Targeting Quorum Sensing: High-Throughput Screening to Identify Novel LsrK Inhibitors


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
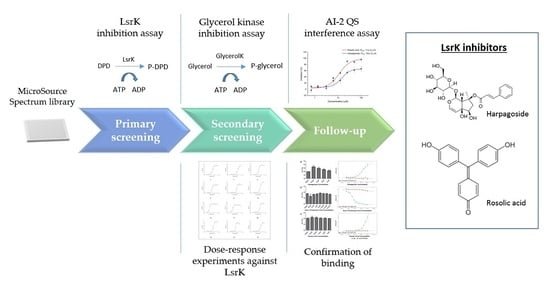
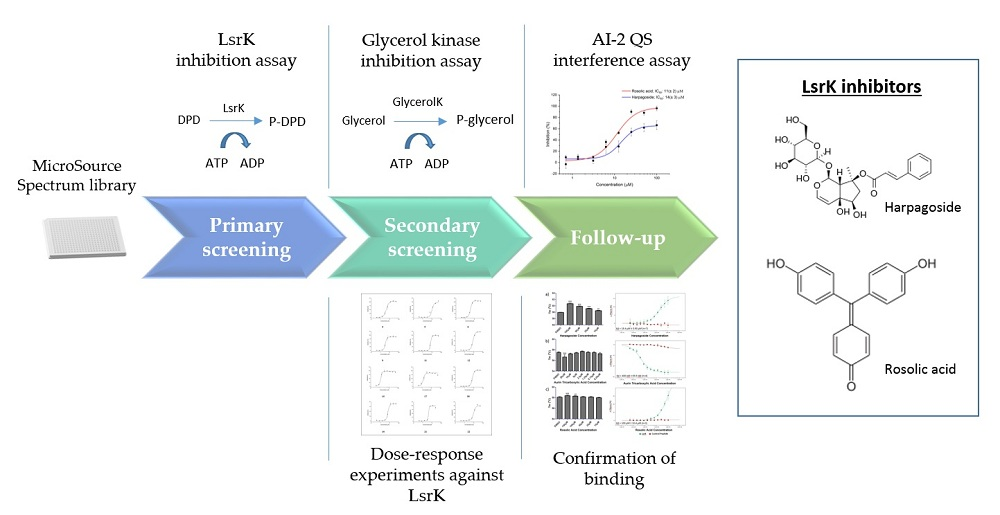
2.1. Assay Selection
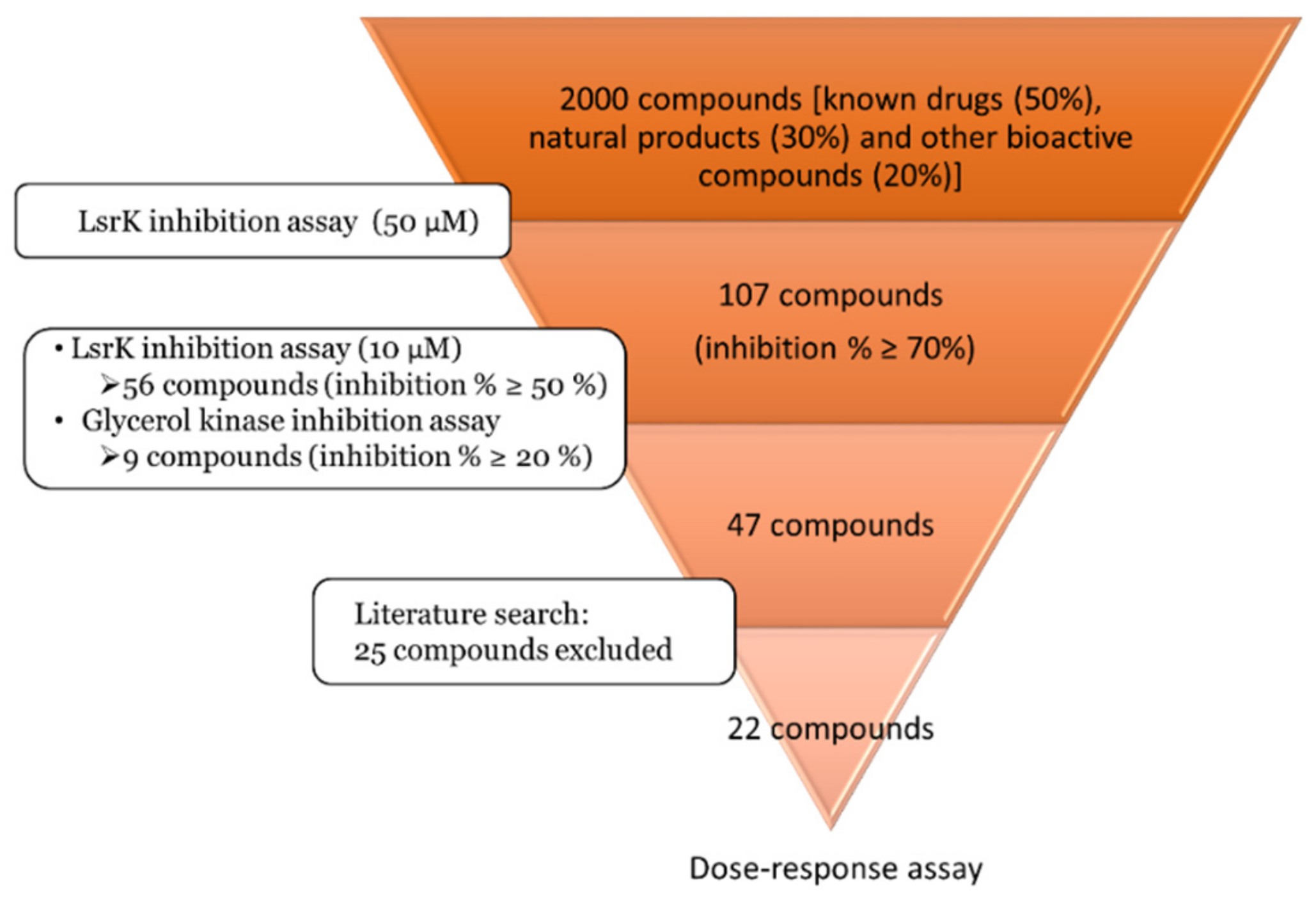
2.2. Pilot Screening
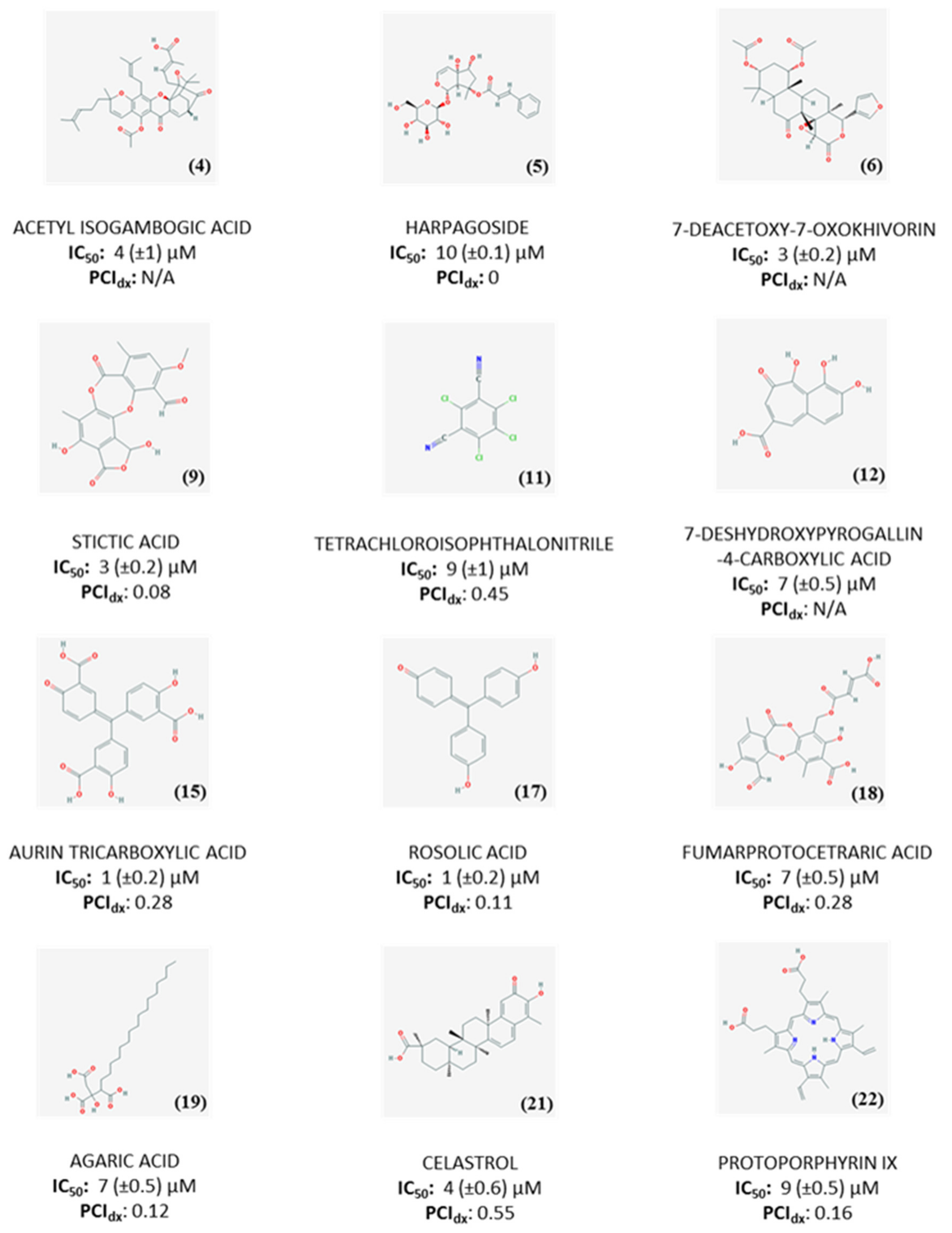
2.3. Dose-Response Experiments and Hit Identification
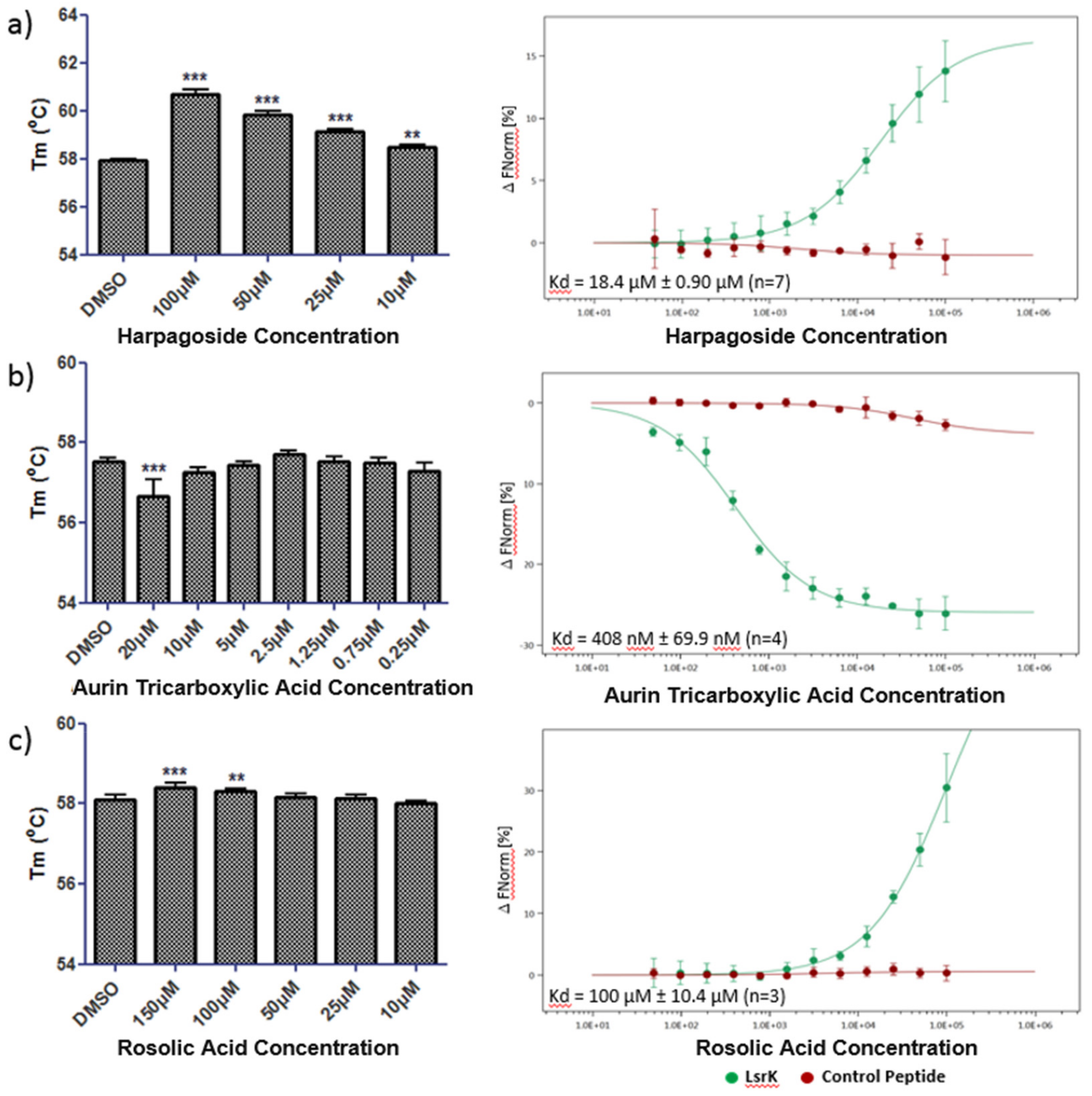
2.4. Thermal Shift and MST Assay
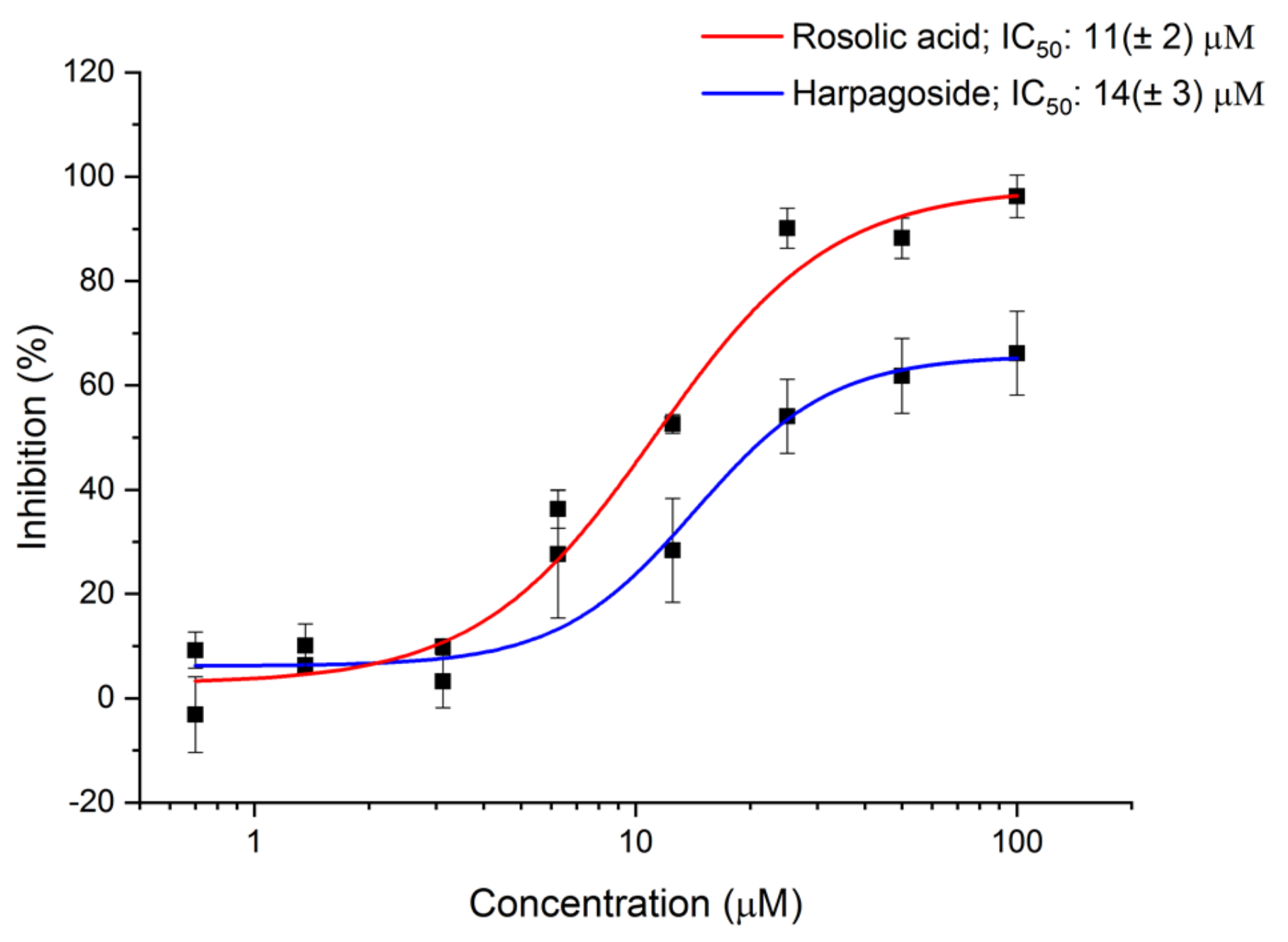
2.5. AI-2 QS Interference Assay
3. Materials and Methods
3.1. Materials
3.2. Overexpression and Purification of LsrK from S. thyphmurium
3.3. Assay Development
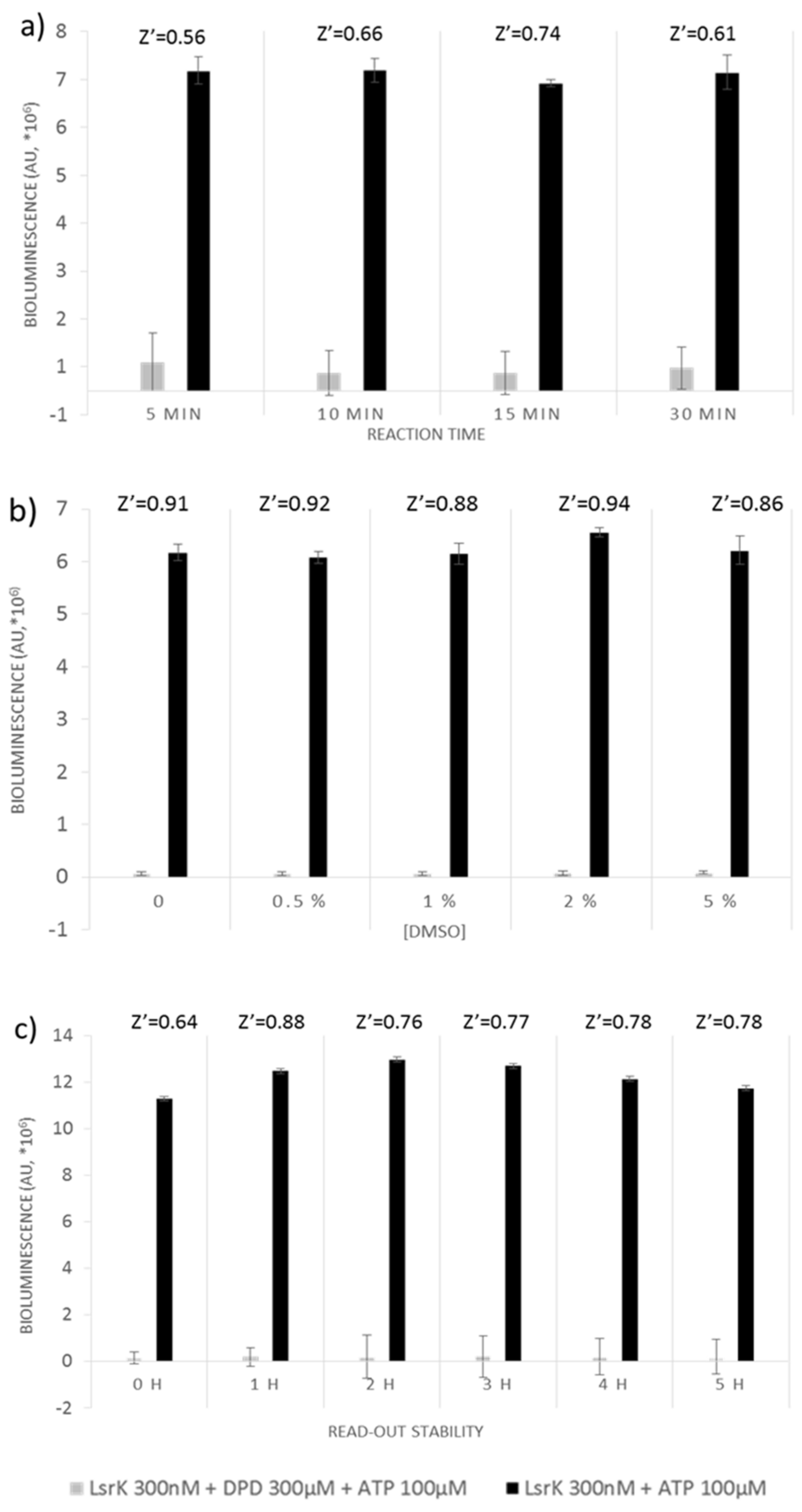
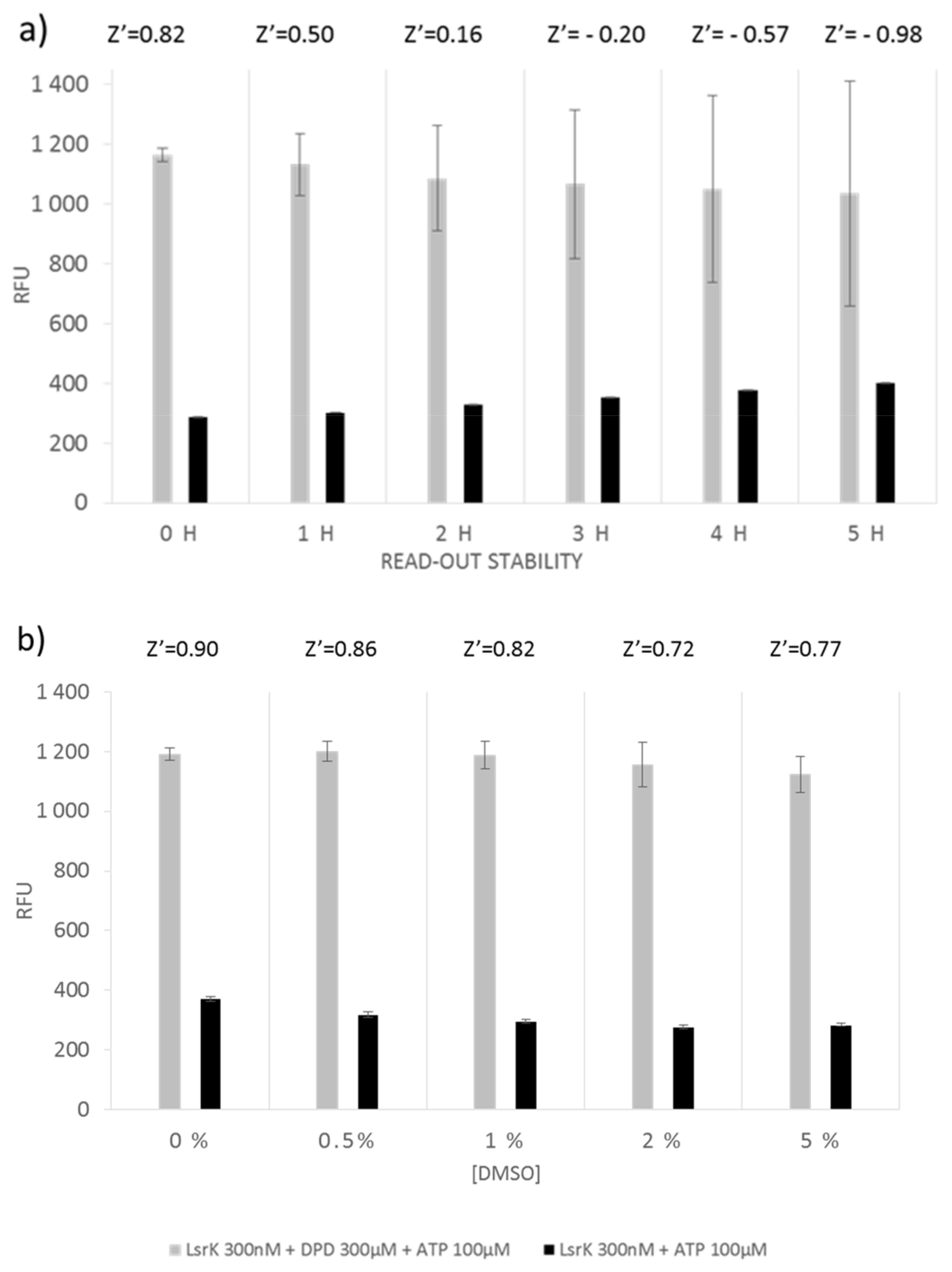
3.3.1. ATP Bioluminescence CLS II Assay
3.3.2. Assay Miniaturization and Optimization for HTS
Kinase-Glo Max Luminescent Kinase Assay
ADP-Quest
3.4. Assay Validation
3.5. Glycerol Kinase Inhibition Assay
3.6. Thermal Shift Assay
3.7. MST Assay
3.8. AI-2 QS Interference Assay
3.9. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| QS | Quorum sensing |
| AI-2 | Autoinducer 2 |
| DPD | (S)-4,5-dihydroxy-2,3-pentanedione |
| LsrK | Autoinducer-2 kinase |
| HTS | High-throughput screening |
| TEA | Triethanolamine |
| BSA | Bovine serum albumin |
| DTT | Dithiothreitol |
| ATP | Adenosine 5′-triphosphate |
| DMSO | Dimethyl sulfoxide |
| ADP | Adenosine diphosphate |
| PcIDx | Promiscuity index |
| MST | Microscale thermophoresis |
| PAβN | phe-arg β-naphtylamide dihydrochloride |
References
- Allen, R.C.; Popat, R.; Diggle, S.P.; Brown, S.P. Targeting virulence: Can we make evolution-proof drugs? Nat. Rev. Microbiol. 2014, 12, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Calvert, M.B.; Jumde, V.R.; Titz, A. Pathoblockers or antivirulence drugs as a new option for the treatment of bacterial infections. Beilstein J. Org. Chem. 2018, 14, 2607–2617. [Google Scholar] [CrossRef] [PubMed]
- Gerdt, J.P.; Blackwell, H.E. Competition studies confirm two major barriers that can preclude the spread of resistance to quorum-sensing inhibitors in bacteria. ACS Chem. Biol. 2014, 9, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.T.; Bassler, B.L. Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef] [PubMed]
- Defoirdt, T. Quorum-sensing systems as targets for antivirulence therapy. Trends Microbiol. 2018, 26, 313–328. [Google Scholar] [CrossRef]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef]
- Xavier, K.B.; Bassler, B.L. Regulation of uptake and processing of the quorum-sensing autoinducer AI-2 in Escherichia coli. J. Bacteriol. 2005, 187, 238–248. [Google Scholar] [CrossRef]
- Roy, V.; Fernandes, R.; Tsao, C.Y.; Bentley, W.E. Cross species quorum quenching using a native AI-2 processing enzyme. ACS Chem. Biol. 2010, 5, 223–232. [Google Scholar] [CrossRef]
- Xavier, K.B.; Miller, S.T.; Lu, W.; Kim, J.H.; Rabinowitz, J.; Pelczer, I.; Semmelhack, M.F.; Bassler, B.L. Phosphorylation and processing of the quorum-sensing molecule autoinducer-2 in enteric bacteria. ACS Chem. Biol. 2007, 2, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; Zhu, J.; Lowery, C.A.; Kaufmann, G.F.; Janda, K.D. C4-alkoxy-HPD: A potent class of synthetic modulators surpassing nature in AI-2 quorum sensing. J. Am. Chem. Soc. 2012, 134, 13562–13564. [Google Scholar] [CrossRef] [PubMed]
- Stotani, S.; Gatta, V.; Medda, F.; Padmanaban, M.; Karawajczyk, A.; Tammela, P.; Giordanetto, F.; Tzalis, D.; Collina, S. A versatile strategy for the synthesis of 4,5-dihydroxy-2,3-pentanedione (DPD) and related compounds as potential modulators of bacterial quorum sensing. Molecules 2018, 23, 2545. [Google Scholar] [CrossRef] [PubMed]
- Stotani, S.; Gatta, V.; Medarametla, P.; Padmanaban, M.; Karawajczyk, A.; Giordanetto, F.; Tammela, P.; Laitinen, T.; Poso, A.; Tzalis, D.; et al. DPD-inspired discovery of novel LsrK kinase inhibitors: An opportunity to fight antimicrobial resistance. J. Med. Chem. 2019, 62, 2720–2737. [Google Scholar] [CrossRef] [PubMed]
- Medarametla, P.; Gatta, V.; Kajander, T.; Laitinen, T.; Tammela, P.; Poso, A. Structure-based virtual screening of LsrK kinase inhibitors to target quorum sensing. ChemMedChem 2018, 13, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Inglese, J.; Johnson, R.L.; Simeonov, A.; Xia, M.; Zheng, W.; Austin, C.P.; Auld, D.S. High-throughput screening assays for the identification of chemical probes. Nat. Chem. Biol. 2007, 3, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Schürer, S.C.; Vempati, U.; Smith, R.; Southern, M.; Lemmon, V. BioAssay ontology annotations facilitate cross-analysis of diverse high-throughput screening data sets. J. Biomol. Screen. 2011, 16, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Haseeb, A.; Ansari, M.Y.; Haqqi, T.M. Harpagoside suppresses IL-6 expression in primary human osteoarthritis chondrocytes. J. Orthop. Res. 2017, 35, 311–320. [Google Scholar] [CrossRef]
- Dinda, B.; Dinda, M.; Kulsi, G.; Chakraborty, A.; Dinda, S. Therapeutic potentials of plant iridoids in Alzheimer’s and Parkinson’s diseases: A review. Eur. J. Med. Chem. 2019, 169, 185–199. [Google Scholar] [CrossRef]
- Foresti, R.; Hoque, M.; Monti, D.; Green, C.J.; Motterlini, R. Differential activation of heme oxygenase-1 by chalcones and rosolic acid in endothelial cells. J. Pharmacol. Exp. Ther. 2005, 312, 686–693. [Google Scholar] [CrossRef]
- García, N.; Zazueta, C.; Pavón, N.; Chávez, E. Agaric acid induces mitochondrial permeability transition through its interaction with the adenine nucleotide translocase. Its dependence on membrane fluidity. Mitochondrion 2005, 5, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Chao, Y.; Chang, Y.H.; Hsu, M.J.; Lin, W.W. Inhibition of cytokine-induced JAK-STAT signalling pathways by an endonuclease inhibitor aurintricarboxylic acid. Br. J. Pharmacol. 2002, 137, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.Z.; Guo, R.J.; Takagi, K.; Miao, Z.Q.; Li, S.D. Chlorothalonil degradation by Ochrobactrum lupini strain TP-D1 and identification of its metabolites. World J. Microb. Biot. 2011, 27, 1755–1764. [Google Scholar] [CrossRef]
- Van Scoy, A.R.; Tjeerdema, R.S. Environmental fate and toxicology of chlorothalonil. Rev. Environ. Contam. Toxicol. 2014, 232, 89–105. [Google Scholar] [PubMed]
- Zhang, Q.; Ji, C.; Yan, L.; Lu, M.; Lu, C.; Zhao, M. The identification of the metabolites of chlorothalonil in zebrafish (Danio rerio) and their embryo toxicity and endocrine effects at environmentally relevant levels. Environ. Pollut. 2016, 218, 8–15. [Google Scholar] [CrossRef]
- Cascão, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A spectrum of treatment opportunities in chronic diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Lee, D.; Eom, Y.B. Anti-biofilm and anti-virulence efficacy of celastrol against Stenotrophomonas maltophilia. Int. J. Med. Sci. 2018, 15, 617–627. [Google Scholar] [CrossRef]
- Zhang, J.; Li, C.Y.; Xu, M.J.; Wu, T.; Chu, J.H.; Liu, S.J.; Ju, W.Z. Oral bioavailability and gender-related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats. J. Ethnopharmacol. 2012, 144, 195–200. [Google Scholar] [CrossRef]
- Konieczny, J.; Jantas, D.; Lenda, T.; Domin, H.; Czarnecka, A.; Kuter, K.; Śmiałowska, M.; Lasoń, W.; Lorenc-Koci, E. Lack of neuroprotective effect of celastrol under conditions of proteasome inhibition by lactacystin in In Vitro and In Vivo studies: Implications for Parkinson’s disease. Neurotox. Res. 2014, 26, 255–273. [Google Scholar] [CrossRef]
- Bai, J.P.; Shi, Y.L.; Fang, X.; Shi, Q.X. Effects of demethylzeylasteral and celastrol on spermatogenic cell Ca2+ channels and progesterone-induced sperm acrosome reaction. Eur. J. Pharmacol. 2003, 464, 9–15. [Google Scholar] [CrossRef]
- Chang, D.; Garcia, R.A.; Akers, K.S.; Mende, K.; Murray, C.K.; Wenke, J.C.; Sanchez, C.J. Activity of gallium meso-and protoporphyrin IX against biofilms of multidrug-resistant Acinetobacter baumannii isolates. Pharmaceuticals 2016, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, S.; Visca, P.; Frangipani, E. Gallium-protoporphyrin IX inhibits Pseudomonas aeruginosa growth by targeting cytochromes. Front. Cell Infect. Microbiol. 2017, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Stojiljkovic, I.; Kumar, V.; Srinivasan, N. Non-iron metalloporphyrins: Potent antibacterial compounds that exploit haem/Hb uptake systems of pathogenic bacteria. Mol. Microbiol. 1999, 31, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Kosanić, M.; Ranković, B. Antioxidant and antimicrobial properties of some lichens and their constituents. J. Med. Food 2011, 14, 1624–1630. [Google Scholar] [CrossRef] [PubMed]
- Lohézic-Le Dévéhat, F.; Tomasi, S.; Elix, J.A.; Bernard, A.; Rouaud, I.; Uriac, P.; Boustie, J. Stictic acid derivatives from the lichen Usnea articulata and their antioxidant activities. J. Nat. Prod. 2007, 70, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Schiebel, J.; Radeva, N.; Köster, H.; Metz, A.; Krotzky, T.; Kuhnert, M.; Diederich, W.E.; Heine, A.; Neumann, L.; Atmanene, C.; et al. One question, multiple answers: Biochemical and biophysical screening methods retrieve deviating fragment hit lists. ChemMedChem 2015, 10, 1511–1521. [Google Scholar] [CrossRef]
- Martin, L.J.; Koegl, M.; Bader, G.; Cockcroft, X.L.; Fedorov, O.; Fiegen, D.; Gerstberger, T.; Hofmann, M.H.; Hohmann, A.F.; Kessler, D.; et al. Structure-based design of an In Vivo active selective BRD9 inhibitor. J. Med. Chem. 2016, 59, 4462–4475. [Google Scholar] [CrossRef]
- Ilina, P.; Ma, X.; Sintim, H.O.; Tammela, P. Miniaturized whole-cell bacterial bioreporter assay for identification of quorum sensing interfering compounds. J. Microbiol. Methods 2018, 154, 40–45. [Google Scholar] [CrossRef]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Lamers, R.P.; Cavallari, J.F.; Burrows, L.L. The efflux inhibitor phenylalanine-arginine β-naphthylamide (PAβN) permeabilizes the outer membrane of Gram-negative bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatta, V.; Ilina, P.; Porter, A.; McElroy, S.; Tammela, P. Targeting Quorum Sensing: High-Throughput Screening to Identify Novel LsrK Inhibitors. Int. J. Mol. Sci. 2019, 20, 3112. https://doi.org/10.3390/ijms20123112
Gatta V, Ilina P, Porter A, McElroy S, Tammela P. Targeting Quorum Sensing: High-Throughput Screening to Identify Novel LsrK Inhibitors. International Journal of Molecular Sciences. 2019; 20(12):3112. https://doi.org/10.3390/ijms20123112
Chicago/Turabian StyleGatta, Viviana, Polina Ilina, Alison Porter, Stuart McElroy, and Päivi Tammela. 2019. "Targeting Quorum Sensing: High-Throughput Screening to Identify Novel LsrK Inhibitors" International Journal of Molecular Sciences 20, no. 12: 3112. https://doi.org/10.3390/ijms20123112
APA StyleGatta, V., Ilina, P., Porter, A., McElroy, S., & Tammela, P. (2019). Targeting Quorum Sensing: High-Throughput Screening to Identify Novel LsrK Inhibitors. International Journal of Molecular Sciences, 20(12), 3112. https://doi.org/10.3390/ijms20123112