal was generated by complementation of strands 1–7 of one “flopped” sfGFP molecule, fused to one β2-AR, with strands 8–11 of another “flopped” sGFP molecule, fused to a second β2-AR. The advantage of this approach lies in the fact that only one receptor construct needs to be generated, to allow the assessment of homodimerization.
2.1.3. Cyan Fluorescent Protein (CFP)
Apart from using the aforementioned split Venus, Vidi et al. [59,68] also used split fragments of Cerulean to show that A2A homodimerizes in CAD cells (see Section 2.2.1). The same fluorescent protein fragments were also used in 2010 by Przybyla and Watts [62] to illustrate the homodimerization of D2LR.
2.1.4. Red, Far-Red and Near-Infrared Fluorescent Proteins
Red fluorescent proteins encompass a couple of fluorescent proteins that emit in the red region of the visible spectrum. They have evolved through time to overcome issues such as slow maturation or the tendency to oligomerize (as exhibited by Discosoma Red (DsRed)) [69]. Red variants such as monomer red fluorescent protein 1 (mRFP1) are not appropriate for BiFC due to their low extinction coefficient, quantum yield, and low photostability, despite faster maturation than DsRed and better tissue penetration. Although several variants of RFP have been used for FRET, BiFC assays with the split parts have not been reported for demonstrating GPCR–GPCR interactions.
For imaging deep tissues in animals, fluorescent proteins with emission in the far-red region of the spectrum are chosen. RFP, mcherry and DsRed, have limited application in BiFC due to the requirement of low temperatures for their maturation [70,71]. This led to the development of mKate, a far-red, monomeric form of Katushka fluorescent protein, that matures faster and is photostable [72]. BiFC based on split fragments of mKate fused to transcription factors was successfully applied in COS-7 cells [73], but has not been applied yet for studying GPCR oligomerization.
2.2. BiFC Assays
2.2.1. MBiFC
Multicolor BiFC (MBiFC) has gained a lot of attention in recent years as it essentially gives the freedom to explore multiple protein–protein interactions inside the same living cell [74]. It has been studied with split parts of Venus and Cerulean, wherein the C terminal part of Cerulean is tagged to protein A (CC), the N-terminal part of Venus is tagged to protein B (VN), and finally, the N-terminal fragment of Cerulean is fused to protein C (CN). The C-terminal fragment of Cerulean (CC), comprising residues 155–238 (C155), can functionally complement with the N-terminal fragment of VN and CN, comprising residues 1–172 (N173), to produce a Venus (VN+CC) signal or Cerulean (CN+CC) signal. Since these parts are non-fluorescent per se, simple employment of different excitation and emission wavelengths allows to study simultaneously the interaction between A–B and A–C within the same cell [59,75,76].
Multicolor BiFC has aided in the understanding of GPCR oligomerization amongst all its other applications. Vidi et al. [59] (2008) used MBiFC to visualize the hetero- and homodimerization capacity of A2A in the CAD neuronal cell line. To this end, D2LR-VN was co-transfected with A2A-CN and A2A-CC in CAD cells. The detection of Venus (D2LR-VN/A2A-CC) and Cerulean (A2A-CN/A2A-CC) fluorescence signals revealed the presence of the A2A–D2LR heteromer and A2A homomer, respectively. In addition, fluorescence microscopy also revealed that the dimers were localized at the plasma membrane as well as in intracellular compartments.
Przybyla and Watts [62] in 2010 showed oligomerization of CB1 and D2LR. To achieve this, they used CAD cells that expressed CB1-VN and D2LR fused to the split fragments of Cerulean (D2LR-CN, D2LR-CC) and these cells were imaged using fluorescence microscopy. The heterodimers in the cell formed by CB1-VN–D2LR-CC produced a Venus signal that was mainly localized in the intracellular compartment, whereas the homodimers of D2LR produced a Cerulean signal that was localized both in the intracellular compartment and on the plasma membrane.
2.2.2. BiFC-RET
The fluorescent proteins used in BiFC could also be coupled to FRET or BRET to study GPCR oligomerization, e.g., functionally complemented YFP could act as a FRET/BRET acceptor. BiFC-BRET [77] employs the energy transfer between GPCR-A fused to a luciferase and the complemented fragments of YFP that are fused to GPCR-B and GPCR-C that are as close as 10 nm apart, indicating a trivalent complex of GPCRs.
Navarro et al. [78] in 2008 used BiFC-BRET to demonstrate the interaction between D2R, A2A, and CB1 receptors in HEK cells. The split fragments of YFP fused to CB1 and A2A acted as the acceptor fluorophore in BRET, and Renilla luciferase (RLuc) fused to D2R (D2R-RLuc) acting as the donor.
BiFC-FRET [79] is similar, offering the additional benefit that the complex can be visualized within the cell. BiFC-FRET was used to understand the existence of higher order oligomers in the case of A2A by using fusion constructs with Cerulean and split parts of Venus. Prior to this, to test if the system could record FRET, A2A dimerization was tested by fusing A2A to Cerulean and Venus. Based on this, CAD neuronal cells were co-transfected with A2A-Cerulean and A2A fused to split parts of Venus. FRET signals were detected, suggesting the existence of A2A oligomers, as opposed to a negative control wherein cells transfected with M4 as the acceptor only showed very low FRET [60].
In 2017, Bagher et al. [80] combined BRET with BiFC which forms the basis of “sequential resonance energy transfer 2” (SRET2). Herein, the donor for BRET was the Renilla luciferase (RLuc) fused to D2LR (D2LR-RLuc), which upon addition of the substrate coelenterazine 400a, excited the acceptor GFP2 fused to D2LR (D2LR-GFP2), thereby confirming the homodimerization of D2LR. Sequentially, the GFP2 acted as FRET donor for the CB1-VN/CB1-VC homodimer, thus providing evidence for heterotetramerization. With increasing concentrations of CB1-VN/CB1-VC, there was a hyperbolic increase in net SRET2, as opposed to negative control cells that were transfected with D2LR-RLuc and mGLuR6-GFP2 and increasing concentrations of CB1-VN/CB1-VC, confirming that the homodimers of D2LR and CB1 associate to form oligomers.
2.3. Ligand-Dependent Modulation of Dimerization
The level of GPCR dimer formation could possibly be altered by ligands interacting with the receptor(s). The potential of split-protein sensors to monitor the dynamic changes in dimerization, provoked by chemical inducers or inhibitors, was demonstrated using the rapamycin-dependent FKBP/FRB Chemically Induced Dimerization (CID) system [81]. The first demonstration for the use of MBiFC to monitor drug-modulated GPCR oligomerization was published by Vidi et al. [59] in 2008 for the A2A–D2R interaction. Stimulation of D2R with the D2R agonist quinpirole led to internalization of D2R homodimers as well as of A2A − D2R oligomers, which was blocked by the D2R antagonist, sulpiride [59]. Furthermore, treatment with quinpirole decreased the formation of A2A − D2R heterodimers, as compared to A2A homodimers. Also, treatment with 5-N-methylcarboxamidoadenosine (MECA), an agonist for the adenosine receptor, increased the proportion of A2A/D2LR heterodimers, as compared to homodimers of A2A, while treatment with its antagonist (CGS15943) had no effect on BiFC. Interestingly, these drug-induced alterations on the formation of oligomers could not be supported by changes in the receptor density.
As discussed above, Przybyla and Watts [62] demonstrated heterodimerization of CB1 − D2LR using MBiFC. Also, the influence of ligands on the balance between hetero- and homodimers was analyzed. Quinpirole and CP55,940 shifted the balance towards the formation of CB1 − D2LR heterodimers, as opposed to D2LR − D2LR homodimers. The D2R antagonist sulpiride favored D2LR homodimerization. Consequently, it was shown that sustained treatment with quinpirole influenced the expression of D2LR, which in turn might affect the dimerization status inside the cell as: D2LR − CB1 > D2LR − D2LR > A2A − D2LR. In a similar way, studies also indicated that a CB1 antagonist (1-(7-(2-chlorophenyl)-8-(4-chlorophenyl)-2-methylpyrazolo (1,5-a)-(1,3,5)triazin-4-yl)-3-ethylaminoazetidine-3-carboxylic acid amide benzenesulfonate) could have a potential therapeutic role in Parkinsonism by enhancing the activity of L-DOPA [82].
In 2018, Navarro et al. [65] evaluated if activation of A1 or A2A alone or by a combination of agonists could modulate the TM interface of the heteromer formed by A2A-nYFP and A1-cYFP. Selective agonists for A1, N6-cyclopentyladenosine (CPA), and A2A, 4-(2-((6-Amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-yl) amino) ethyl) benzenepropanoic acid (CGS-21680), were used alone or in combination. None of the agonists, either used in combination or alone, could modify the TM interface of the A1 − A2A heterodimer.
Various ligands were tested for their capacity to affect β2AR oligomerization by Song et al. [67] in 2019. A panel of compounds consisting of five agonists, two antagonists, and two inverse agonists were screened using the abovementioned homo-FC assay. They observed that agonists induced oligomerization, while inverse agonists reduced the signals, and a combination of both also resulted in reduced intensity of signal, thus confirming the same.
3. Luminescence-Based Complementation Assays
Luciferases, which have seen an expansive growth in use as reporter proteins in biological research, are attractive due to the high signal-to-background ratio associated with their usage, as no excitation light is required to generate a signal [83]. Similar to fluorescent proteins, these enzymes can also be used in applications where the luminescent protein itself is split into two fragments, which are conjugated to proteins of interest (Figure 4). Several luminescent proteins have served this purpose, with Firefly luciferase (FLuc) [84,85] and Renilla luciferase (Rluc) [86,87] being the two most commonly used bioluminescent proteins. However, many other novel luciferases have also been developed with favorable characteristics in terms of stability, substrate requirement, brightness, and emission spectrum, e.g., Gaussia princeps luciferase (GLuc) [88] and NanoLuciferase [89,90,91]. All mentioned split-protein reporters were initially validated using the aforementioned FKBP/FRB CID system [84,88,92,93,94].
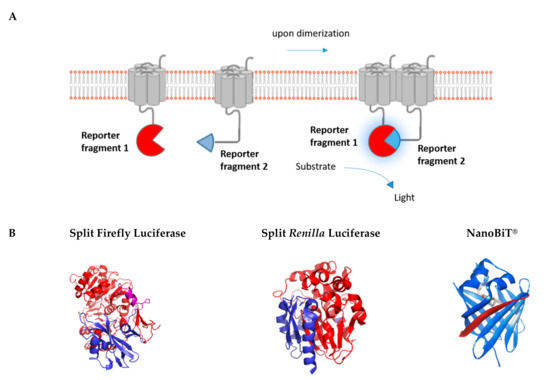
Figure 4.
Luminescence-based protein complementation assay. (A) The principle of the luminescent complementation assay is shown schematically. (B) Split versions of the luminescent reporters Firefly, Renilla, and Nanoluciferase are shown in blue and red. Purple refers to overlapping sections. Split luminescent biosensors are depicted in proportion to their size (Fluc: 62 kDa, Rluc: 36 kDa, and NanoBiT®: 19 kDa) (PDB Accession no. 1LCI for Firefly luciferase, PDB Accession no. 2PSD for Renilla luciferase) (NanoBiT®: source Promega).
3.1. Renilla/Firefly Luciferase
FLuc is the least optimal reporter for employment as a bioluminescent tag, due to its size (±60 kDa) and its dependence on ATP, molecular oxygen, and magnesium for activity. Luciferases that use coelenterazine as a substrate, such as the luciferase from the sea pansy Renilla reniformis RLuc [95,96], have an advantage over FLuc in that they are not ATP dependent and only require the presence of molecular oxygen for the enzyme-catalyzed conversion of a substrate to a luminescent reaction product. As the bioluminescent activity of formerly used non-truncated luciferases is a limiting factor to permit their application in high throughput screenings, specific mutations of certain luciferases, like in RLuc, were selected using a consensus sequence-driven strategy and screened for their ability to confer stability in activity as well as for their light output [97].
For GPCR dimerization purposes, the split-luciferase reporter approaches have not been applied to the same extent as their fluorescence counterparts. Nevertheless, these reporter systems also allow the rapid detection of macromolecular GPCR–GPCR interactions. For instance, the split FLuc methodology was used in cell culture models as well as in tumor xenograft models of breast cancer (see Section 8) to measure changes in chemokine receptor CXCR4 (and CXCR7) homodimerization in response to pharmacological agents [98]. Likewise, the CXCR4 homodimer has also been demonstrated with a split RLuc assay [99]. For this Renilla luciferase complementation assay, RlucII was implemented, which is derived from RLuc, wherein two mutations (C124A and M185V) were introduced to make it brighter.
For studying dopamine D2R oligomers, an alternative RLuc construct was implemented, namely, Rluc8 [100]. Owing to eight favorable mutations, this RLuc8 has a 4-fold improved light output, compared with the parental enzyme [97]. More recently, the same complementation assay was also used by Casado–Anguera et al. [101] (2016) to demonstrate the existence of a therapeutically relevant GPCR dimer, namely, the A2a − D2R dimer. This dimer and its inter-protomer allosteric mechanisms have been proposed as a new model that could contribute to our knowledge concerning drug dosage for the treatment of Parkinson’s disease.
3.2. NanoLuciferase
One of the most recent and undoubtedly the most optimized luminescent PCA so far is “NanoLuciferase Binary Technology” or the NanoBiT® system, developed by Promega. The assay is based on complementation of the split fragments of NanoLuciferase or NanoLuc. This engineered luciferase reporter is a small (19 kDa), ATP-independent luminescent protein, originating from a luminous deep-sea shrimp, Oplophorus gracilirostris [102]. In combination with the development of a novel cell-permeable imidazopyrazinone substrate, furimazine, this bioluminescence system generates a glow-type luminescent signal that is over 150-fold greater compared to that of the former Renilla and Firefly luciferases. NanoLuc exhibits high physical stability, in a wide range of environmental conditions such as temperature, pH, urea, and ionic strength [103]. As opposed to luminescent protein fragments described earlier, the NanoBiT® subunits do not consist of two fragments similar in size but correspond to a small 1.3 kDa subunit (Small BiT; SmBiT) and an 18 kDa subunit (Large BiT; LgBiT). Both subunits have been thoroughly characterized and have a low affinity (KD = 190 µM) for one another, thus providing the ability to follow kinetics of PPIs.
Although the NanoBiT® PCA has only recently been developed [92], its broad applicability has proven successful in numerous research fields. NanoBiT® has, for instance, been applied to develop bio-assays based on the recruitment of β-arrestins [104,105,106,107,108,109,110] and G-proteins [111] for the detection or activity profiling of certain compounds or to elucidate the molecular interaction between the transducer and a GPCR. For GPCR dimerization purposes, NanoBiT® has been implemented to detect ligand-dependent modulation of D2LR − D2LR homodimers [40,112]. The methodology has also been used to perform live-cell monitoring of the dynamics of the interaction between the Melanocortin 4 receptor (MC4R), and the Melanocortin 2 receptor accessory protein 2 (MRAP2), as well as of MC4R homodimerization [113].
The NanoBiT® system has been further optimized into a tri-part protein fragment complementation assay by two independent groups [114,115]. For this purpose, the LgBiT was dissected into a smaller C-terminal part of 11 amino acids (LcBiT) and an N-terminal derivative (LnBiT), serving as a “detector” protein of 16.5 kDa, similar to the Tripartite GFP mentioned in Section 2.1.1. Initially, this assay was designed to facilitate the purification of fusion proteins to procure significant quantities and to avoid the lapse of detection of the PPI of interest due to the steric hindrance of LgBiT, due to its higher molecular weight. In addition, another novel split-luciferase reporter based on NanoBiT® was developed that implements GFP- and mCherry-recognizing nanobodies fused to LgBiT and SmBiT [116]. Using this experimental set-up, GFP- or mCherry-tagged dimers or higher order oligomers can be detected. As fluorescently tagged proteins are already often in use, this luminescent PCA can be directly used and consequently bypasses the tedious protein re-cloning to explore the different possible configurations for setting up a PCA (also see Section 6). Furthermore, due to the strong affinity of the GFP- and mCherry-recognizing nanobodies, a low limit of detection of PPIs or protein aggregations, from micromolar up to low nanomolar, can be achieved.
3.3. BiLC-RET
The application of BiLC has already shown its efficiency in combination with a BRET assay, allowing to detect ternary protein complexes. Sahlholm et al. [117] (2018) demonstrated, by combining BiLC of D2R − A2a heterodimers with a YFP-tagged β-arrestin, a BRET signal could be obtained. Furthermore, it was postulated that D2R agonists quinpirole or UNC9994 require the formation of D2R − A2aR heterodimers, to promote β-arrestin2 recruitment.
4. Combinatorial Assays: BiFC and BiLC
For the detection of higher order oligomerization of GPCRs, a combinatorial assay with both BiFC and BiLC can be implemented (Table 2). Rebois et al. [77] (2008) introduced the concept of detection of a tetravalent complex using a combination of BiFC and BiLC, wherein four β2ARs were fused to split parts of Venus or GLuc and the homodimerized β2AR-GLuc acted as the donor for the homodimerized β2AR-Venus acceptor, the presence of a BRET signal indicating the presence of the tetramer. In the same year (2008), Guo and colleagues [100] showed the existence of a D2R homo-oligomer, using the same technique.
Table 2.
Combinatorial assays with BiFC and BiLC for GPCR oligomerization purposes.
A complemented donor–acceptor resonance energy transfer (CODA-RET) assay showed that the A2A − D2R dimer not only forms dimers but can also assemble to form a heterotetramer, composed of two receptor homodimers [101,118]. For this experimental setup, a complemented YFP was implemented and combined with Rluc8.
5. Comparison of Split Protein Approaches
Protein complementation techniques have shown their applicability in many fields to unravel PPIs. However, every technique has its advantages and limitations, as summarized in Table 3 for BiFC and BiLC.
Table 3.
Advantages and disadvantages of BiFC and BiLC techniques.
5.1. Advantages of PCA
A key advantage of BiFC and BiLC lies in the fact that complementation assays offer a robust and straightforward approach to evaluate PPIs in living cells. Consequently, their applicability in high-throughput experiments is a valuable quality. The development of MBiFC, BiFC-RET, or CODA-RET has made the detection of multiple protein–protein interactions feasible, such as GPCR oligomers, without the need for cell lysis or fixation. Due to the simplicity of performing these assays, they can be used as a screening platform for drugs [59]. When used with a plate reader, BiFC and BiLC also offer quantitative and rapid results on relatively large cell populations.
The most valuable aspect of BiLC is that kinetic measurements can be performed due to its sensitivity and the limited propensity of self-association events, at least for certain BiLC reporters (e.g., NanoBiT®). For the purpose of studying GPCR dimerization, the NanoBiT® assay has been compared to related bioluminescence and fluorescence PCAs, including split Venus and RLuc [112]. As a benchmark, the generally accepted D2R − D2R dimer was compared in the different PCAs. The NanoBiT® assay presented the best signal-to-noise ratio and was considered the most optimal candidate assay for targeting GPCR dimers. In addition, this method can also be implemented to analyze the kinetics of ligand-dependent modulation of dimerization, broadening its application potential [40]. Even high-throughput screenings can be performed, which is highly relevant, given the growing interest and effort to identify small molecule drugs that can target disease-relevant dimers (or even selectively alter GPCR dimer function). Moreover, BiLC has also proven its applicability in an in vivo setup [85,87].
On the other hand, BiFC has the capacity to visualize and localize the interaction, also offering insight into potential protein aggregation artefacts inside the cell. BiFC does not require any exogenous stains or substrate. Thus, depending on the intrinsic fluorescence, it allows the direct measurement of the interaction between proteins.
5.2. Limitations of PCA
BiFC and BiLC also come with their own set of drawbacks. First of all, both techniques require the fusion of the target proteins with split fragments of a fluorescent or luminescent protein, which could affect the original proteins’ interaction dynamics. Moreover, the amount of proteins expressed by the cell may potentially cause false positive readouts. Therefore, a good set of negative controls is obligatory (see Section 6.3). In addition, one should bear in mind that these techniques, though indicating the close proximity of the proteins being studied, are not an actual assessment of the physical contact between these proteins.
BiFC needs a longer maturation time, has an irreversible nature of complementation, shows a high degree of self-assembly of the split fragments of the fluorescent protein, and requires molecular oxygen for the maturation of the fluorophore, thereby making it unsuitable for obligate anaerobes. Since maturation is a time-dependent process, the data procured from BiFC are not in real time. The irreversibility of the technique also makes it more difficult to study the dynamics of interactions inside the cell and the influence of drugs on the interaction. Despite these limitations, BiFC is still the only protein complementation technique that gives information about the location of PPIs in living cells. For BiLC, its limitations are the need of a substrate and the rather limited possibility to detect the localization of the interaction.
6. Guidelines to Perform Accurate PCA-Based Assays
A potential but inherent drawback of PCA-based assays is the requirement to fuse fragments derived from fluorescent or luminescent proteins to the GPCR, which inherently may alter the GPCR’s function or cellular localization. Therefore, a list of recommendations for proper implementation of PCA-based assays can be put forward: (1) examine all possible combinations of fusion proteins; (2) verify the functionality and localization of the fused proteins; (3) include proper controls, i.e., non-interacting partners, to control for self-assembly; (4) include a normalization reporter, to allow compensation for differences in transfection efficiencies; (5) preferentially transfect an amount of DNA near to endogenous expression levels of GPCRs, to avoid false positives due to the random collisions or potentially generate stable cell lines; and (6) follow the kinetics of the GPCR interaction (Figure 5).
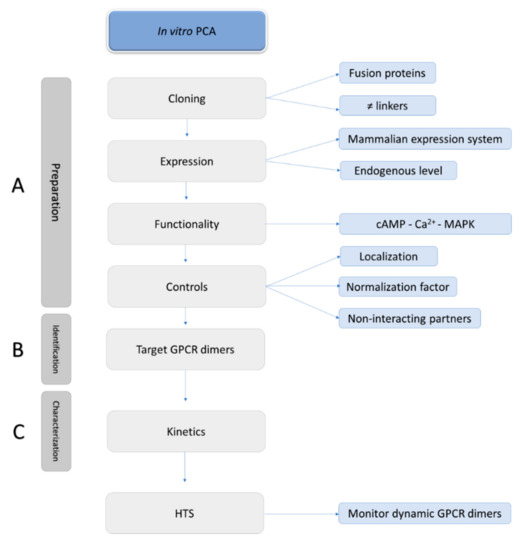
Figure 5.
A guideline for the set-up of an optimized PCA.
6.1. Possible Fusions
The development of PCAs requires multiple rounds of cloning, as one needs to design the most optimal “split” system for each GPCR of interest. Several configurations should be explored, being (i) different reporter systems, (ii) (N- or) C-terminally tagged proteins, and (iii) diverse linkers (i.e., length and composition). While this is a time-consuming step, having a certain workload, it is of paramount importance for the development of a robust, sensitive, and successful complementation assay.
6.1.1. Selection of the Reporter System
A broad variety of fluorescent and luminescent split reporter fragments is available to unravel PPIs. For studying GPCR dimerization, different split reporter systems might be advised, depending on the characteristics of the GPCR dimer of interest (if known). For obligate GPCR dimers, the rules could be less stringent for the choice of the reporter system to be used, whereas for transient GPCR dimers, a dynamic reporter assay with kinetic measurements close to real-time is required. When BiFC is preferred, the truncated YFP fragments YN155 and YC155 are recommended, due to the high signal-to-noise ratio [119]. Venus, a brighter version of GFP, split at 155 or 173, is also commonly used for studying protein interactions. The benefit of using Venus fragments over YFP is that the complementation can be read at 37 °C, thus avoiding the incubation at 30 °C which is necessary when dealing with split-YFP.
When BiLC is preferred, the NanoBiT® system offers advantages in flexibility and in the low affinity of the SmBiT–LgBiT fragments for each other, so dynamic interactions can be monitored in living cells in real time [92].
6.1.2. (N- or) C-Terminally Tagged GPCRs
The site at which the tag is fused to a GPCR plays an important role in the functionality of the GPCR under study and the purpose of the study (Figure 6). All the published research on PCAs with GPCR dimers involves fusion proteins where the split biosensors are tagged to the C-terminal end of GPCRs, so transport and expression at the membrane surface are not hampered. Especially for monitoring interactions of Class C GPCRs, like the metabotropic glutamate receptors, C-terminal tagging of biosensors is recommended, as modification of the long N-terminus of the receptor could lead to deviating outcomes.
Figure 6.
N- or C-terminally split biosensor GPCRs.
On the other hand, split fragments tagged to the N-termini of GPCRs, though not yet implemented for research on GPCR dimerization, might be an interesting tool as well, as these fusion proteins may enable the simultaneous monitoring of GPCR-GPCR interactions and the signaling properties of the dimer. For example, when a split biosensor is fused to the N-terminus of the GPCRs to monitor the GPCR-GPCR interaction, another split biosensor fused to the C-terminus of the GPCR can be applied to monitor G-protein or β-arrestin recruitment.
6.1.3. Linkers
GPCRs of interest and the fragments of the split luminescent or fluorescent partners are usually separated by a linker sequence to assure the flexibility required for the proper folding and “settling” of the biosensor. Multiple linkers with alternating sequences and conformations have been designed, the design of these linkers often being empirical but based on linker sequences derived from natural multi-domain proteins [120].
The most frequently applied linkers in PCAs are composed of non-polar glycine and polar serine amino acids (“GS” linker), developed by Argos [121]. The flexibility of these linkers originates from the incorporation of these small residues. Moreover, the use of serine (or threonine) residues in these linkers contributes to the stability in aqueous conditions by the formation of hydrogen bonds with water molecules [122]. A model linker design involves the (GGGGS)n template, where n represents the copy number. By adjusting this copy number, one might obtain the most optimal “GS” linker length to achieve appropriate separation of the GPCR and the split biosensor. For PCA purposes, often utilized GS linkers comprise roughly eight to fifteen amino acids, such as DGGSGGGS [123], GGGSGGGS [99,124] or GSSGGGGSGGGGSSG [105]. Other -more rigid linkers have also been implemented in PCA, for example ATGLDLELKASNSAVDGTAGPVAT [117]. The proline residues in linkers might increase stiffness to keep fusion moieties at a distance, whereas lysine residues are often added to improve solubility [122]. Furthermore, proper codon usage (i.e., avoiding rare ones) is advised to not compromise expression levels. Also, depending on the expression system, sequences which could represent potential protease recognition sites should be avoided.
Overall, the selection of the linker length is crucial to allow proper folding and accomplish optimal biological activity of the fusion proteins. For fusion proteins, the linker length is an essential feature of the PCA setup since linkers that are too short often result in impaired biological activity due to the inability to accommodate the complementation of the two split proteins, whereas linkers that are too long can create false positives. Therefore, the linker length will vary on a case-by-case basis. With this design in mind, the NanoBiT® system offers different restriction sites in the provided biosensor templates, resulting in various GS linkers, ranging from 15 to 21 amino acids [112]. Some examples of linkers that have been applied in PCA assays to detect GPCR dimers are given in Table 4.
Table 4.
Linker sequences applied in PCA assays for studying GPCR dimerization.
6.2. Functionality and Localization of the Fusion Proteins
Occasionally, the fusion of biosensors to a protein of interest (POI) may interfere with the expression level, activity or function of the latter [126,127]. This may, for example, highly depend on the sequence and length of the linker between the POI and the biosensor (as discussed in Section 6.1). To verify whether the functional integrity of the fusion POI is not affected, several techniques should ideally be implemented, such as ligand binding as well as functional assays (e.g., calcium signaling, cAMP, β-arrestin recruitment, MAPK activation, etc.). An immunofluorescence assay can also be used to confirm correct localization of the recombinant proteins at the plasma membrane.
6.3. Non-Interacting Partners
Studying GPCR–GPCR interactions by complementation assays is relatively straightforward. However, the importance of including appropriate controls (i.e., GPCRs that do not bind to the GPCR of interest) should not be underestimated. It is also essential that the split reporter proteins do not spontaneously associate in the absence of binding partners (that normally drive the complementation). If so, a high number of false positives would be generated [112,128,129]. Ideally, when the site of interaction is known, a mutation at the specific interaction site of the receptor, is performed to evaluate whether this indeed disrupts the interaction. In those cases where the interaction site is unknown, a screening could be performed to elucidate the perfect non-interacting GPCR partner. Negative controls implemented in already published research articles are shown in Table 5 for the BiFC and in Table 6 for BiLC.
Table 5.
An overview of GPCR–GPCR interactions, shown by fluorescence complementation assays.
Table 6.
An overview of GPCR–GPCR interactions, shown by luminescence complementation assays.
6.4. Normalization Factor
To allow better comparison between different experimental setups, it is advisable to include a normalization factor, i.e., a reporter gene such as a fluorescent marker, as previously described [112,147]. In a transient expression system, co-transfection of this fluorescent marker, which should not cause cross-excitation, is recommended. A list of PCAs with corresponding fluorescent markers is given in Table 7. For BiLC in principle, all fluorescent reporters can be used, although it is recommended to avoid cross-excitation by choosing an excitation wavelength more than 20 nm before or after the emission wavelength of the luminescent protein. A potential strategy to cope with alterations in expression levels of the fusion proteins, is the implementation of ratiometric expression systems, consisting of two GPCRs of interest and a fluorescent marker for optical expression control. The so-called ratiometric BiFC (rBiFC) makes it feasible to control the expression through FACS or flow cytometry analysis [147].
Table 7.
The application of normalization factors. * Given as examples.
6.5. Endogenous Expression Levels
In an arbitrary system, PCAs often involve overexpression of split biosensors since these reporter genes are typically introduced in the cells by transfection. To circumvent excessively high levels of membrane expression, which may ultimately result in random collisions rather than a real interaction, low levels of biosensors should ideally be expressed, preferably close to endogenous expression levels. White et al. [148] (2017) have demonstrated that PPIs can be endogenously monitored by BRET when a Nanoluciferase reporter is genetically fused to a natively expressed GPCR of interest by CRISPR/Cas9-mediated homology-directed repair. This methodology also offers the potential to study GPCR–GPCR interactions at endogenous levels.
6.6. Kinetics
To follow the dynamics of GPCR interactions in living cells in real time or to monitor the influence of ligand-dependent modulation on the level of dimerization, one can use a real-time PCA-based method for a certain amount of time. For example, to evaluate the effect of the α-melanocyte-stimulating hormone (α-MSH) on the interaction between MC4R and MRAP2, Habara et al. [113] (2018) measured this interaction with a NanoBiT®-based PCA for up to 120 s before and after the addition of α-MSH. By doing so, the authors could demonstrate that the stimulation of MC4R with α-MSH slightly decreased the NanoBiT® signal, which led to the postulation that the activated structural change of MC4R negatively impacts the interaction with MRAP2. Overall, depending on the half-life of the substrate and the PPI of interest, different timeframes of real-time measurement can be implemented.
7. HTS with Cell-Based PCAs
High-throughput screening (HTS) assays are powerful ways to assess the influence of ligands that affect the protein complexes, thereby providing a method to measure spatial and temporal changes in the protein association in response to drugs (Figure 7). This section will focus on the potential use of HTS assays based on BiLC and BiFC for GPCRs.
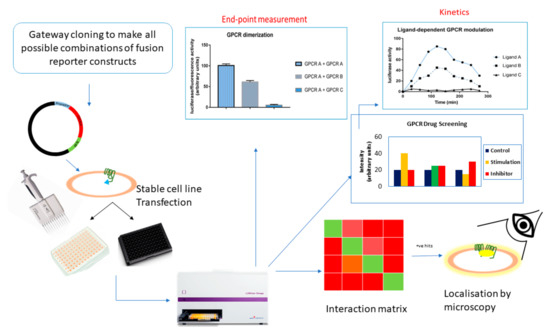
Figure 7.
High-throughput screening.
7.1. GPCR Oligomerization Screening
Given that the protein interaction partners are known, PCAs based on BiLC and BiFC could aid in studying the strength of the interaction compared to a negative control. To find new (unknown) interactions, a screening assay would be handy, in which case the bait protein is fused to a split luminescent/fluorescent fragment and screened against a cDNA library fused to the other half of the reporter fragment. This way, one can identify which GPCRs interact with each other. In conjunction to this is the functional validation of the detected interactions. False readouts can be sidelined upon employment of fusion proteins with mutations at the site of interaction, which consequently inhibit the interaction and decrease the level of complementation of the luminescent or fluorescent fragments. Clearly, this is only feasible if the interaction sites are known, which may be unraveled by alanine screening. For such screening assays, a robust plate reader format would be the best choice.
A non-GPCR example of such a workflow was published by Zych et al. in 2013 [149]. These authors established a high-throughput imaging based-screening that was based on the principles of BiFC, with as the aim the study of the dynamics of Vpr (a nonstructural protein encoded by HIV-1) dimerization.
Screening based on BiFC coupled to flow cytometry is a fast way to capture weak interactions, and hence, this has been used to screen for mutations that modulate the affinity and specificity of PPIs. For example, in 2013, Morell et al. [150] coupled flow cytometry to BiFC to study the weak interaction between the SH3 domain of C-Abl kinase and its natural or mutated binding partners. This combination helps to select for good interacting partners, even if they were deficient in the whole cell population. Hence, this combination has proven to be fast, specific, and sensitive and can capture even weak, transient interactions, thus opening up a new dimension in the field of proteomics.
7.2. GPCR Drug Discovery
Since GPCR–GPCR interactions have been implied in several disease patterns, e.g., neurodegenerative disorders, a strong interest in compounds which could interfere with or alter these GPCR–GPCR interactions has arisen. Monitoring these GPCR–GPCR interactions has the potential of unveiling differences in interaction specifics, such as dynamics, as well as identifying potential therapeutic agents.
For an in vitro HTS assay, PCAs can be used to screen for compounds that inhibit (BiLC) or enhance (BiLC and BiFC) the PPIs. Upon addition of the modulating compound, the recorded signal changes, depending on its potential to influence the GPCR–GPCR interaction. Counter screens should be implemented to negate the possibility of an artefact, such as compounds which possibly interfere with reporter complementation, show autofluorescence, scatter light, quench luminescence or have toxic side-effects [151,152,153]. A key advantage of PCA-based approaches is that these include formats using living cells, so GPCR receptors are in their native environment.
Overall, this type of HTS strategy would be of great help to further enhance the interest in GPCR drug-discovery research.
8. In Vivo Application
Besides the potential as HTS assays, in recent years, BiFC and BiLC complementation techniques have also been applied in vivo, aiming at a better understanding of the dynamics of homo- or heterodimers in living animals. Several studies established an imaging reporter strategy, which can monitor the specific pharmacological regulation of oligomer complexes.
For GPCR dimerization purposes, Luker et al. [98] (2009) applied the FLuc-based PCA (NFLuc-416 and CFLuc-398) to investigate the homo- and heterodimerization of CXCR4 and CXCR7 in a tumor xenograft model of breast cancer. Type 231 cells stably expressing CXCR4-NFLuc and CXCR4-CFLuc or CXCR7-NFLuc and CXCR7-CFLuc were injected bilaterally into 6 to 8 weeks-old female nude mice. To test whether treatment with chemokine ligands produced time- and dose-dependent changes in reporter signal due to the alterations in the level of dimerization, fibroblasts stably expressing the chemokine CXCL12 were co-implanted. This chemokine reduced the level of both CXCR4 homodimerization and CXCR4 − CXCR7 heterodimerization. Blocking the binding of CXCL12 to CXCR4 resulted in a time-dependent increase in the level of CXCR4 homodimer formation, but no increase in the CXCR4 − CXCR7 heterodimerization was detected. In another setup, mice were injected with 231 cells stably expressing CXCR4 or CXCR7 homodimers and with fibroblasts only transduced with GFP. After 1 h of treatment with the CXCR7 modulator CCX754 (100 mg/kg subcutaneous), an increase in CXCR7 homodimer formation, but not CXCR4 homodimer formation, was detected. Overall, it was observed that the chemokine CXCL12 decreased the level of CXCR4 homodimerization and CXCR4 − CXCR7 heterodimerization and the CXCR7 modulator CCX754 increased the level of CXCR7 homodimerization. The application of such newly designed drugs that specifically modify the levels of certain dimers could be beneficial in the treatment of not only breast cancer, but also in rheumatoid arthritis, HIV disease, lung and prostate cancers, where these GPCRs play an important role as well.
Rather than trying to bridge the gap between cell culture studies and in vivo physiology by the injection of cells stably expressing GPCRs fused to the split biosensors, use of the CRISPR/Cas9 methodology (previously described in Section 6.5) to fuse native receptors to split protein fragments would allow the detection of dimerization at endogenous protein levels. This has already been successfully implemented for the β-arrestin2 recruitment to CXCR4 [148], thus, it could likewise be tested for GPCR dimerization.
Most importantly, it has been shown that the bioluminescent reporter is sensitive at all the depths and locations within a live mouse [154], thus making this one of the best platforms to study PPIs as it better mimics a “real-life scenario”.
In the field of fluorescence, Han et al. [155] in 2014 applied BiFC based on split fragments of mNeptune, a far-red monomeric Neptune variant (600 nm/650 nm), for in vivo applications. The main obstacle of imaging in live animals, being the tissue opacity to excitation light below 600 nm, was overcome by this fluorescent protein. This system was tested by imaging the interaction between bFos and bJun in live mice and could also serve to unravel GPCR–GPCR interactions.
Finally, luminescence- or fluorescence-based systems can be applied to study GPCR oligomerization in vivo as these assays allow real-time detection of the interaction and subsequently would help in the screening of pharmacological compounds acting on these oligomers. These non-invasive animal imaging assays, for the quantification of GPCR–GPCR interactions, might aid in the transformation of treatments targeting specific oligomers (e.g., small molecules or bivalent ligands) from cell-based assays to clinical trials.
9. Conclusions
GPCRs are highly dynamic proteins that are subject to multiple spatiotemporal interactions inside the cell to trigger appropriate cellular responses. These interactions involve transducers, kinases, as well as other GPCRs, among others [156,157,158]. While great progress has been made during the past decade in targeting and understanding GPCR–GPCR interactions, some key questions remain unanswered. Although increasing evidence supports the existence of GPCR–GPCR interactions in vitro and in vivo [159], the sophisticated characteristics of these interactions, such as their variation in dynamics, complexity (i.e., dimerization or higher-order oligomerization), binding affinities, and the lack of a consensus sequence in the interfaces, make GPCR–GPCR interactions a challenging research field. Multiple studies based on PCA techniques have shed new light on the composition, localization, and even ligand-induced modulation of these GPCR complexes. Moreover, the potential of these PCA-based methodologies has been further explored by their combination in a variety of ways to investigate not only binary but also ternary or even quaternary partnerships. The current review describes the journey taken by these BiLC- and BiFC-based PCAs and the GPCR–GPCR interactions that have been identified by applying these techniques. PCAs are straightforward and flexible, and provide on the one hand a convenient approach to visualize the oligomerization processes in living cells and on the other hand an ideal tool to monitor the interaction dynamics. Nevertheless, certain milestones could still be achieved, such as the development of reversible split-fluorescent proteins or the discovery of split-luminescent proteins and their respective ligands that allow enhanced imaging of the GPCR–GPCR interactions. Finally, we anticipate a growing interest in the application of PCA-based high-throughput drug screenings, which makes it possible to identify compounds that selectively modulate the composition of GPCR oligomers in living cells.
Funding
This research was funded by IWT/SBO, grant number 140028 and BOF.DCV.20—BOF15/DOS/021 of Ghent University.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| A1/A2A | Adenosine receptor type 1/type 2A |
| α1b | α1b adrenergic receptor |
| AT1/AT2 | Angiotensin II receptor type 1/type 2 |
| β2AR | β2 Adrenergic receptor |
| BiFC | Bimolecular fluorescence complementation assay |
| BiLC | Bimolecular luminescence complementation assay |
| BRET | Bioluminescence resonance energy transfer |
| CAD | Cath.-a-differentiated |
| cAMP | Cyclic adenosine monophosphate |
| CB1 | Cannabinoid receptor 1 |
| CC | C-terminal fragment of split Cerulean |
| CFP | Cerulean fluorescent protein |
| CC2 | C-C chemokine receptor type 2 |
| CFLuc | C-terminal fragment of split Firefly luciferase |
| CGFP | C-terminal fragment of split green fluorescent protein |
| CID | Chemically induced dimerization |
| CN | N-terminal fragment of split Cerulean |
| CODA-RET | Complemented donor–acceptor resonance energy transfer |
| co-IP | Co-immunoprecipitation |
| CPA | N6-cyclopentyladenosine |
| CXCR4/CXCR7 | C-X-C chemokine receptor type 4/type 7 |
| D1R/D2R/D3R | Dopamine receptor type 1/type 2/type 3 |
| D2LR | Dopamine receptor type 2 long isoform |
| D2SR | Dopamine receptor type 2 short isoform |
| DsRed | Discosoma Red |
| ECL | Extracellular loop |
| EGFP | Enhanced green fluorescent protein |
| FLuc | Firefly Luciferase |
| FFAR2/3 | Free fatty acid receptor type 2/type 3 |
| FRB | FKBP-rapamycin binding domain of mTOR |
| FKBP12 | FK506- and rapamycin-binding protein |
| FRET | Förster resonance energy transfer |
| GHSR1a | Growth hormone secretagogue receptor 1α |
| GLuc | Gaussia princeps luciferase |
| GPCR | G protein-coupled receptor |
| GRK | G protein-coupled receptor kinase |
| HEK | Human embryonic kidney |
| Homo-FC | Homo-molecular fluorescence complementation |
| 5-HTR | 5-hydroxytryptamine receptor |
| HTS | High-throughput screening |
| L-DOPA | L-3,4-dihydroxyphenylalanine |
| LcBiT | C-terminal fragment of split large BiT |
| LgBiT | Large BiT, large subunit of Nanoluciferase |
| LnBiT | N-terminal fragment of split large BiT |
| M1/M2/M4 | Muscarinic acetylcholine receptor type 1/type 2/type 4 |
| MAPK | Mitogen activated protein kinase |
| MBiFC | Multicolor Bimolecular fluorescence complementation assay |
| MC4R | Melanocortin 4 receptor |
| MOP | Mu-opioid receptor |
| MRAP2 | Melanocortin 2 receptor accessory protein 2 |
| α-MSH | α-Melanocyte- stimulating hormone |
| mGluR2/mGLuR5/mGluR6 | Metabotropic glutamate receptor type 2/type 5/type 6 |
| mRFP | Monomer red fluorescent protein |
| NanoBiT® | NanoLuc Binary Technology® |
| NFLuc | N-terminal fragment of split Firefly luciferase |
| NGFP | N-terminal fragment of split green fluorescent protein |
| NPFF2 | Neuropeptide FF receptor 2 |
| NPY | Neuropeptide Y |
| OX1R | Orexin 1 receptor |
| PCA | Protein complementation assay |
| PLA | Proximity ligation assay |
| PPI | Protein–protein interaction |
| P2RY1 | P2Y purinoceptor 1 |
| RLuc | Renilla Luciferase |
| SDS-PAGE | Sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| SH3 | SRC Homology 3 Domain |
| SmBiT | Small BiT, small subunit of Nanoluciferase |
| SpIDA | Spatial intensity distribution analysis |
| SRET | Sequential resonance energy transfer |
| TAT | Transactivator of transcription |
| TM | Transmembrane |
| TSHr | Thyrotropin receptor |
| VC | C-terminal fragment of split Venus |
| VN | N-terminal fragment of split Venus |
| YC | C-terminal fragment of split yellow fluorescent protein |
| YFP | Yellow fluorescent protein |
| YN | N-terminal fragment of split yellow fluorescent protein |
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Foord, S.M.; Bonner, T.I.; Neubig, R.R.; Rosser, E.M.; Pin, J.P.; Davenport, A.P.; Spedding, M.; Harmar, A.J. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol. Rev. 2005, 57, 279–288. [Google Scholar] [CrossRef]
- Bjarnadottir, T.K.; Gloriam, D.E.; Hellstrand, S.H.; Kristiansson, H.; Fredriksson, R.; Schioth, H.B. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 2006, 88, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Resina, I.; Aguinaga, D.; Labandeira-Garcia, J.L.; Lanciego, J.L.; Navarro, G.; Franco, R. Usefulness of identifying G-protein-coupled receptor dimers for diagnosis and therapy of neurodegenerative diseases and of gliomas. Histol. Histopathol. 2018, 11963. [Google Scholar] [CrossRef]
- Zhao, J.; Deng, Y.; Jiang, Z.; Qing, H. G Protein-Coupled Receptors (GPCRs) in Alzheimer’s Disease: A Focus on BACE1 Related GPCRs. Front. Aging Neurosci. 2016, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Ziegler, M.E.; Kim, H.B.; Evans, S.J.; Choudary, P.V.; Li, J.Z.; Meng, F.; Dai, M.; Myers, R.M.; Neal, C.R.; et al. G protein-linked signaling pathways in bipolar and major depressive disorders. Front. Genet. 2013, 4, 297. [Google Scholar] [CrossRef]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-Coupled Receptors in Cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef]
- Nieto Gutierrez, A.; McDonald, P.H. GPCRs: Emerging anti-cancer drug targets. Cell. Signal. 2018, 41, 65–74. [Google Scholar] [CrossRef]
- Huang, Y.H.; Todd, N.; Thathiah, A. The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2017, 32, 96–110. [Google Scholar] [CrossRef]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41.e19–54.e19. [Google Scholar] [CrossRef] [PubMed]
- Waldhoer, M.; Fong, J.; Jones, R.M.; Lunzer, M.M.; Sharma, S.K.; Kostenis, E.; Portoghese, P.S.; Whistler, J.L. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. USA 2005, 102, 9050–9055. [Google Scholar] [CrossRef] [PubMed]
- Vinals, X.; Moreno, E.; Lanfumey, L.; Cordomi, A.; Pastor, A.; de La Torre, R.; Gasperini, P.; Navarro, G.; Howell, L.A.; Pardo, L.; et al. Cognitive Impairment Induced by Delta9-tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors. PLoS Biol. 2015, 13, e1002194. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.C.; Vasudevan, L.; Huysentruyt, J.; Risseeuw, M.D.P.; Stove, C.; Vanderheyden, P.M.L.; Van Craenenbroeck, K.; Van Calenbergh, S. Design, Synthesis, and Biological Evaluation of Bivalent Ligands Targeting Dopamine D-2-Like Receptors and the -Opioid Receptor. Chemmedchem 2018, 13, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Wouters, E.; Dalton, J.A.R.; Risseeuw, M.D.P.; Crans, R.A.J.; Stove, C.; Giraldo, J.; Van Craenenbroeck, K.; Van Calenbergh, S. Synthesis toward Bivalent Ligands for the Dopamine D2 and Metabotropic Glutamate 5 Receptors. J. Med. Chem. 2018, 61, 8212–8225. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, K.; Rual, J.F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.F.; Goh, K.I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef] [PubMed]
- AbdAlla, S.; Lother, H.; el Massiery, A.; Quitterer, U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat. Med. 2001, 7, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Dopamine D2 receptors as treatment targets in schizophrenia. Clin. Schizophr. Relat. Psychoses 2010, 4, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Renner, U.; Zeug, A.; Woehler, A.; Niebert, M.; Dityatev, A.; Dityateva, G.; Gorinski, N.; Guseva, D.; Abdel-Galil, D.; Frohlich, M.; et al. Heterodimerization of serotonin receptors 5-HT1A and 5-HT7 differentially regulates receptor signalling and trafficking. J. Cell Sci. 2012, 125, 2486–2499. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Romero-Fernandez, W.; Perez-Alea, M.; Narvaez, M.; Tarakanov, A.O.; Mudo, G.; Agnati, L.F.; Ciruela, F.; Belluardo, N.; Fuxe, K. The existence of FGFR1-5-HT1A receptor heterocomplexes in midbrain 5-HT neurons of the rat: relevance for neuroplasticity. J. Neurosci. 2012, 32, 6295–6303. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Corrales, F.; Narvaez, M.; Oflijan, J.; Agnati, L.F.; Palkovits, M.; Fuxe, K. Dynamic modulation of FGFR1-5-HT1A heteroreceptor complexes. Agonist treatment enhances participation of FGFR1 and 5-HT1A homodimers and recruitment of beta-arrestin2. Biochem. Biophys. Res. Commun. 2013, 441, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Perez-Alea, M.; Narvaez, M.; Tarakanov, A.O.; Mudo, G.; Jimenez-Beristain, A.; Agnati, L.F.; Ciruela, F.; Belluardo, N.; Fuxe, K. Enhancement of the FGFR1 signaling in the FGFR1-5-HT1A heteroreceptor complex in midbrain raphe 5-HT neuron systems. Relevance for neuroplasticity and depression. Biochem. Biophys. Res. Commun. 2015, 463, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Tarakanov, A.O.; Fuxe, K. FGFR1-5-HT1A Heteroreceptor Complexes: Implications for Understanding and Treating Major Depression. Trends Neurosci. 2016, 39, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Millon, C.; Flores-Burgess, A.; Narvaez, M.; Borroto-Escuela, D.O.; Santin, L.; Parrado, C.; Narvaez, J.A.; Fuxe, K.; Diaz-Cabiale, Z. A role for galanin N-terminal fragment (1-15) in anxiety- and depression-related behaviors in rats. Int. J. Neuropsychopharmacol. 2014, 18, pyu064. [Google Scholar] [CrossRef] [PubMed]
- Millon, C.; Flores-Burgess, A.; Narvaez, M.; Borroto-Escuela, D.O.; Santin, L.; Gago, B.; Narvaez, J.A.; Fuxe, K.; Diaz-Cabiale, Z. Galanin (1-15) enhances the antidepressant effects of the 5-HT1A receptor agonist 8-OH-DPAT: involvement of the raphe-hippocampal 5-HT neuron system. Brain Struct. Funct. 2016, 221, 4491–4504. [Google Scholar] [CrossRef] [PubMed]
- Rondard, P.; Huang, S.; Monnier, C.; Tu, H.; Blanchard, B.; Oueslati, N.; Malhaire, F.; Li, Y.; Trinquet, E.; Labesse, G.; et al. Functioning of the dimeric GABA(B) receptor extracellular domain revealed by glycan wedge scanning. EMBO J. 2008, 27, 1321–1332. [Google Scholar] [CrossRef]
- Maggio, R.; Millan, M.J. Dopamine D2-D3 receptor heteromers: Pharmacological properties and therapeutic significance. Curr. Opin. Pharmacol. 2010, 10, 100–107. [Google Scholar] [CrossRef]
- So, C.H.; Varghese, G.; Curley, K.J.; Kong, M.M.; Alijaniaram, M.; Ji, X.; Nguyen, T.; O’Dowd B, F.; George, S.R. D1 and D2 dopamine receptors form heterooligomers and cointernalize after selective activation of either receptor. Mol. Pharmacol. 2005, 68, 568–578. [Google Scholar]
- Petrin, D.; Hebert, T.E. The functional size of GPCRs - monomers, dimers or tetramers? Subcell. Biochem. 2012, 63, 67–81. [Google Scholar]
- Bouvier, M.; Hébert, T.E. CrossTalk proposal: Weighing the evidence for Class A GPCR dimers, the evidence favours dimers. J. Physiol. 2014, 592, 2439–2441. [Google Scholar] [CrossRef] [PubMed]
- Lambert, N.A.; Javitch, J.A. CrossTalk opposing view: Weighing the evidence for class A GPCR dimers, the jury is still out. J. Physiol. 2014, 592, 2443–2445. [Google Scholar] [CrossRef] [PubMed]
- Felce, J.H.; Latty, S.L.; Knox, R.G.; Mattick, S.R.; Lui, Y.; Lee, S.F.; Klenerman, D.; Davis, S.J. Receptor Quaternary Organization Explains G Protein-Coupled Receptor Family Structure. Cell Rep. 2017, 20, 2654–2665. [Google Scholar] [CrossRef] [PubMed]
- Herrick-Davis, K.; Milligan, G.; Di Giovanni, G. G-Protein-Coupled Receptor Dimers; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Marsango, S.; Caltabiano, G.; Jimenez-Roses, M.; Millan, M.J.; Pediani, J.D.; Ward, R.J.; Milligan, G. A Molecular Basis for Selective Antagonist Destabilization of Dopamine D3 Receptor Quaternary Organization. Sci. Rep. 2017, 7, 2134. [Google Scholar] [CrossRef] [PubMed]
- Nenasheva, T.A.; Neary, M.; Mashanov, G.I.; Birdsall, N.J.; Breckenridge, R.A.; Molloy, J.E. Abundance, distribution, mobility and oligomeric state of M(2) muscarinic acetylcholine receptors in live cardiac muscle. J. Mol. Cell. Cardiol. 2013, 57, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hern, J.A.; Baig, A.H.; Mashanov, G.I.; Birdsall, B.; Corrie, J.E.; Lazareno, S.; Molloy, J.E.; Birdsall, N.J. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 2693–2698. [Google Scholar] [CrossRef]
- Kasai, R.S.; Kusumi, A. Single-molecule imaging revealed dynamic GPCR dimerization. Curr. Opin. Cell Biol. 2014, 27, 78–86. [Google Scholar] [CrossRef]
- Kasai, R.S.; Ito, S.V.; Awane, R.M.; Fujiwara, T.K.; Kusumi, A. The Class-A GPCR Dopamine D2 Receptor Forms Transient Dimers Stabilized by Agonists: Detection by Single-Molecule Tracking. Cell Biochem. Biophys. 2018, 76, 29–37. [Google Scholar] [CrossRef]
- Wouters, E.; Marin, A.R.; Dalton, J.A.R.; Giraldo, J.; Stove, C. Distinct Dopamine D(2) Receptor Antagonists Differentially Impact D(2) Receptor Oligomerization. Int. J. Mol. Sci. 2019, 20, 1686. [Google Scholar] [CrossRef]
- Van Craenenbroeck, K. GPCR oligomerization: contribution to receptor biogenesis. Sub-Cell. Biochem. 2012, 63, 43–65. [Google Scholar]
- Baragli, A.; Alturaihi, H.; Watt, H.L.; Abdallah, A.; Kumar, U. Heterooligomerization of human dopamine receptor 2 and somatostatin receptor 2 Co-immunoprecipitation and fluorescence resonance energy transfer analysis. Cell. Signal. 2007, 19, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Suzuki, T.; Kishimoto, Y.; Hirasawa, N. Biochemical assay of G protein-coupled receptor oligomerization: Adenosine A1 and thromboxane A2 receptors form the novel functional hetero-oligomer. Methods Cell Biol. 2013, 117, 213–227. [Google Scholar] [PubMed]
- Skieterska, K.; Duchou, J.; Lintermans, B.; Van Craenenbroeck, K. Detection of G protein-coupled receptor (GPCR) dimerization by coimmunoprecipitation. Methods Cell Biol. 2013, 117, 323–340. [Google Scholar] [PubMed]
- Ciruela, F.; Vilardaga, J.P.; Fernandez-Duenas, V. Lighting up multiprotein complexes: lessons from GPCR oligomerization. Trends Biotechnol. 2010, 28, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Pediani, J.D.; Godin, A.G.; Milligan, G. Regulation of oligomeric organization of the serotonin 5-hydroxytryptamine 2C (5-HT2C) receptor observed by spatial intensity distribution analysis. J. Biol. Chem. 2015, 290, 12844–12857. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Pediani, J.D.; Harikumar, K.G.; Miller, L.J.; Milligan, G. Spatial intensity distribution analysis quantifies the extent and regulation of homodimerization of the secretin receptor. Biochem. J. 2017, 474, 1879–1895. [Google Scholar] [CrossRef] [PubMed]
- Pediani, J.D.; Ward, R.J.; Godin, A.G.; Marsango, S.; Milligan, G. Dynamic Regulation of Quaternary Organization of the M1 Muscarinic Receptor by Subtype-selective Antagonist Drugs. J. Biol. Chem. 2016, 291, 13132–13146. [Google Scholar] [CrossRef]
- Pediani, J.D.; Ward, R.J.; Marsango, S.; Milligan, G. Spatial Intensity Distribution Analysis: Studies of G Protein-Coupled Receptor Oligomerisation. Trends Pharmacol. Sci. 2018, 39, 175–186. [Google Scholar] [CrossRef]
- Wehr, M.C.; Rossner, M.J. Split protein biosensor assays in molecular pharmacological studies. Drug Discov. Today 2016, 21, 415–429. [Google Scholar] [CrossRef]
- Ghosh, I.; Hamilton, A.D.; Regan, L. Antiparallel leucine zipper-directed protein reassembly: Application to the green fluorescent protein. J. Am. Chem. Soc. 2000, 122, 5658–5659. [Google Scholar] [CrossRef]
- Hu, C.D.; Chinenov, Y.; Kerppola, T.K. Visualization of interactions among bZip and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 2002, 9, 789–798. [Google Scholar] [CrossRef]
- Shyu, Y.J.; Liu, H.; Deng, X.H.; Hu, C.D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques 2006, 40, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Cevheroglu, O.; Kumas, G.; Hauser, M.; Becker, J.M.; Son, C.D. The yeast Ste2p G protein-coupled receptor dimerizes on the cell plasma membrane. Bba-Biomembranes 2017, 1859, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Nguyen, H.B.; Pedelacq, J.D.; Koraichi, F.; Chaudhary, A.; Ganguly, K.; Lockard, M.A.; Favre, G.; Terwilliger, T.C.; Waldo, G.S. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci. Rep. 2013, 3, 2854. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001, 276, 29188–29194. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef]
- Vidi, P.A.; Chemel, B.R.; Hu, C.D.; Watts, V.J. Ligand-dependent oligomerization of dopamine D(2) and adenosine A(2A) receptors in living neuronal cells. Mol. Pharmacol. 2008, 74, 544–551. [Google Scholar] [CrossRef]
- Vidi, P.-A.; Chen, J.; Irudayaraj, J.M.K.; Watts, V.J. Adenosine A2A receptors assemble into higher-order oligomers at the plasma membrane. FEBS Lett. 2008, 582, 3985–3990. [Google Scholar] [CrossRef]
- Kilpatrick, L.E.; Humphrys, L.J.; Holliday, N.D. A G protein-coupled receptor dimer imaging assay reveals selectively modified pharmacology of neuropeptide Y Y1/Y5 receptor heterodimers. Mol. Pharmacol. 2015, 87, 718–732. [Google Scholar] [CrossRef]
- Przybyla, J.A.; Watts, V.J. Ligand-induced regulation and localization of cannabinoid CB1 and dopamine D2L receptor heterodimers. J. Pharmacol. Exp. Ther. 2010, 332, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Ang, Z.; Xiong, D.; Wu, M.; Ding, J.L. FFAR2-FFAR3 receptor heteromerization modulates short-chain fatty acid sensing. FASEB J. 2018, 32, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Bai, B.; Ji, B.; Chen, X.; Wang, C.; Wang, P.; Yang, C.; Zhang, R.; Jiang, Y.; Pan, Y.; et al. Ghrelin Through GHSR1a and OX1R Heterodimers Reveals a Galphas-cAMP-cAMP Response Element Binding Protein Signaling Pathway in Vitro. Front. Mol. Neurosci. 2018, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Cordomi, A.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Perez-Benito, L.; Ferre, S.; Cortes, A.; Casado, V.; Mallol, J.; et al. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Navarro, G.; Borroto-Escuela, D.; Seibt, B.F.; Ammon, Y.C.; de Filippo, E.; Danish, A.; Lacher, S.K.; Cervinkova, B.; Rafehi, M.; et al. Adenosine A2A receptor ligand recognition and signaling is blocked by A2B receptors. Oncotarget 2018, 9, 13593–13611. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Jung, Y. Homo-molecular Fluorescence Complementation for Direct Visualization of Receptor Oligomerization in Living Cells. Angew. Chem. (Int. Ed. Engl.) 2019, 58, 2045–2049. [Google Scholar] [CrossRef] [PubMed]
- Vidi, P.A.; Chemel, B.R.; Watts, V.J. Direct visualization of adenosine A(2A) and dopamine D-2L receptor oligomers in a neuronal cell model. FASEB J. 2008, 22. [Google Scholar]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 2000, 97, 11984–11989. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.Y.; Cui, Z.Q.; Wei, H.P.; Zhang, Z.P.; Zhou, Y.F.; Wang, Y.P.; Zhang, X.E. Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein-protein interactions in living cells. Biochem. Biophys. Res. Commun. 2008, 367, 47–53. [Google Scholar] [CrossRef]
- Jach, G.; Pesch, M.; Richter, K.; Frings, S.; Uhrig, J.F. An improved mRFP1 adds red to bimolecular fluorescence complementation. Nat. Methods 2006, 3, 597–600. [Google Scholar] [CrossRef]
- Shcherbo, D.; Merzlyak, E.M.; Chepurnykh, T.V.; Fradkov, A.F.; Ermakova, G.V.; Solovieva, E.A.; Lukyanov, K.A.; Bogdanova, E.A.; Zaraisky, A.G.; Lukyanov, S.; et al. Bright far-red fluorescent protein for whole-body imaging. Nat. Methods 2007, 4, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Zhang, Z.; Zheng, Y.; Yang, J.; Qin, L.; Lu, J.; Huang, Z.L.; Zeng, S.; Luo, Q. A novel far-red bimolecular fluorescence complementation system that allows for efficient visualization of protein interactions under physiological conditions. Biosens. Bioelectron. 2009, 25, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.D.; Kerppola, T.K. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 2003, 21, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Vidi, P.A.; Przybyla, J.A.; Hu, C.D.; Watts, V.J. Visualization of G protein-coupled receptor (GPCR) interactions in living cells using bimolecular fluorescence complementation (BiFC). Curr. Protoc. Neurosci. 2010, 51, 5–29. [Google Scholar] [CrossRef]
- Vidi, P.A.; Watts, V.J. Fluorescent and bioluminescent protein-fragment complementation assays in the study of G protein-coupled receptor oligomerization and signaling. Mol. Pharmacol. 2009, 75, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Rebois, R.V.; Robitaille, M.; Petrin, D.; Zylbergold, P.; Trieu, P.; Hebert, T.E. Combining protein complementation assays with resonance energy transfer to detect multipartner protein complexes in living cells. Methods (San Diego, Calif.) 2008, 45, 214–218. [Google Scholar] [CrossRef]
- Navarro, G.; Carriba, P.; Gandia, J.; Ciruela, F.; Casado, V.; Cortes, A.; Mallol, J.; Canela, E.I.; Lluis, C.; Franco, R. Detection of heteromers formed by cannabinoid CB1, dopamine D2, and adenosine A2A G-protein-coupled receptors by combining bimolecular fluorescence complementation and bioluminescence energy transfer. Sci. World J. 2008, 8, 1088–1097. [Google Scholar] [CrossRef]
- Shyu, Y.J.; Suarez, C.D.; Hu, C.D. Visualization of AP-1 NF-kappaB ternary complexes in living cells by using a BiFC-based FRET. Proc. Natl. Acad. Sci. USA 2008, 105, 151–156. [Google Scholar] [CrossRef]
- Bagher, A.M.; Laprairie, R.B.; Toguri, J.T.; Kelly, M.E.M.; Denovan-Wright, E.M. Bidirectional allosteric interactions between cannabinoid receptor 1 (CB1) and dopamine receptor 2 long (D2L) heterotetramers. Eur. J. Pharmacol. 2017, 813, 66–83. [Google Scholar] [CrossRef]
- Spencer, D.M.; Wandless, T.J.; Schreiber, S.L.; Crabtree, G.R. Controlling Signal-Transduction with Synthetic Ligands. Science 1993, 262, 1019–1024. [Google Scholar] [CrossRef]
- Cao, X.; Liang, L.; Hadcock, J.R.; Iredale, P.A.; Griffith, D.A.; Menniti, F.S.; Factor, S.; Greenamyre, J.T.; Papa, S.M. Blockade of cannabinoid type 1 receptors augments the antiparkinsonian action of levodopa without affecting dyskinesias in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated rhesus monkeys. J. Pharmacol. Exp. Ther. 2007, 323, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Wood, K.V. Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 2007, 5, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Smith, M.C.; Luker, G.D.; Gammon, S.T.; Piwnica-Worms, H.; Piwnica-Worms, D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. USA 2004, 101, 12288–12293. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Umezawa, Y.; Gambhir, S.S. Noninvasive imaging of protein-protein interactions in living subjects by using reporter protein complementation and reconstitution strategies. Proc. Natl. Acad. Sci. USA 2002, 99, 15608–15613. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Gambhir, S.S. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal. Chem. 2003, 75, 1584–1589. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Massoud, T.F.; Huang, J.; Gambhir, S.S. Molecular imaging of drug-modulated protein-protein interactions in living subjects. Cancer Res. 2004, 64, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Remy, I.; Michnick, S.W. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat. Methods 2006, 3, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Bodle, C.R.; Hayes, M.P.; O’Brien, J.B.; Roman, D.L. Development of a bimolecular luminescence complementation assay for RGS: G protein interactions in cells. Anal. Biochem. 2017, 522, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Oh-Hashi, K.; Hirata, Y.; Kiuchi, K. SOD1 dimerization monitoring using a novel split NanoLuc, NanoBit. Cell Biochem. Funct. 2016, 34, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, L.G.; Mattioli, M.; Ricci, F.; Li, Y.C.; Wade, M. Multiplex detection of protein-protein interactions using a next generation luciferase reporter. Biochim. Biophys. Acta 2016, 1863, 284–292. [Google Scholar] [CrossRef]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, A.; Primeau, M.; Trudeau, L.E.; Michnick, S.W. beta-Lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein-protein interactions. Nat. Biotechnol. 2002, 20, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Gambhir, S.S. Novel fusion protein approach for efficient high-throughput screening of small molecule-mediating protein-protein interactions in cells and living animals. Cancer Res. 2005, 65, 7413–7420. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matthews, J.C.; Hori, K.; Cormier, M.J. Substrate and substrate analogue binding properties of Renilla luciferase. Biochemistry US 1977, 16, 5217–5220. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.C.; Hori, K.; Cormier, M.J. Purification and properties of Renilla reniformis luciferase. Biochemistry US 1977, 16, 85–91. [Google Scholar] [CrossRef]
- Loening, A.M.; Fenn, T.D.; Wu, A.M.; Gambhir, S.S. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng. Des. Sel. 2006, 19, 391–400. [Google Scholar] [CrossRef]
- Luker, K.E.; Gupta, M.; Luker, G.D. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009, 23, 823–834. [Google Scholar] [CrossRef]
- Armando, S.; Quoyer, J.; Lukashova, V.; Maiga, A.; Percherancier, Y.; Heveker, N.; Pin, J.P.; Prezeau, L.; Bouvier, M. The chemokine CXC4 and CC2 receptors form homo- and heterooligomers that can engage their signaling G-protein effectors and betaarrestin. FASEB J. 2014, 28, 4509–4523. [Google Scholar] [CrossRef]
- Guo, W.; Urizar, E.; Kralikova, M.; Mobarec, J.C.; Shi, L.; Filizola, M.; Javitch, J.A. Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. 2008, 27, 2293–2304. [Google Scholar] [CrossRef]
- Casado-Anguera, V.; Bonaventura, J.; Moreno, E.; Navarro, G.; Cortes, A.; Ferre, S.; Casado, V. Evidence for the heterotetrameric structure of the adenosine A2A-dopamine D2 receptor complex. Biochem. Soc. Trans. 2016, 44, 595–600. [Google Scholar] [CrossRef]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- England, C.G.; Ehlerding, E.B.; Cai, W. NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence. Bioconjug. Chem. 2016, 27, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Cannaert, A.; Franz, F.; Auwarter, V.; Stove, C.P. Activity-Based Detection of Consumption of Synthetic Cannabinoids in Authentic Urine Samples Using a Stable Cannabinoid Reporter System. Anal. Chem. 2017, 89, 9527–9536. [Google Scholar] [CrossRef] [PubMed]
- Cannaert, A.; Storme, J.; Franz, F.; Auwarter, V.; Stove, C.P. Detection and Activity Profiling of Synthetic Cannabinoids and Their Metabolites with a Newly Developed Bioassay. Anal. Chem. 2016, 88, 11476–11485. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, N.; Laschet, C.; Franssen, D.; Szpakowska, M.; Gilissen, J.; Geubelle, P.; Soni, A.; Parent, A.S.; Pirotte, B.; Chevigne, A.; et al. Activation of the Orphan G Protein-Coupled Receptor GPR27 by Surrogate Ligands Promotes beta-Arrestin 2 Recruitment. Mol. Pharmacol. 2017, 91, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.; Cannaert, A.; Linnet, K.; Stove, C.P. Application of an activity-based receptor bioassay to investigate the in vitro activity of selected indole- and indazole-3-carboxamide-based synthetic cannabinoids at CB1 and CB2 receptors. Drug Test. Anal. 2019, 11, 501–511. [Google Scholar] [CrossRef]
- Storme, J.; Cannaert, A.; Van Craenenbroeck, K.; Stove, C.P. Molecular dissection of the human A3 adenosine receptor coupling with beta-arrestin2. Biochem. Pharmacol. 2018, 148, 298–307. [Google Scholar] [CrossRef]
- Wouters, E.; Mogler, L.; Cannaert, A.; Auwärter, V.; Stove, C. Functional evaluation of carboxy metabolites of synthetic cannabinoid receptor agonists featuring scaffolds based on L-valine or L-tert-leucine. Drug Test. Anal. 2019. [Google Scholar] [CrossRef]
- Cannaert, A.; Vasudevan, L.; Friscia, M.; Mohr, A.L.A.; Wille, S.M.R.; Stove, C.P. Activity-Based Concept to Screen Biological Matrices for Opiates and (Synthetic) Opioids. Clin. Chem. 2018, 64, 1221–1229. [Google Scholar] [CrossRef]
- Laschet, C.; Dupuis, N.; Hanson, J. A dynamic and screening-compatible nanoluciferase-based complementation assay enables profiling of individual GPCR-G protein interactions. J. Biol. Chem. 2018. [Google Scholar] [CrossRef]
- Wouters, E.; Vasudevan, L.; Ciruela, F.; Saini, D.K.; Stove, C.; Van Craenenbroeck, K. Assessing GPCR Dimerization in Living Cells: Comparison of the NanoBiT Assay with Related Bioluminescence- and Fluorescence-Based Approaches. In Receptor-Receptor Interactions in the Central Nervous System; Fuxe, K., Borroto-Escuela, D.O., Eds.; Springer: New York, NY, USA, 2018; pp. 239–250. [Google Scholar] [CrossRef]
- Habara, M.; Mori, N.; Okada, Y.; Kawasumi, K.; Nakao, N.; Tanaka, Y.; Arai, T.; Yamamoto, I. Molecular characterization of feline melanocortin 4 receptor and melanocortin 2 receptor accessory protein 2. Gen. Comp. Endocrinol. 2018, 261, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.S.; Kim, S.J.; Baumgartner, B.K.; Krippner, S.; Owen, S.C. A Tri-part Protein Complementation System Using AntibodySmall Peptide Fusions Enables Homogeneous Immunoassays. Sci. Rep.-UK 2017, 7, 8186. [Google Scholar] [CrossRef] [PubMed]
- Ohmuro-Matsuyama, Y.; Ueda, H. Homogeneous Noncompetitive Luminescent Immunodetection of Small Molecules by Ternary Protein Fragment Complementation. Anal. Chem. 2018, 90, 3001–3004. [Google Scholar] [CrossRef] [PubMed]
- Moustaqil, M.; Bhumkar, A.; Gonzalez, L.; Raoul, L.; Hunter, D.J.B.; Carrive, P.; Sierecki, E.; Gambin, Y. A Split-Luciferase Reporter Recognizing GFP and mCherry Tags to Facilitate Studies of Protein-Protein Interactions. Int. J. Mol. Sci. 2017, 18, 2681. [Google Scholar] [CrossRef] [PubMed]
- Sahlholm, K.; Gomez-Soler, M.; Valle-Leon, M.; Lopez-Cano, M.; Taura, J.J.; Ciruela, F.; Fernandez-Duenas, V. Antipsychotic-Like Efficacy of Dopamine D2 Receptor-Biased Ligands is Dependent on Adenosine A2A Receptor Expression. Mol. Neurobiol. 2018, 55, 4952–4958. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, J.; Navarro, G.; Casado-Anguera, V.; Azdad, K.; Rea, W.; Moreno, E.; Brugarolas, M.; Mallol, J.; Canela, E.I.; Lluis, C.; et al. Allosteric interactions between agonists and antagonists within the adenosine A2A receptor-dopamine D2 receptor heterotetramer. Proc. Natl. Acad. Sci. USA 2015, 112, E3609–E3618. [Google Scholar] [CrossRef] [PubMed]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar] [CrossRef]
- Gokhale, R.S.; Khosla, C. Role of linkers in communication between protein modules. Curr. Opin. Chem. Biol. 2000, 4, 22–27. [Google Scholar] [CrossRef]
- Argos, P. An investigation of oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J. Mol. Biol. 1990, 211, 943–958. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef]
- Carayon, K.; Mouledous, L.; Combedazou, A.; Mazeres, S.; Haanappel, E.; Salome, L.; Mollereau, C. Heterologous regulation of Mu-opioid (MOP) receptor mobility in the membrane of SH-SY5Y cells. J. Biol. Chem. 2014, 289, 28697–28706. [Google Scholar] [CrossRef] [PubMed]
- Stefan, E.; Aquin, S.; Berger, N.; Landry, C.R.; Nyfeler, B.; Bouvier, M.; Michnick, S.W. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 16916–16921. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Pfleger, K.D.; Seeber, R.M.; Qian, H.; Oro, C.; Abogadie, F.; Delbridge, L.M.; Thomas, W.G. Heteromerization of angiotensin receptors changes trafficking and arrestin recruitment profiles. Cell. Signal. 2011, 23, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, S.; Weise, A.; Walsh, R.P.; Ward, J.M.; Frommer, W.B. Fusion to GFP blocks intercellular trafficking of the sucrose transporter SUT1 leading to accumulation in companion cells. BMC Plant Biol. 2003, 3, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meyer, E.; Fromherz, P. Ca2+ activation of hSlo K+ channel is suppressed by N-terminal GFP tag. Eur. J. Neurosci. 1999, 11, 1105–1108. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol 2005, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Ventura, S.; Aviles, F.X. Protein complementation assays: approaches for the in vivo analysis of protein interactions. FEBS Lett. 2009, 583, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Cabello, N.; Gandia, J.; Bertarelli, D.C.; Watanabe, M.; Lluis, C.; Franco, R.; Ferre, S.; Lujan, R.; Ciruela, F. Metabotropic glutamate type 5, dopamine D2 and adenosine A2a receptors form higher-order oligomers in living cells. J. Neurochem. 2009, 109, 1497–1507. [Google Scholar] [CrossRef]
- Wang, M.; Pei, L.; Fletcher, P.J.; Kapur, S.; Seeman, P.; Liu, F. Schizophrenia, amphetamine-induced sensitized state and acute amphetamine exposure all show a common alteration: increased dopamine D2 receptor dimerization. Mol. Brain 2010, 3, 25. [Google Scholar] [CrossRef]
- Zawarynski, P.; Tallerico, T.; Seeman, P.; Lee, S.P.; O’Dowd, B.F.; George, S.R. Dopamine D2 receptor dimers in human and rat brain. FEBS Lett. 1998, 441, 383–386. [Google Scholar] [CrossRef]
- Bonaventura, J.; Rico, A.J.; Moreno, E.; Sierra, S.; Sanchez, M.; Luquin, N.; Farre, D.; Muller, C.E.; Martinez-Pinilla, E.; Cortes, A.; et al. L-DOPA-treatment in primates disrupts the expression of A(2A) adenosine-CB1 cannabinoid-D-2 dopamine receptor heteromers in the caudate nucleus. Neuropharmacology 2014, 79, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.S.; Weaver, D.R.; Rivkees, S.A.; Peterfreund, R.A.; Pollack, A.E.; Adler, E.M.; Reppert, S.M. Molecular-Cloning of the Rat Adenosine-A2 Receptor—Selective Coexpression with D2-Dopamine Receptors in Rat Striatum. Mol. Brain Res. 1992, 14, 186–195. [Google Scholar] [CrossRef]
- Fuxe, K.; Ferre, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A(2A) and dopamine D-2 heteromeric receptor complexes and their function. J. Mol. Neurosci. 2005, 26, 209–219. [Google Scholar] [CrossRef]
- Hillion, J.; Canals, M.; Torvinen, M.; Casado, V.; Scott, R.; Terasmaa, A.; Hansson, A.; Watson, S.; Olah, M.E.; Mallol, J.; et al. Coaggregation, cointernalization, and codesensitization of adenosine A(2A) receptors and dopamine D-2 receptors. J. Biol. Chem. 2002, 277, 18091–18097. [Google Scholar] [CrossRef] [PubMed]
- Soriano, A.; Ventura, R.; Molero, A.; Hoen, R.; Casado, V.; Cortes, A.; Fanelli, F.; Albericio, F.; Lluis, C.; Franco, R.; et al. Adenosine A(2A) Receptor-Antagonist/Dopamine D-2 Receptor-Agonist Bivalent Ligands as Pharmacological Tools to Detect A(2A)-D-2 Receptor Heteromers. J. Med. Chem. 2009, 52, 5590–5602. [Google Scholar] [CrossRef]
- Riggleman, A.; Harvey, J.; Baylis, C. Endothelin mediates some of the renal actions of acutely administered angiotensin II. Hypertension 2001, 38, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.Y.; Hopfer, U.; Asico, L.D.; Eisner, G.M.; Felder, R.A.; Jose, P.A. Altered AT(1) receptor regulation of ETB receptors in renal proximal tubule cells of spontaneously hypertensive rats. Hypertension 2005, 46, 926–931. [Google Scholar] [CrossRef]
- Zeng, C.Y.; Wang, Z.; Asico, L.D.; Hopfer, U.; Eisner, G.M.; Felder, R.A.; Jose, P.A. Aberrant ETB receptor regulation of AT(1) receptors in immortalized renal proximal tubule cells of spontaneously hypertensive rats. Kidney Int. 2005, 68, 623–631. [Google Scholar] [CrossRef]
- Ciruela, F.; Casado, V.; Rodrigues, R.J.; Lujan, R.; Burgueno, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A(1)-A(2A) receptor heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef]
- Moreno, J.L.; Miranda-Azpiazu, P.; Garcia-Bea, A.; Younkin, J.; Cui, M.; Kozlenkov, A.; Ben-Ezra, A.; Voloudakis, G.; Fakira, A.K.; Baki, L.; et al. Allosteric signaling through an mGlu2 and 5-HT2A heteromeric receptor complex and its potential contribution to schizophrenia. Sci. Signal. 2016, 9, ra5. [Google Scholar] [CrossRef]
- Lopez-Gimenez, J.F.; Canals, M.; Pediani, J.D.; Milligan, G. The alpha(1b)-adrenoceptor exists as a higher-order oligomer: Effective oligomerization is required for receptor maturation, surface delivery, and function. Mol. Pharmacol. 2007, 71, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Frade, J.M.; del Real, G.; Serrano, A.; Hernanz-Falcon, P.; Soriano, S.F.; Vila-Coro, A.J.; de Ana, A.M.; Lucas, P.; Prieto, I.; Martinez-A, C.; et al. Blocking HIV-1 infection via CCR5 and CXCR4 receptors by acting in trans on the CCR2 chemokine receptor. EMBO J. 2004, 23, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Sohy, D.; Parmentier, M.; Springael, J.Y. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J. Biol. Chem. 2007, 282, 30062–30069. [Google Scholar] [CrossRef] [PubMed]
- Sohy, D.; Yano, H.; de Nadai, P.; Urizar, E.; Guillabert, A.; Javitch, J.A.; Parmentier, M.; Springael, J.Y. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the Protean Effects of "Selective" Antagonists. J. Biol. Chem. 2009, 284, 31270–31279. [Google Scholar] [CrossRef] [PubMed]
- Grefen, C.; Blatt, M.R. A 2in1 cloning system enables ratiometric bimolecular fluorescence complementation (rBiFC). BioTechniques 2012, 53, 311–314. [Google Scholar] [CrossRef] [PubMed]
- White, C.W.; Vanyai, H.K.; See, H.B.; Johnstone, E.K.M.; Pfleger, K.D.G. Using nanoBRET and CRISPR/Cas9 to monitor proximity to a genome-edited protein in real-time. Sci. Rep. 2017, 7, 3187. [Google Scholar] [CrossRef] [PubMed]
- Zych, C.; Domling, A.; Ayyavoo, V. Development of a robust cell-based high-throughput screening assay to identify targets of HIV-1 viral protein R dimerization. Drug Des. Dev. Ther. 2013, 7, 403–412. [Google Scholar]
- Morell, M.; Espargaro, A.; Aviles, F.X.; Ventura, S. Detection of transient protein-protein interactions by bimolecular fluorescence complementation: The Abl-SH3 case. Proteomics 2007, 7, 1023–1036. [Google Scholar] [CrossRef]
- Auld, D.S.; Inglese, J. Interferences with Luciferase Reporter Enzymes. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Simeonov, A.; Davis, M.I. Interference with Fluorescence and Absorbance. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Caaveiro, J.M.M., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Wade, M.; Mendez, J.; Coussens, N.P.; Arkin, M.R.; Glicksman, M.A. Inhibition of Protein-Protein Interactions: Cell-Based Assays. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Caaveiro, J.M.M., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2017. [Google Scholar]
- Wu, J.C.; Sundaresan, G.; Iyer, M.; Gambhir, S.S. Noninvasive optical imaging of firefly luciferase reporter gene expression in skeletal muscles of living mice. Mol. Ther. J. Am. Soc. Gene Ther. 2001, 4, 297–306. [Google Scholar] [CrossRef]
- Han, Y.; Wang, S.F.; Zhang, Z.P.; Ma, X.H.; Li, W.; Zhang, X.W.; Deng, J.Y.; Wei, H.P.; Li, Z.Y.; Zhang, X.E.; et al. In vivo imaging of protein-protein and RNA-protein interactions using novel far-red fluorescence complementation systems. Nucleic Acids Res. 2014, 42, e103. [Google Scholar] [CrossRef]
- Faron-Gorecka, A.; Szlachta, M.; Kolasa, M.; Solich, J.; Gorecki, A.; Kusmider, M.; Zurawek, D.; Dziedzicka-Wasylewska, M. Understanding GPCR dimerization. Methods Cell Biol. 2019, 149, 155–178. [Google Scholar] [PubMed]
- Maurice, P.; Guillaume, J.L.; Benleulmi-Chaachoua, A.; Daulat, A.M.; Kamal, M.; Jockers, R. GPCR-interacting proteins, major players of GPCR function. Adv. Pharmacol. (San Diego, Calif.) 2011, 62, 349–380. [Google Scholar]
- Milligan, G.; Ward, R.J.; Marsango, S. GPCR homo-oligomerization. Curr. Opin. Cell Biol. 2018, 57, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Ayoub, M.A.; Fujita, W.; Jaeger, W.C.; Pfleger, K.D.; Devi, L.A. G Protein-Coupled Receptor Heteromers. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 403–425. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).