The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases

Abstract
1. Introduction-What Is Neurodegeneration?
1.1. Aging and Age-Associated Changes in the Brain
1.2. Approaches to Drug Discovery for Neurodegenerative Diseases
1.3. What to Start with
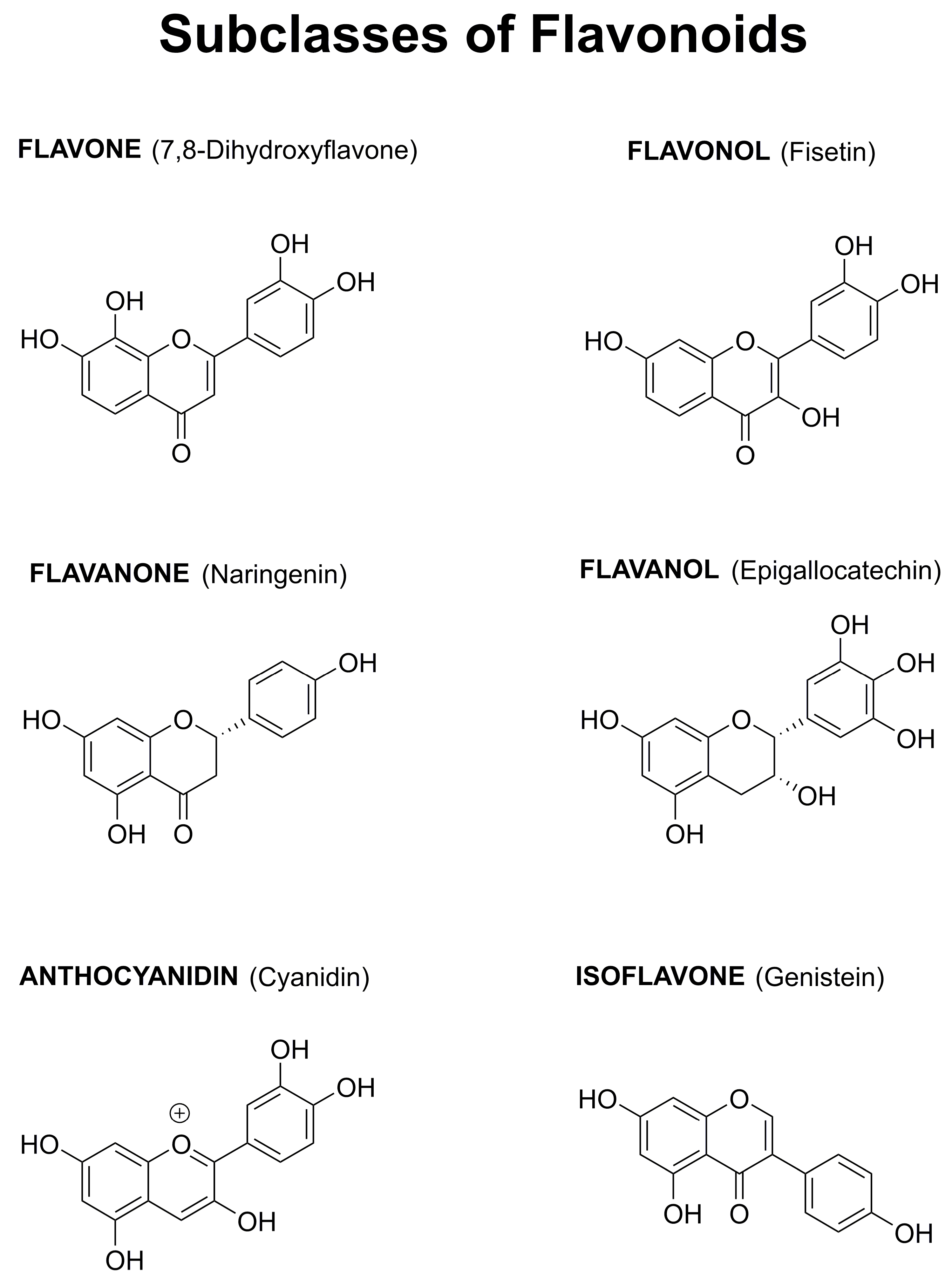
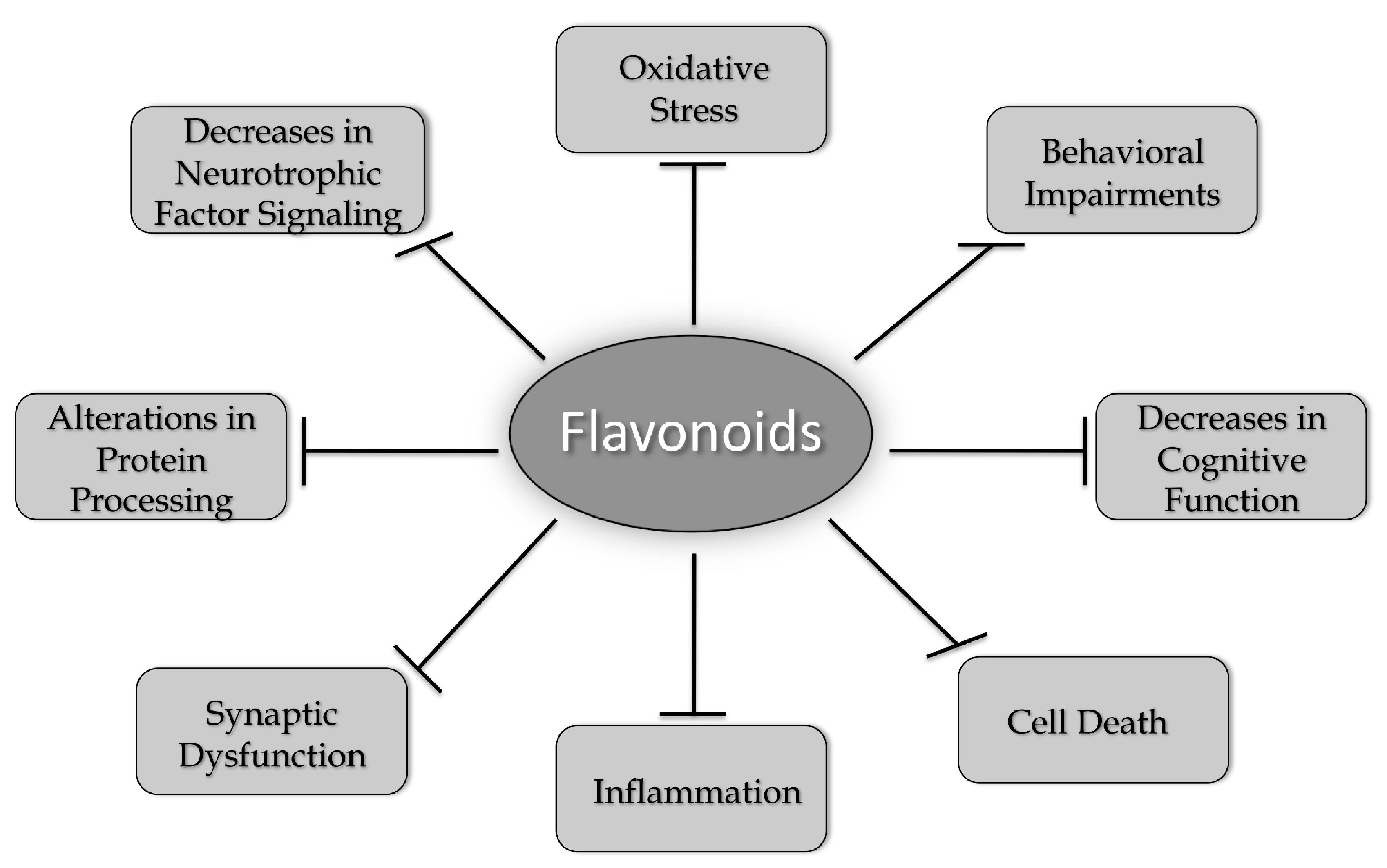
1.4. Why Focus on Flavonoids
1.5. Flavonoids and Alzheimer’s Disease (AD)
1.6. Flavonoids and Parkinson’s Disease (PD)
1.7. Flavonoids and Huntington’s Disease (HD)
1.8. Flavonoids and Amyotrophic Lateral Sclerosis (ALS)
2. Summary and Outlook
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid beta peptide |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| APP | Amyloid precursor protein |
| CNS | Central nervous system |
| CREB | cAMP response element binding protein |
| DA | dopaminergic |
| DHF | dihydroflavone |
| EGCG | (−)-Epigallocatechin gallate |
| ERK | Extracellular signal regulated kinase |
| FAD | Familial Alzheimer’s disease |
| FTD | Fronto-temporal dementia |
| GSH | glutathione |
| HD | Huntington’s disease |
| ip | Intraperitoneal |
| icv | Intracerebroventricular |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| 3-NP | 3-nitropropionic acid |
| 6-OHDA | 6-hydroxydopamine |
| PD | Parkinson’s disease |
| PS1 | Presenilin 1 |
| ROS | Reactive oxygen species |
| SNc | Substantia nigra pars compacta |
| SOD | Superoxide dismutase |
| TH | Tyrosine hydroxylase |
| TrkB | Tyrosine receptor kinase B |
References
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Young, J.J.; Lavakumar, M.; Tampi, D.; Balachandran, S.; Tampi, R.R. Frontotemporal dementia: Latest evidence and clinical implications. Ther. Adv. Psychopharmacol. 2018, 8, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 21 June 2019).
- Available online: https://parkinson.org/Understanding-Parkinsons/Statistics (accessed on 21 June 2019).
- Available online: https://rarediseases.org/rare-diseases/huntingtons-disease (accessed on 21 June 2019).
- Available online: https://www.cdc.gov/mmwr/volumes/67/wr/mm6707a3.htm (accessed on 21 June 2019).
- Prior, M.; Chiruta, C.; Currais, A.; Goldberg, J.; Ramsey, J.; Dargusch, R.; Maher, P.A.; Schubert, D. Back to the future with phenotypic screening. ACS Chem. Neurosci. 2014, 5, 503–513. [Google Scholar]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Maher, P. An alternative approach to drug discovery for Alzheimer’s disease dementia. Future Med. Chem. 2012, 4, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- 1Schubert, D.; Currais, A.; Goldberg, J.; Finley, K.; Petrascheck, M.; Maher, P. Geroneuroprotectors: Effective geroprotectors for the brain. Trends Pharmacol. Sci. 2018, 39, 1004–1007. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Zheng, X.; Wang, G. Insights into drug discovery from natural medicines using reverse pharmacokinetics. Trends Pharmacol. Sci. 2014, 35, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C. Phenotypic vs. target-based drug discovery for firrst-in-class medicines. Clin. Pharm. Therap. 2013, 93, 299–301. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef]
- Yun, B.-W.; Yan, Z.; Amir, R.; Hong, S.; Jin, Y.-W.; Lee, E.-K.; Loake, G.J. Plant natural products: History, limitations and the potential of cambial meristematic cells. Biotech. Gen. Eng. Rev. 2013, 28, 47–60. [Google Scholar] [CrossRef]
- Bakoyiannis, I.; Daskalopoulou, A.; Pergialiotis, V.; Perrea, D. Phytochemicals and cognitive health: Are flavonoids doing the trick? Biomed. Pharmacother. 2019, 109, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Beking, K.; Vieira, A. Flavonoid intake and disability-adjuested life years due to Alzheimer’s and related dementias: A population-based study involving twenty-three developed countries. Public Health Nutr. 2010, 13, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Commenges, D.; Scotet, V.; Renaud, S.; Jacqmin-Gadda, H.; Barberger-Gateau, P.; Dartigues, J.F. Intake of flavonoids and risk of dementia. Eur. J. Epidemiol. 2000, 16, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cassidy, A.; Schwarzschild, M.A.; Rimm, E.B.; Ascherio, A. Habitual intake of dietary flavonoids and risk of Parkinson’s disease. Neurology 2012, 78, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Jones, Q.R.D.; Warford, J.; Rupasinghe, H.P.V.; Robertson, G.S. Target-based selection of flavonoids for neurodegenerative disorders. Trends Pharmacol. Sci. 2012, 33, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.C.B.; Santana, M.G.S.; Ofrfali, G.; Parisi de Oliveira, C.T.; Priolli, D.G. Literature evidence and ARRIVE assessment on neuroprotective effects of flavonols in neurodegenerative diseases’ models. CNS Neurolog. Dis. Drug Targets 2018, 17, 34–42. [Google Scholar] [CrossRef] [PubMed]
- de Andrade Teles, R.B.; Diniz, T.C.; Costa Pinto, T.C.; de Oliviera, R.G.; Gama e Silva, M.; de Lavor, E.M.; Fernandes, A.W.C.; de Oliviera, A.P.; de Almeida Ribeiro, F.P.R.; da Silva, A.A.M.; et al. Flavonoids as therapeutic agents in Alzheimer’s and Parkinson’s diseases: A systematice review of preclinical evidences. Oxid. Med. Cell. Longev. 2018, 2018, 7043213. [Google Scholar] [CrossRef]
- Kujawska, M.; Jodynis-Liebert, J. Polyphenols in Parkinson’s disease: A systematic review of in vivo studies. Nutrients 2018, 10, 642. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Irish, M.; van Eersel, J.; Ittner, A.; Ke, Y.D.; Volkerling, A.; van der Hoven, J.; Tanaka, K.; Karl, T.; Kassiou, M.; et al. Mouse models of frontotemporal dementia: A comparison of phenotypes with clinical symptomatology. Neurosc. Biobehav. Rev. 2017, 74, 126–138. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA work group under the auspices of Department of Health and Humans Services Task Force on Alzheimer’s disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.; Cummings, J.L. Behavioral changes and psychological symptoms in dementia disorders. Lancet Neurol. 2005, 4, 735–742. [Google Scholar] [CrossRef]
- Haas, C. Strategies, development and pitfalls of therapeutic options for Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 28, 241–281. [Google Scholar] [CrossRef] [PubMed]
- Rafil, M.S.; Aisen, P.S. Recent developments in Alzheimer’s disease therapeutics. BMC Med. 2009, 7, 7. [Google Scholar]
- Gold, M. Phase II clinical trials of anti-amyloid β antibodies: When is enough, enough? Alzheimer’s Dement. 2017, 3, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.alzforum.org/research-models/alzheimers-disease (accessed on 21 June 2019).
- Swerdlow, R.H. Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiol. Aging 2007, 28, 1465–1480. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Pallas, M. Senescence-accelerated mice P8: A tool to study brain aging and Alzheimer’s disease in a mouse model. ISRN Cell Biol. 2012, 2012, 917167. [Google Scholar] [CrossRef]
- Morley, J.E.; Armbrecht, H.J.; Farr, S.A.; Kumar, V.B. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 650–656. [Google Scholar] [CrossRef]
- Cheng, X.R.; Zhou, W.X.; Zhang, Y.X. The behavioral, pathological and therapeutic features of the senescence-accelerated mouse prone 8 strain as an Alzheimer’s disease animal model. Ageing Res. Rev. 2014, 13, 13–37. [Google Scholar] [CrossRef]
- Currais, A.; Goldberg, J.; Farrokhi, C.; Chang, M.; Prior, M.; Dargusch, R.; Daugherty, D.; Armando, A.; Quehenberger, O.; Maher, P.; et al. A comprehensive multiomics approach toward understanding the relationship between aging and dementia. Aging 2015, 7, 937–955. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.A.; Kozhevnikova, O.S.; Vitovtov, A.O.; Logvinov, S.V.; Rudnitskaya, E.A.; Korbolina, E.E.; Muraleva, N.A.; Kolosova, N.G. Senescence-accelerated OXYS rats: A model of age-related cognitive decline with relevance to Alzheimer disease. Cell Cycle 2014, 13, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. 7,8 Dihydroxyflavone, a small molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2012, 37, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Schroeder, J.P.; Chan, C.-B.; Song, M.; Yu, S.P.; Weinshenker, D.; Ye, K. 7,8 Dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 638–650. [Google Scholar] [CrossRef]
- Zhou, W.X.; Li, X.; Huang, D.; Zhou, W.X.; Li, T.; Song, W. No significant effect of 7,8 dihydroxyflavone on APP processing and Alzheimer-associated phenotypes. Curr. Alzheimer Res. 2015, 12, 47–52. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.-L.; Liu, R.; Li, X.-X.; Li, J.-F.; Zhang, L.-L. Neuroprotective, anti-amyloidgenic and neurotrophic effects of apigenin in an Alzheimer’s disease model. Molecules 2013, 18, 9949–9965. [Google Scholar] [CrossRef] [PubMed]
- Onozuka, H.; Nkajima, A.; Matsuzaki, K.; Shin, R.W.; Ogino, K.; Tetsu, N.; Yokosuka, A.; Sashida, Y.; Mimaki, Y.; Yamakuni, T.; et al. Nobiletin, a citrus flavonoid, improves memory impairment and Abeta pathology in a transgenic model of Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2008, 326, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Aoyama, Y.; Shin, E.-J.; Nam, Y.; Kim, H.-C.; Nagai, T.; Yokosuka, A.; Mimaki, Y.; Yokoi, T.; Ohizumi, Y.; et al. Nobiletin, a citrus flavonoid, improves cognitive impairment and reduces soluble Aβ levels in a triple transgenic mouse model of Alzheimer’s disease (3XTg-AD). Behav. Brain Res. 2015, 289, 69–77. [Google Scholar] [CrossRef]
- Nakajima, A.; Aoyama, Y.; Nguyen, T.-T.L.; Shin, E.-J.; Kim, H.-C.; Yamada, S.; Nakai, T.; Nagai, T.; Yokosuka, A.; Mimaki, Y.; et al. Nobiletin, a citrus flavonoid, ameliorates cognitive impairment, oxidative burden and hyperphosphorylation of tau in senescence-accelerated mouse. Behav. Brain Res. 2013, 250, 351–360. [Google Scholar] [CrossRef]
- Zhang, S.-Q.; Obregon, D.F.; Ehrhart, J.; Deng, J.Y.; Tian, J.; Hou, H.; Giunta, B.; Sawmiller, D.; Tan, J. Baicalein reduces b-amyloid and promotes nonamyloidgenic amyloid precursor protein processing in an Alzheimer’s disease transgenic mouse model. J. Neurosci. Res. 2013, 91, 1239–1246. [Google Scholar] [CrossRef]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Quehenberger, O.; Maher, P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Farrokhi, C.; Dargusch, R.; Armando, A.; Quehenberger, O.; Schubert, D.; Maher, P. Fisetin reduces the impact of aging on behavior and physiology in the rapidly aging SAMP8 mouse. J. Gerentol. Biol. Sci. Med. Sci. 2018, 73, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ali, T.; Park, H.Y.; Badshah, H.; Rehman, S.U.; Kim, M.O. Neuroprotective effect of fisetin against amyloid-beta-induced cognitive/synaptic dysfunction, neuroinflammation and neurodegeneration in mice. Mol. Neurobiol. 2017, 54, 2269–2285. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 2004, 14, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Sagara, Y.; Vahnnasy, J.; Maher, P. Induction of PC12 cell differentiation by flavonoids is dependent upon extracellular signal-regulated kinase activation. J. Neurochem. 2004, 90, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Zaplatic, E.; Bule, M.; Shah, S.Z.A.; Uddin, M.S.; Niaz, K. Molecular mechanisms underlying protective role of quercetin in attenuating Alzheimer disease. Life Sci. 2019, 224, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Gayoso e Ibiapina Moreno, L.C.; Puerta, E.; Suarez-Santiago, J.E.; Santos-Magalhaes, N.S.; Ramirez, M.J.; Irache, J.M. Effect of the oral administration of nanoencapsulated quercetin on a mouse model of Alzheimer’s disease. Int. J. Pharmaceut. 2017, 517, 50–57. [Google Scholar] [CrossRef]
- Sabogal-Guaqueta, A.M.; Munoz-Manco, J.I.; Ramirez-Pineda, J.R.; Lamprea-Rodriguez, M.; Osorio, E.; Cardona-Gomez, G.P. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropsychopharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef]
- Moghbelinejad, S.; Nassiri-Asi, M.; Farivar, T.N.; Abbasi, E.; Sheikhi, M.; Taghiloo, M.; Farsad, F.; Samimi, A.; Haijali, F. Rutin activates the MAPK pathway and BDNF gene expression on beta-amyloid induce neurotoxicity in rats. Toxicol. Lett. 2014, 224, 108–113. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.J.; Rehman, S.U.; Ahmad, A.; Kim, M.O. Anthocyanin-loaded PEG-gold nanoparticles enhanced the neuroprotection of anthocyanins in Aβ1-42 mouse model of Alzheimer’s disease. Mol. Neurobiol. 2017, 54, 6490–6506. [Google Scholar] [CrossRef]
- Kim, M.J.; Rehman, S.U.; Amin, F.U.; Kim, M.O. Enhanced neuroprotection of anthocyanin-loaded PEG-gold nanoparticles against Aβ1-42-induced neuroinflammation and neurodegeneration via the NF-kB/JNK/GSK3β signaling pathway. Nanomed. Nanotech. Biol. Med. 2017, 13, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Klakotskaia, D.; Ajit, D.; Weisman, G.A.; Wood, W.G.; Sun, G.Y.; Serfozo, P.; Simonyi, A.; Schachtman, T.R. Beneficial effects of dietary EGCG and voluntary exercise on behavior in an Alzheimer’s disease mouse model. J. Alzheimer’s Dis. 2015, 44, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhao, Y.; Nan, Y.; Wang, X.; Chen, Y.; Wang, S. (−)-Epigallocatechin-3-gallate ameliorates memory impairment and rescues the abnormal synpatic protein levels in the frontal cortex and hippocampus on a mouse model of Alzheimer’s disease. Neuroreport 2017, 28, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Ide, K.; Matsuoka, N.; Yamada, H.; Furushima, D.; Kawakami, K. Effects of tea catechins on Alzheimer’s disease: Recent updates and perspectives. Molecules 2018, 23, 2357. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zug, C.; Qu, H.; Schluesener, H.; Zhang, Z. Hesperidin ameliorates behavioral impairments and neuropathology of transgenic APP/PS1 mice. Behav. Brain Res. 2015, 281, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Hajialyani, M.; Farzaei, M.H.; Echeverria, J.; Nabavi, S.M.; Uriarte, E.; Sobarzo-Sanchez, E. Hesperidin as a neuroprotective agent: A review of animal and clinical evidence. Molecules 2019, 24, 648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Q.; Wang, J.; Zhao, H.; Luo, Y. Effects of three flavonoids from an ancient Chinese medicine Radix puerariae on geriatric diseases. Brain Circ. 2018, 4, 174–184. [Google Scholar]
- Weintraub, D.; Comella, C.L.; Horn, S. Parkinson’s disease. Am. J. Manag. Care 2008, 14, S40–S69. [Google Scholar]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef]
- Schulz, J.B.; Falkenburger, B.H. Neuronal pathology in Parkinson’s disease. Cell Tissue Res. 2004, 318, 135–147. [Google Scholar] [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.-C.; Huang, H.-J.; Wang, Y.-T.; Lin, A.M.-Y. Baicalein attenuates α-synuclein aggregation, inflammasome activation and autophagy in the MPP+-treated nigrostriatal dopaminergic system in vivo. J. Ethnopharmcol. 2016, 194, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, G.; Mu, X.; Zhang, T.; Li, X.; Hu, J.; Xu, B.; Du, G. Neuroprotective effect of baicalein against MPTP neurotoxicity: Behavioral, biochemical and immunohistochemical profile. Neurosci. Lett. 2008, 441, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Du, L.; Zhang, W.; Yang, Y.; Zhou, Q.; Du, G. Therapeutic effects of baicalein on rotenone-induced Parkinson’s disease through protecting mitochondrial function and biogenesis. Sci. Rep. 2017, 7, 9968. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Shi, Y.; Wang, J.; Lin, Q.; Sun, Y.; Ye, K.; Yan, Q.; Zhang, H. 7,8 Dihydroxyflavone protects 6-OHDA and MPTP induced dpaminergic neurons degeneration through activation of TrkB in rodents. Neurosci. Lett. 2016, 620, 43–49. [Google Scholar] [CrossRef]
- Li, X.H.; Dai, C.F.; Chen, L.; Zhou, W.T.; Han, H.L.; Dong, Z.F. 7,8 Dihydroxyflavone ameliorates motor deficits via supressing a-synuclein expression and oxidative stress in the MPTP-indiuced mouse model of Parkinson’s disease. CNS Neurosci. Ther. 2016, 22, 617–624. [Google Scholar] [CrossRef]
- Sconce, M.D.; Churchill, M.J.; Moore, C.; Meshul, C.K. Intervention with 7,8 dihydorxyflavone blocks further striatal terminal loss and restores motor deficits in a progressive mouse model of Parkinson’s disease. Neuroscience 2015, 290, 454–471. [Google Scholar] [CrossRef] [PubMed]
- He, J.C.; Xiang, Z.; Zhu, X.; Ai, Z.; Shen, J.; Huang, T.; Liu, L.; Ji, W.; Li, T. Neuroprotective effects of 7,8, dihydroxyflavone on midbrain dopaminergic neurons in MPP+-treated monkeys. Sci. Rep. 2016, 6, 34339. [Google Scholar] [CrossRef] [PubMed]
- Anusha, C.; Sumathi, T.; Joseph, L.D. Protective role of apigenin on rotenone induced rat model of Parkinson’s disease: Suppression of neuroinflammation and oxidative stress mediated apoptosis. Chem.-Biol. Interactions 2017, 269, 67–79. [Google Scholar] [CrossRef]
- Patil, S.P.; Jain, P.D.; Sancheti, J.S.; Ghumatkar, P.J.; Tambe, R.; Sathye, S. Neuroprotective and neurotrophic effects of apigenin and luteolin in MPTP induced parkinsonism in mice. Neuropharmacology 2014, 86, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Goes, A.T.R.; Jesse, C.R.; Antunes, M.S.; Ladd, F.V.L.; Ladd, A.A.B.L.; Luchese, C.; Paroul, N.; Boeira, S.P. Protective role of chrysin on 6-hydroxydopamine-induced neurodegeneration a mouse model of Parkinson’s disease: Involvement of neuroinflammation and neurotrophins. Chem.-Biol. Interact. 2018, 279, 111–120. [Google Scholar] [CrossRef]
- Jeong, K.H.; Jeon, M.T.; Kim, H.D.; Jung, U.J.; Jang, M.C.; Chu, J.W.; Yang, S.J.; Choi, I.Y.; Choi, M.S.; Kim, S.R. Nobiletin protects dopaminergic neurons in the 1-mehtyl-4-phenylpyridinium rat model of Parkinson’s disease. J. Med. Food 2015, 18, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Lee, Y.J.; Chun, A.H.; Kim, A.H.; Kim, J.Y.; Lee, J.Y.; Ishigami, A.; Lee, J.H. Neuroprotective and anti-inflammatory effects of morin in a murine model of Parkinson’s disease. J. Neurosci. Res. 2016, 94, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Zbarsky, V.; Datla, K.P.; Parkar, S.; Rai, D.K.; Arouma, O.I.; Dexter, D.T. Neuroprotective properties of the natural phenolic antioxidants curcumin and naringenin but not quercetin and fisetin in a 6-OHDA model of Parkinson’s disease. Free Rad. Res. 2005, 39, 1119–1125. [Google Scholar] [CrossRef]
- Haleagrahara, N.; Siew, C.J.; Ponnusamy, K. Effect of quercetin and desferriooxamine on 6-hydroxydopamine (6-OHDA) induced neurotoxicity in striatum of rats. J. Toxicol. Sci. 2013, 38, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Karuppagounder, S.S.; Madathil, S.K.; Pandey, M.; Haobam, R.; Rajamma, U.; Mohanakumar, K.P. Quercetin up-regulates mitochondrial complex-I activity to protect against programmed cell death in rotenone model of Parkinson’s disease in rats. Neuroscience 2013, 236, 136–148. [Google Scholar] [CrossRef]
- Lv, C.; Hong, T.; Yang, Z.; Zhang, Y.; Wang, L.; Dong, M.; Zhao, J.; Mu, J.; Meng, Y. Effect of quercetin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease. Evid.-Based Complemen. Alternat. Med. 2012, 2012, 928643. [Google Scholar] [CrossRef] [PubMed]
- Ay, M.; Luo, J.; Langley, M.; Jin, H.; Anatharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Molecular mechanisms underlying protective effects of quercetin agianst mitochondrial dysfunction and progressive dopaminergic neurodegeneration in cell culture and MitoPark transgenic mouse models of Parkinson’s disease. J. Neurochem. 2017, 141, 766–782. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Galter, D. The MitoPark Mouse-An animal model of Parkinson’s diease with impaired respiratory chain function in dopamine neurons. Parkinsonism Rel. Dis. 2009, 1553, S185–S188. [Google Scholar] [CrossRef]
- Khan, M.M.; Raza, S.S.; Javed, H.; Ahmad, A.; Khan, A.; Islam, F.; Safhi, M.M.; Islam, F. Rutin protects dopaminergic neurons from oxidative stress in an animal model of Parkinson’s disease. Neurotox. Res. 2012, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. Protective effects of fisetin and other berry flavonoids in Parkinson’s disease. Food Func. 2017, 8, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Pu, X.-P. Neuroprotective effect of kaempferol against 1-methyl-4-phenyl-1,2,3,6,-tetrahydropyridine-induced mouse model of Parkinson’s disease. Biol. Pharm. Bull. 2011, 34, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.D.A.; Souza, C.M.; Menezes, A.P.F.; Carmo, M.R.S.; Fonteles, A.A.; Gurgel, J.P.; Lima, F.A.V.; Viana, G.S.B.; Andrade, G.M. Catechin attenuates behavioral neurotoxicity induced by 6-OHDA in rats. Pharmacol. Biochem. Behav. 2013, 110, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Langley, M.; Kanthasamy, A.G.; Reddy, M.B. Epigallocatechin gallate has a neurorescue effect in a mouse model of Parkinson Disease. J. Nutr. 2017, 147, 1926–1931. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Osornio, M.; Gorostieta-Salas, E.; Montes, S.; Perez-Severiano, F.; Rubio, C.; Gomez, C.; Rios, C.; Guevara, J. Epicatechin reduces striatal MPP+-induce damage in rats through slight increases in SOD-Cu,Zu Activity. Oxid. Med. Cell. Longev. 2015, 2015, 276039. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Sekar, S.; Barathidasan, R.; Manivasagam, T.; Thenmozhi, A.J.; Sevanan, M.; Chidambaram, S.B.; Essa, M.M.; Guillemin, G.J.; Sakharkar, M.K. Naringenin decreases a-synuclein expression and neuroinflammation in MPTP-induced Parkinson’s disease model in mice. Neurotox. Res. 2018, 33, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Kiasalari, Z.; Khalili, M.; Baluchnejadmojarad, T.; Roghani, M. Protective effect of oral hesperetin against unilateral striatal 6-hydorxydopamine damage in the rat. Neurochem. Res. 2016, 41, 1065–1072. [Google Scholar] [CrossRef]
- Kim, H.D.; Jeong, K.H.; Jung, U.J.; Kim, S.R. Naringin treatment induces neuroprotective effects in a mouse model of Parkinson’s disease in vivo, but not enough to restore the lesioned dopaminergic system. J. Nutr. Biochem. 2016, 28, 140–146. [Google Scholar] [CrossRef]
- Antunes, M.S.; Goes, A.T.R.; Boeira, S.P.; Prigol, M.; Jesse, C.R. Protective effect of hesperidin in a model of Parkinson’s disease induced by 6-hydroxydopmaine in aged mice. Nutrition 2014, 30, 1415–1422. [Google Scholar] [CrossRef]
- Borrell-Pages, M.; Zala, D.; Humbert, S.; Saudou, F. Huntington’s disease: From huntingtin function and dysfunction to therapeutic strategies. Cell. Mol. Life Sci. 2006, 63, 2642–2660. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Shannon, K.M.; Kordower, J.H. Huntington’s disease: Pathological mechanisms and therapeutic strategies. Cell Transplant. 2007, 16, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Imarisio, S.; Carmichael, J.; Korolchuk, V.; Chen, C.-W.; Saiki, S.; Rose, C.; Krishna, G.; Davies, J.E.; Ttofi, E.; Underwood, B.R.; et al. Huntington’s disease: From pathology and genetics to potential therapeutics. Biochem. J. 2008, 412, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.M.; Rego, A.C. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 2008, 27, 2803–2820. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Barajas, C.; Rebec, G.V. Overview of Huntington’s disease models: Neuropathological, molecular and behavioral differences. Curr. Protoc. Neurosci. 2018, 83, e47. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Koutouzis, T.K.; Sanberg, P.R. 3-Nitropropionic acid animal model and Huntington’s disease. Neurosci. Biobehav. Rev. 1997, 21, 289–293. [Google Scholar] [CrossRef]
- Thangarajan, S.; Ramachandran, S.; Krishnamurthy, P. Chrysin exerts neuroprotective effects against 3-nitropropionic acid induced behavioral despair-mitochondrial dysfunction and striatal apoptosis via upregulating Bcl-2 gene and downregulating Bax-Bad gnes in male wistar rats. Biomed. Pharmacother. 2016, 84, 514–525. [Google Scholar] [CrossRef]
- Barriga, G.G.-D.; Giralt, A.; Anglada-Huguet, M.; Gaja-Capdevila, N.; Orlandi, J.G.; Soriano, J.; Canals, J.-M.; Alberch, J. 7,8 Dihydroxyflavone ameliorates cognitive and motor deficits in a Huntington’s disease mouse model through specific activation of the PLCγ1 pathway. Hum. Mol. Gen. 2017, 26, 3144–3160. [Google Scholar]
- Maher, P.; Dargusch, R.; Bodai, L.; Gerard, P.; Purcell, J.M.; Marsh, J.L. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum. Mol. Gen. 2011, 20, 261–270. [Google Scholar] [CrossRef]
- Sandhir, R.; Mehrotra, A. Quercetin supplementation is effective in improving mitochondrial dysfunctions induced by 3-nitropropionic acid: Implications in Huntington’s disease. Biochim. Biophys. Acta 2013, 1832, 421–430. [Google Scholar] [CrossRef]
- Chakraborty, J.; Singh, R.J.; Dutta, D.; Naskar, A.; Rajamma, U.; Mohanakumar, K.P. Quercetin improves behavioral deficiencies, restores astrocytes and microglia, and reduces serotonin metabolism in 3-nitropropionic acid-induced rat model of Huntington’s disease. CNS Neurosci. Ther. 2014, 20, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Suganya, S.N.; Sumathi, T. Effect of rutin against a mitochondrial toxin, 3-nitropropionic acid induced biochemical, behavioral and histological alterations-a pilot study on Huntington’s disease model in rats. Metab. Brain Dis. 2017, 32, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Lagoa, R.; Lopez-Sanchez, C.; Samhan-Arias, A.K.; Ganan, C.M.; Garcia-Martinez, V.; Gutierrez-Merino, C. Kaempferol protects against rat striatal degeneration induced by 3-nitropropionic acid. J. Neurochem. 2009, 111, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Menze, E.T.; Tadros, M.G.; Abdel-Tawab, A.M.; Khalifa, A.E. Potential neuroprotective effects of hesperidin on 3-nitropropionic acid-induced neurotoxicity in rats. Neurotoxicology 2012, 33, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Menze, E.T.; Esmat, A.; Tadros, M.G.; Khalifa, A.E.; Abdel-Naim, A.B. Genistein improves sensorimotor gating: Mechanisms related to its neuroprotective effects on the striatum. Neuropharmacology 2016, 105, 35–46. [Google Scholar] [CrossRef]
- Kreilaus, F.; Spiro, A.S.; Hannan, A.J.; Garner, B.; Jenner, A.M. Therapeutic effects of anthocyanins and environmental enrichment in R6/1 Huntongton’s disease mice. J. Huntington’s Dis. 2016, 5, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Gil-Bea, F.J.; Santurtan, A.; Lopez de Munain, A. Amytotrophic lateral sclerosis: A complex syndrome that needs an integrated research approach. Neural Regen. Res. 2019, 14, 193–196. [Google Scholar] [CrossRef]
- Lutz, C. Mouse models of ALS. Brain Res. 2018, 1693, 1–10. [Google Scholar] [CrossRef]
- Ittner, L.M.; Halliday, G.M.; Kril, J.J.; Gotz, J.; Hodges, J.R.; Kiernan, M.C. FTD and ALS-translating mouse studies into clinical trials. Nat. Rev. Neurol. 2015, 11, 360–366. [Google Scholar] [CrossRef]
- Korkmaz, O.T.; Aytan, N.; Carreras, I.; Choi, J.-K.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. 7,8 Dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2014, 566, 286–291. [Google Scholar] [CrossRef]
- Wang, T.H.; Wang, S.Y.; Wang, X.D.; Jiang, H.Q.; Yang, Y.Q.; Wang, Y.; Cheng, J.L.; Zhang, C.T.; Liang, W.W.; Feng, H.L. Fisetin exerts antioxidant and neuroprotective effects in mulitple mutant hSOD1 modles of amyotrophic lateral sclerosis by activating ERK. Neuroscience 2018, 379, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, S.; Li, X.; Luo, G.; Li, L.; Le, W. Neuroprotective effects of (−)-epigallocatechin-3-gallate in a transgenic mouse model of amyotrophic lateral sclerosis. Neurochem. Res. 2006, 31, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.H.; Lee, S.M.; Kim, H.Y.; Lee, K.Y.; Lee, Y.J.; Kim, H.T.; Kim, J.; Kim, M.H.; Hwang, M.S.; Song, C.; et al. The effect of epigallocatechin gallate on suppressing disease progression of ALS model mice. Neurosci. Lett. 2006, 395, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, R. Bioavailable curcumin formulations: A review of pharmacokinetic studies in healthy volunteers. J. Integr. Med. 2018, 16, 367–374. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| AD | PD | HD | ALS | |
|---|---|---|---|---|
| Flavones | ||||
| 7,8-DHF | X | X | X | X |
| Apigenin | X | X | ||
| Baicalein | X | X | ||
| Chrysin | X | X | ||
| Luteolin | X | |||
| Morin | X | |||
| Nobiletin | X | X | ||
| Flavonols | ||||
| Fisetin | X | X | X | X |
| Kaempferol | X | |||
| Myricetin | X | |||
| Myricitrin | X | |||
| Quercetin | X | X | X | |
| Rutin | X | X | X | |
| Flavanols | ||||
| Catechin | X | |||
| Epicatechin | X | |||
| ECGC | X | X | X | |
| Flavanones | ||||
| Hesperetin | X | |||
| Hesperidin | X | X | X | |
| Naringenin | X | |||
| Naringin | X | |||
| Anthocyanidins | ||||
| Anthocyanins | X | X | ||
| Isoflavones | ||||
| Genistein | X | X | ||
| Puerarin | X | X |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3056. https://doi.org/10.3390/ijms20123056
Maher P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. International Journal of Molecular Sciences. 2019; 20(12):3056. https://doi.org/10.3390/ijms20123056
Chicago/Turabian StyleMaher, Pamela. 2019. "The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases" International Journal of Molecular Sciences 20, no. 12: 3056. https://doi.org/10.3390/ijms20123056
APA StyleMaher, P. (2019). The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. International Journal of Molecular Sciences, 20(12), 3056. https://doi.org/10.3390/ijms20123056