Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development



 , , ,
, , ,
Abstract
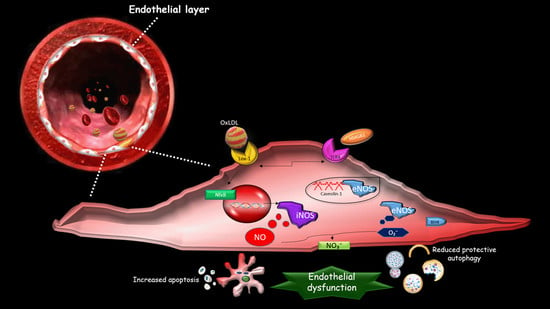
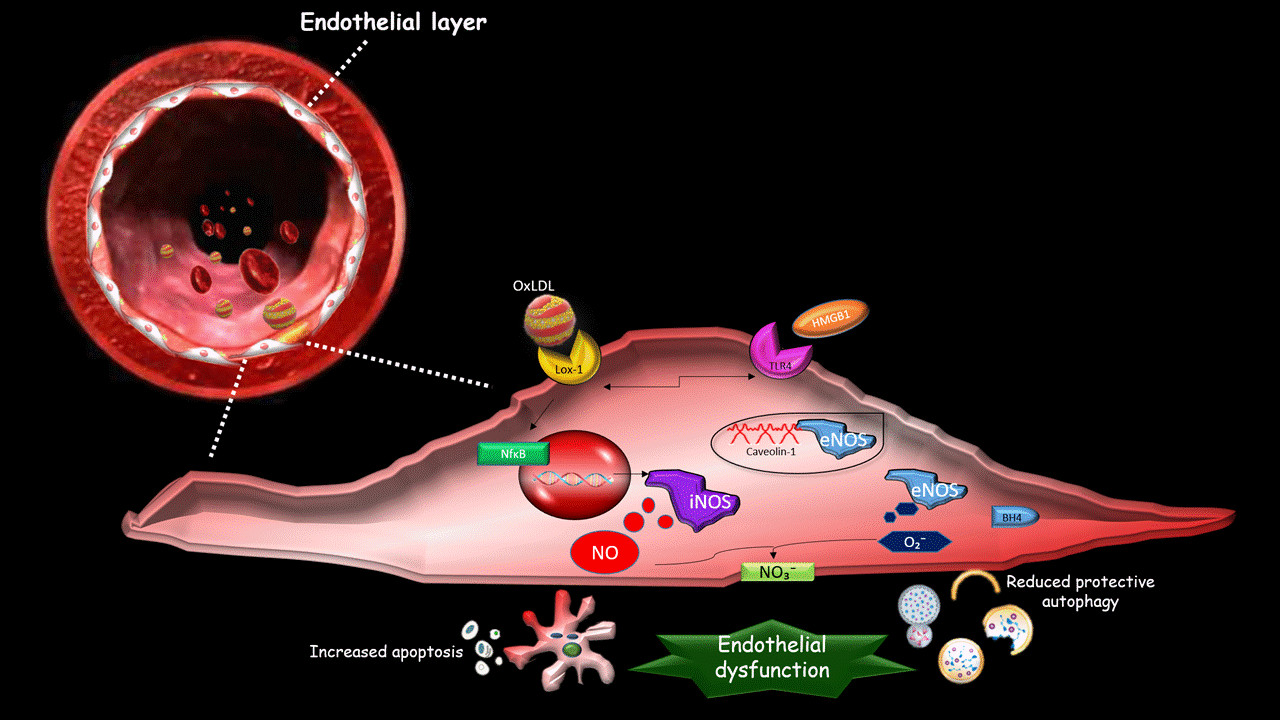
1. Introduction
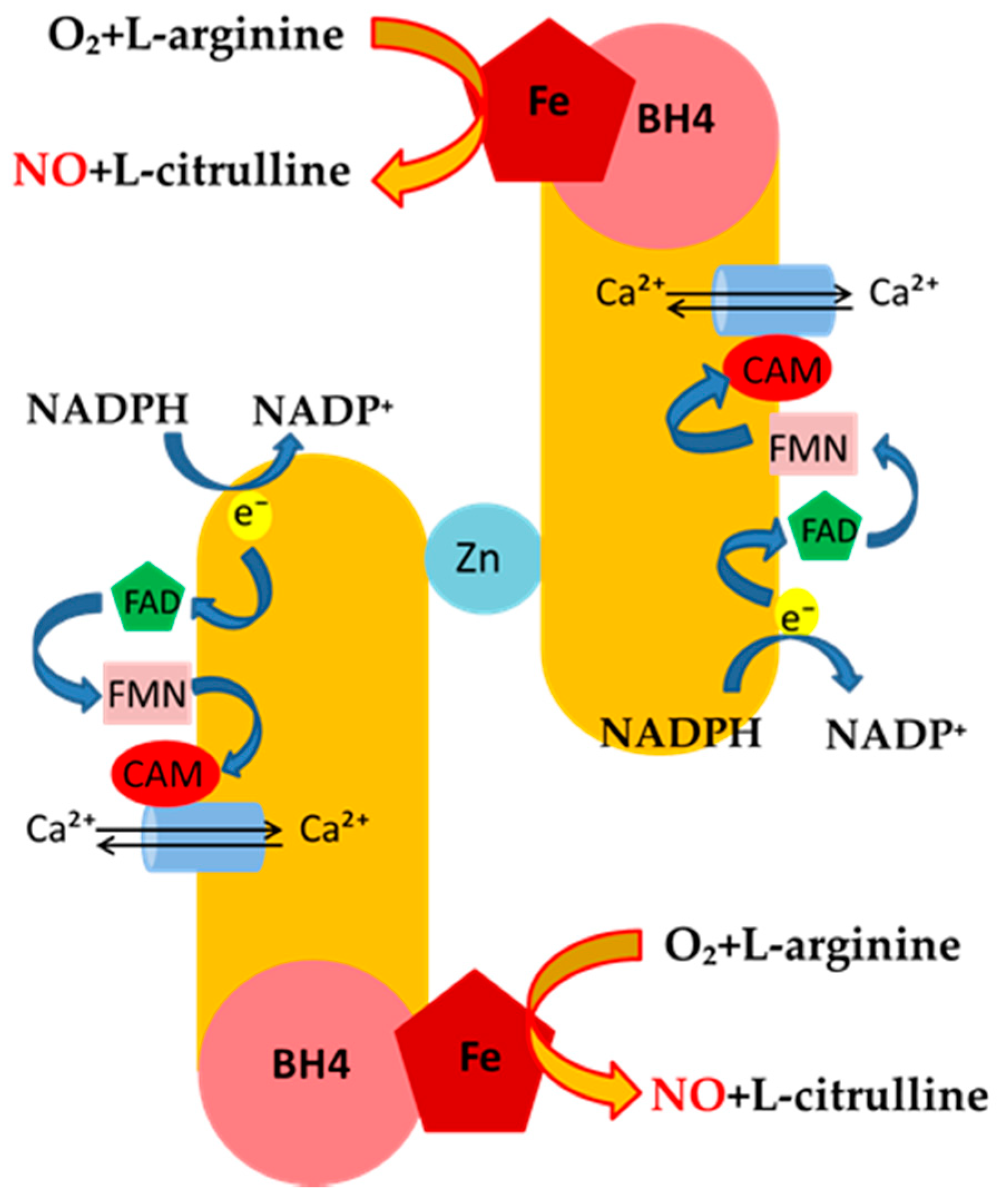
Mechanisms of NO Release and NOS Regulation
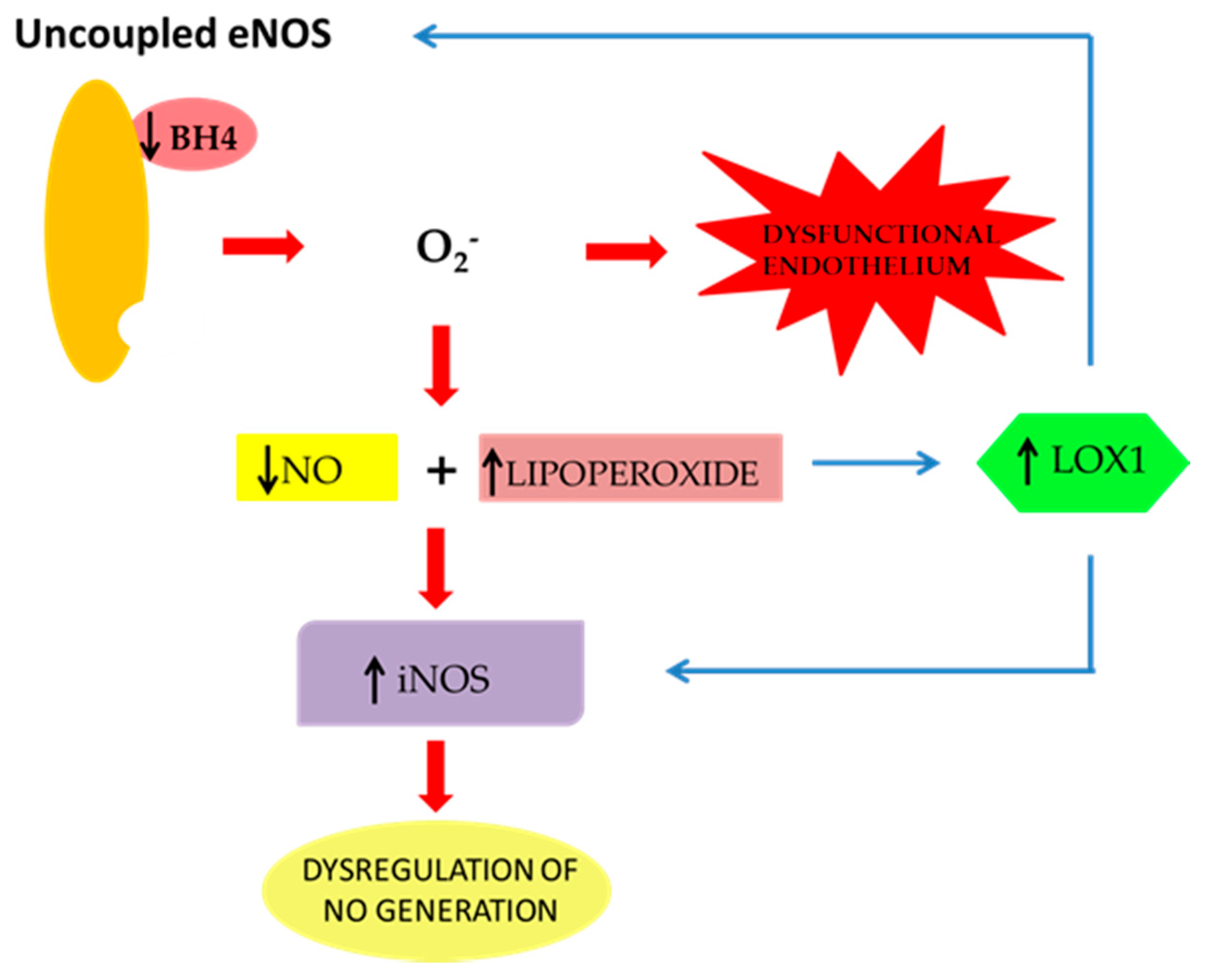
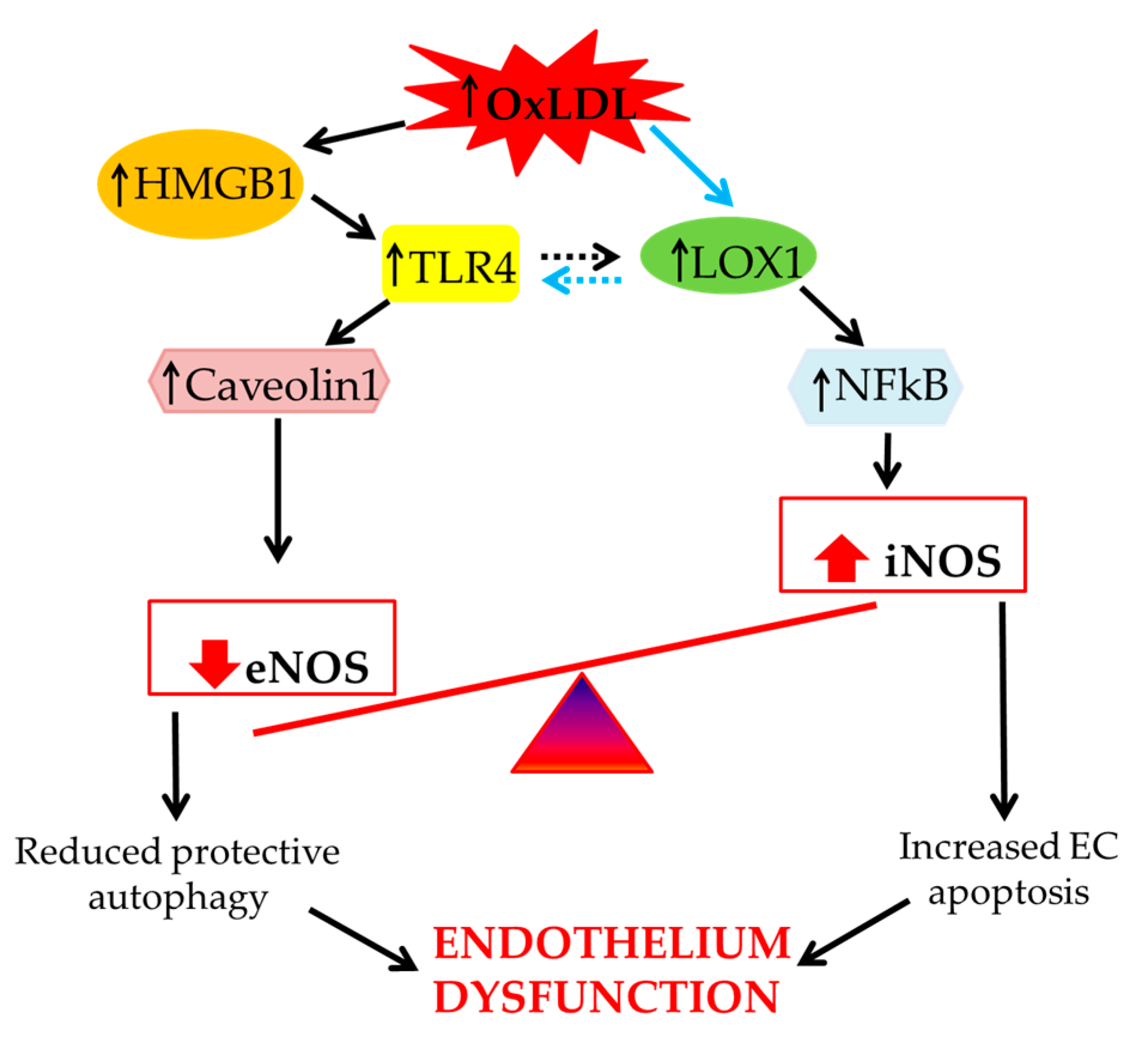
2. The Effect of oxLDLs in the Modulation of NOS Isoforms
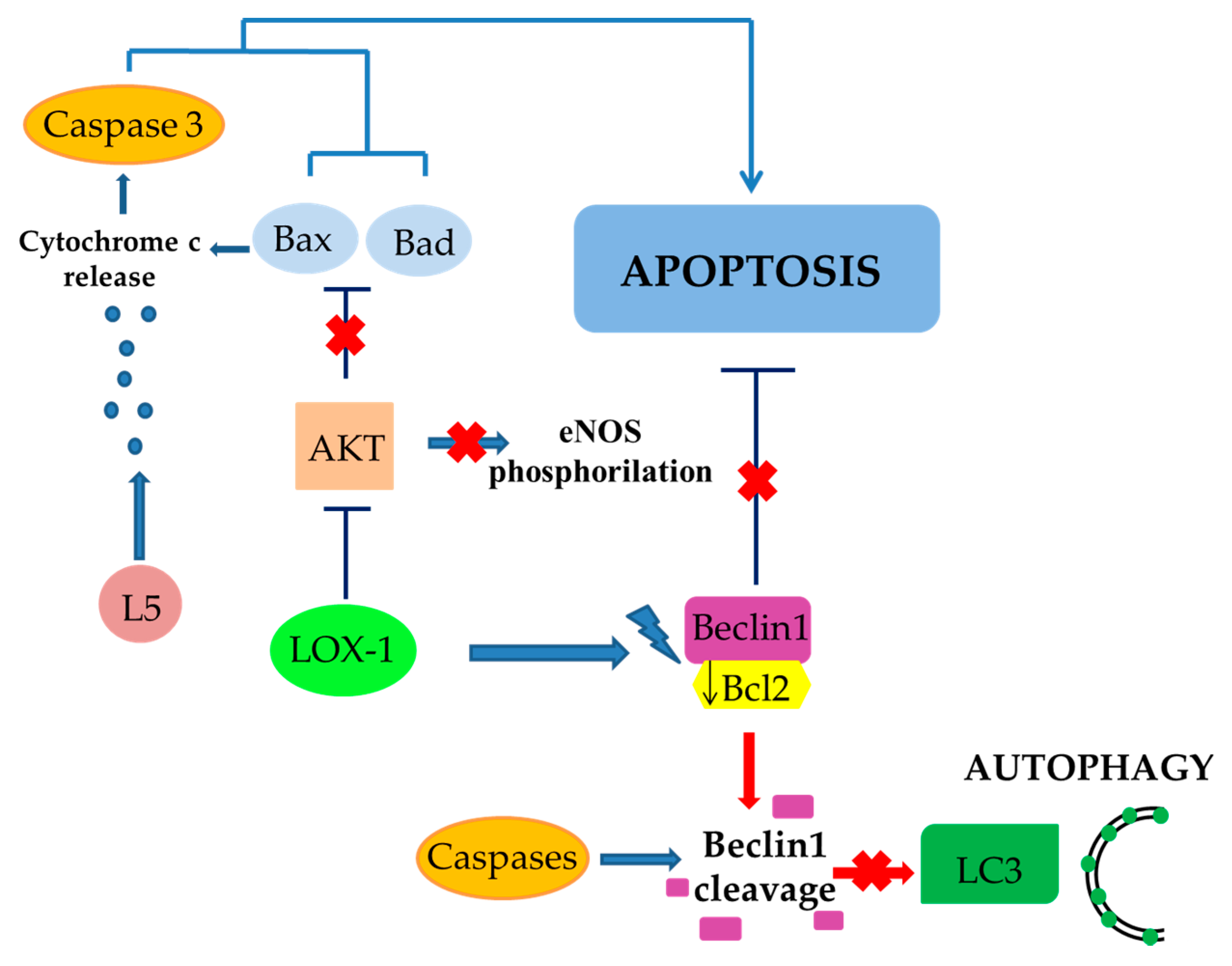
3. Role of Scavenger Receptor LOX-1 in iNOS Modulation
4. The Role of LOX-1/iNOS Activation in the Crosstalk between Apoptosis and Autophagy of ECs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Scicchitano, M.; Carresi, C.; Scarano, F.; Bosco, F.; Nucera, S.; Ruga, S.; Zito, M.C.; et al. The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. Int. J. Mol. Sci. 2018, 19, 2693. [Google Scholar] [CrossRef] [PubMed]
- Lanuti, P.; Rotta, G.; Almici, C.; Avvisati, G.; Budillon, A.; Doretto, P.; Malara, N.; Marini, M.; Neva, A.; Simeone, P.; et al. Endothelial progenitor cells, defined by the simultaneous surface expression of VEGFR2 and CD133, are not detectable in healthy peripheral and cord blood. Cytom. A 2016, 89, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Malara, N.M.; Trunzo, V.; Musolino, G.; Aprigliano, S.; Rotta, G.; Macrina, L.; Limongi, T.; Gratteri, S.; di Fabrizio, E.; Renzulli, A.; et al. Soluble CD54 induces human endothelial cells ex vivo expansion useful for cardiovascular regeneration and tissue engineering application. Int. J. Cardiol. Heart Vasc. 2015, 6, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Di, X.; Tang, X.; di, X. Montelukast inhibits oxidized low-density lipoproteins (ox-LDL) induced vascular endothelial attachment: An implication for the treatment of atherosclerosis. Biochem. Biophys Res. Commun. 2017, 486, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Aw, N.H.; Canetti, E.; Suzuki, K.; Goh, J. Monocyte Subsets in Atherosclerosis and Modification with Exercise in Humans. Antioxidants 2018, 19, 196. [Google Scholar] [CrossRef]
- Colasanti, M.; Suzuki, H. The dual personality of NO. Trends Pharm. Sci. 2000, 21, 249–252. [Google Scholar] [CrossRef]
- Stancu, C.S.; Toma, L.; Sima, A.V. Dual role of lipoproteins in endothelial cell dysfunction in atherosclerosis. Cell Tissue Res. 2012, 349, 433–446. [Google Scholar] [CrossRef]
- Mollace, V.; Sacco, I.; Janda, E.; Malara, C.; Ventrice, D.; Colica, C.; Visalli, V.; Muscoli, S.; Ragusa, S.; Muscoli, C.; et al. Hypolipemic and hypoglycaemic activity of bergamot polyphenols: From animal models to human studies. Fitoterapia 2011, 82, 309–316. [Google Scholar] [CrossRef]
- Sigala, F.; Kotsinas, A.; Savari, P.; Filis, K.; Markantonis, S.; Iliodromitis, E.K.; Gorgoulis, V.G.; Andreadou, I. Oxidized LDL in human carotid plaques is related to symptomatic carotid disease and lesion instability. J. Vasc. Surg. 2010, 52, 704–713. [Google Scholar] [CrossRef]
- Sigala, F.; Efentakis, P.; Karageorgiadi, D.; Filis, K.; Zampas, P.; Iliodromitis, E.K.; Zografos, G.; Papapetropoulos, A.; Andreadou, I. Reciprocal regulation of eNOS, H2S and CO-synthesizing enzymes in human atheroma: Correlation with plaque stability and effects of simvastatin. Redox Biol. 2017, 12, 70–81. [Google Scholar] [CrossRef]
- Holowatz, L.A.; Kenney, W.L. Acute localized administration of tetrahydrobiopterin and chronic systemic atorvastatin treatment restore cutaneous microvascular function in hypercholesterolaemic humans. J. Physiol 2011, 589, 4787–4797. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Macrì, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int. J. Mol. Sci. 2019, 20, 3022. [Google Scholar] [CrossRef] [PubMed]
- Akalin, C.G.; Ertorun, İ.; Akalin, A.; Alataş, İ.Ö.; Musmul, A. The effects of atorvastatin on antioxidant/antiinflammatory properties of HDLs in hypercholesterolemics. Turk. J. Med. Sci. 2015, 45, 345–351. [Google Scholar] [CrossRef]
- Rossi, R.; Cioni, E.; Nuzzo, A.; Origliani, G.; Modena, M.G. Endothelial-Dependent Vasodilation and Incidence of Type 2 Diabetes in a Population of Healthy Postmenopausal Women. Diabetes Care 2005, 28, 702–707. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, W.T.; Efird, J.T.; Yeboah, J.; Nazarian, S.; Alonso, A.; Heckbert, S.R.; Soliman, E.Z. Brachial Flow-Mediated Dilation and Incident Atrial Fibrillation: The Multi-Ethnic Study of Atherosclerosis. Arter. Thromb. Vasc. Biol. 2014, 34, 2717–2720. [Google Scholar] [CrossRef] [PubMed]
- Kenney, W.L.; Cannon, J.G.; Alexander, L.M. Cutaneous microvascular dysfunction correlates with serum LDL and sLOX-1 receptor concentrations. Microvasc. Res. 2013, 85, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; Balzan, S. Roles of LOX-1 in microvascular dysfunction. Microvasc. Res. 2016, 105, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Ma, G.; Chen, X. Lipopolysaccharide induced LOX-1 expression via TLR4/MyD88/ROS activated p38MAPK-NF-κB pathway. Vasc. Pharm. 2014, 63, 162–172. [Google Scholar] [CrossRef]
- Lubrano, V.; Balzan, S. LOX-1 and ROS, inseparable factors in the process of endothelial damage. Free Radic Res. 2014, 48, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Handy, D.E.; Loscalzo, J. Role of oxidative stress and nitric oxide in atherothrombosis. Front. Biosci. 2008, 13, 5323–5344. [Google Scholar] [CrossRef]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and Atherosclerosis. Mediators Inflamm. 2013. [CrossRef] [PubMed]
- Lubrano, V.; Gabriele, M.; Puntoni, M.R.; Longo, V.; Pucci, L. Relationship among IL-6, LDL cholesterol and lipid peroxidation. Cell Mol. Biol. Lett. 2015, 20, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, M.; Konopinski, R.; Krishnan, M.; Roman, L.; Bera, A.; Hongying, Z.; Habib, S.L.; Mohan, S. Inhibitor-κB kinase attenuates Hsp90-dependent endothelial nitric oxide synthase function in vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2015, 308, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, K.A., Jr.; Ackerman, A.W.; Gross, E.R.; Stepp, D.W.; Shi, Y.; Fontana, J.T.; Baker, J.E.; Sessa, W.C. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J. Biol. Chem. 2001, 276, 17621–17624. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Gliozzi, M. The potential role of TLR4/caveolin-1/NOS pathway in oxyLDL-modulation of autophagic/apoptotic responses in endothelial cells. Int. J. Cardiol. 2016, 15, 457–458. [Google Scholar] [CrossRef]
- Heiss, C.; Rodriguez-Mateos, A.; Kelm, M. Central Role of eNOS in the Maintenance of Endothelial Homeostasis. Antioxid. Redox Signal. 2015, 22, 1230–1242. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, pharmacology. Pharm. Rev. 1991, 43, 109–142. [Google Scholar]
- Lamoke, F.; Mazzone, V.; Persichini, T.; Maraschi, A.; Harris, M.B.; Venema, C.; Marco Colasanti, M.; Gliozzi, M.; Muscoli, C.; Bartoli, M.; et al. Amyloid β peptide-induced inhibition of endothelial nitric oxide production involves oxidative stress-mediated constitutive eNOS/HSP90 interaction and disruption of agonist-mediated Akt activation. J. Neuroinflamm. 2015, 12, 84. [Google Scholar]
- Mollace, V.; Salvemini, D.; Anggård, E.; Vane, J. Cultured astrocytoma cells inhibit platelet aggregation by releasing a nitric oxide-like factor. Biochem. Biophys Res. Commun. 1990, 172, 564–569. [Google Scholar] [CrossRef]
- Mollace, V.; Muscoli, C.; Rotiroti, D.; Nisticó, G. Spontaneous induction of nitric oxide- and prostaglandin E2-release by hypoxic astroglial cells is modulated by interleukin 1 beta. Biochem. Biophys Res. Commun. 1997, 238, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Mollace, V.; Pistelli, A.; Anggård, E.; Vane, J. Cultured astrocytoma cells generate a nitric oxide-like factor from endogenous L-arginine and glyceryl trinitrate: Effect of E. coli lipopolysaccharide. Br. J. Pharm. 1992, 106, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Rodino, P.; Massoud, R.; Rotiroti, D.; Nistico, G. Age-dependent changes of NO synthase activity in the rat brain. Biochem. Biophys Res. Commun. 1995, 215, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Currie, M.G.; Mollace, V. Nitric oxide-mediated cyclooxygenase activation. A key event in the antiplatelet effects of nitrovasodilators. J. Clin. Investig. 1996, 97, 2562–2568. [Google Scholar] [PubMed]
- Garthwaite, J. NO as a multimodal transmitter in the brain: Discovery and current status. Br. J. Pharm. 2019, 176, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Li, P.Y.; Wang, X.; Stetler, R.A.; Chen, J.; Yu, W.F. Anti-inflammatory signaling: The point of convergence for medical gases in neuroprotection against ischemic stroke. Med. Gas. Res. 2016, 6, 227–231. [Google Scholar] [PubMed]
- Colasanti, M.; Cavalieri, E.; Persichini, T.; Mollace, V.; Mariotto, S.; Suzuki, H.; Lauro, G.M. Bacterial lipopolysaccharide plus interferon-gamma elicit a very fast inhibition of a Ca2+-dependent nitric-oxide synthase activity in human astrocytoma cells. J. Biol. Chem. 1997, 272, 7582–7585. [Google Scholar] [CrossRef] [PubMed]
- Mollace, R.; Gliozzi, M.; Tavernese, A.M.; Musolino, V.; Carresi, C.; Scicchitano, M.; Palma, E.; Nucera, S.; Bosco, F.; Scarano, F.; et al. Bergamot Polyphenolic Fraction supplementation improves metabolic balance, endothelial function and maximal oxygen uptake in athletes. J. Sports Med. 2018, 3, 53–61. [Google Scholar]
- Treuer, A.V.; Gonzalez, D.R. Nitric oxide synthases, S-nitrosylation and cardiovascular health: From molecular mechanisms to therapeutic opportunities. Mol. Med. Rep. 2015, 11, 1555–1565. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, J.; Kwak, S.N.; Lee, K.S.; Lee, D.K.; Ha, K.S.; Won, M.H.; Jeoung, D.; Lee, H.; Kwon, Y.G.; et al. Functional role of NF-κB in expression of human endothelial nitric oxide synthase. Biochem. Biophys Res. Commun. 2014, 448, 101–107. [Google Scholar] [CrossRef]
- Janda, E.; Visalli, V.; Colica, C.; Aprigliano, S.; Musolino, V.; Vadalà, N.; Muscoli, C.; Sacco, I.; Iannone, M.; Rotiroti, D.; et al. The protective effect of tianeptine on Gp120-induced apoptosis in astroglial cells: Role of GS and NOS, and NF-κB suppression. Br. J. Pharm. 2011, 164, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Jiang, X.; Zhu, Z.; Qin, H.; Dinkins, M.B.; Kong, J.N.; Leanhart, S.; Wang, R.; Elsherbini, A.; Bieberich, E.; et al. Lipid transporter Spns2 promotes microglia pro-inflammatory activation in response to amyloid-beta peptide. Glia 2019, 67, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Massa, P.T.; Wu, C. Increased inducible activation of NF-KB and responsive genes in astrocytes deficient in the protein tyrosine phosphatase SHP-1. J. Interferon Cytokine Res. 1998, 18, 499–507. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, A.; Schroeder, R.A.; Bartlett, S.T.; Kuo, P.C. Differential effects of nitric oxide-mediated S-nitrosylation on p50 and c-jun DNA binding. Surgery 1998, 124, 137–141. [Google Scholar] [CrossRef]
- Baig, M.S.; Zaichick, S.V.; Mao, M.; de Abreu, A.L.; Bakhshi, F.R.; Hart, P.C.; Saqib, U.; Deng, J.; Chatterjee, S.; Block, M.L.; et al. NOS1-derived nitric oxide promotes NF-κB transcriptional activity through inhibition of suppressor of cytokine signaling-1. J. Exp. Med. 2015, 212, 1725–1738. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Muscoli, C.; Masini, E.; Cuzzocrea, S.; Salvemini, D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharm. Rev. 2005, 57, 217–252. [Google Scholar] [CrossRef]
- Braverman, J.; Stanley, S.A. Nitric Oxide Modulates Macrophage Responses to Mycobacterium tuberculosis Infection through Activation of HIF-1α and Repression of NF-κB. J. Immunol. 2017, 199, 1805–1816. [Google Scholar] [CrossRef]
- Aquaro, S.; Muscoli, C.; Ranazzi, A.; Pollicita, M.; Granato, T.; Masuelli, L.; Modesti, A.; Perno, C.F.; Mollace, V. The contribution of peroxynitrite generation in HIV replication in human primary macrophages. Retrovirology 2007, 4, 76. [Google Scholar] [CrossRef]
- Gliozzi, M.; Carresi, C.; Musolino, V.; Palma, E.; Muscoli, C.; Vitale, C.; Gratteri, S.; Muscianisi, G.; Janda, E.; Muscoli, S.; et al. The Effect of Bergamot-Derived Polyphenolic Fraction on LDL Small Dense articles and Non Alcoholic Fatty Liver Disease in Patients with Metabolic Syndrome. Adv. Biol. Chem. 2014, 4, 129–137. [Google Scholar] [CrossRef]
- Walker, R.; Janda, E.; Mollace, V. The Use of Bergamot-Derived Polyphenol Fraction in Cardiometabolic Risk Prevention and its Possible Mechanisms of Action. Polyphen. Hum. Health Disease 2014, 2, 1087–1105. [Google Scholar]
- Tassone, E.J.; Perticone, M.; Sciacqua, A.; Mafrici, S.F.; Settino, C.; Malara, N.; Mollace, V.; Sesti, G.; Perticone, F. Low dose of acetylsalicylic acid and oxidative stress-mediated endothelial dysfunction in diabetes: A short-term evaluation. Acta. Diabetol. 2015, 52, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Gutierrez-Pajares, J.L.; Katiyar, S.; Jasmin, J.F.; Mercier, I.; Walters, R.; Pavlides, C.; Pestell, R.G.; Lisanti, M.P.; Frank, P.G. Caveolin-1 regulates the anti-atherogenic properties of macrophages. Cell Tissue Res. 2014, 358, 821–831. [Google Scholar] [CrossRef]
- Pojoga, L.H.; Yao, T.M.; Opsasnick, L.A.; Siddiqui, W.T.; Reslan, O.M.; Adler, G.K.; Williams, G.H.; Khalil, R.A. Cooperative Role of Mineralocorticoid Receptor and Caveolin-1 in Regulating the Vascular Response to Low Nitric Oxide-High Angiotensin II-Induced Cardiovascular Injury. J. Pharm. Exp. 2015, 355, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Feron, O. Endothelial nitric oxide synthase expression and its functionality. Curr. Opin. Clin. Nutr. Metab. Care 1999, 2, 291–296. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mollace, V.; Gliozzi, M.; Carresi, C.; Musolino, V.; Oppedisano, F. Re-assessing the mechanism of action of n-3 PUFAs. Int. J. Cardiol. 2013, 170, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.L.; Yang, X.Y.; Li, J.Y.; Li, Y.; Jia, X.; Xiong, Z.F.; Wang, Y.M.; Jin, S. Cavin-1 regulates caveolae-mediated LDL transcytosis: Crosstalk in an AMPK/eNOS/ NF-κB/Sp1 loop. Oncotarget 2017, 8, 103985–103995. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Zhang, Y.; Yan, Z.; Wang, Z.G.; Liu, G.; Minshall, R.D.; Malik, A.B.; Hu, G. Caveolin-1 Tyr14 phosphorylation induces interaction with TLR4 in endothelial cells and mediates MyD88-dependent signalingand sepsis-induced lung inflammation. J. Immunol. 2013, 191, 6191–6199. [Google Scholar] [CrossRef]
- Qin, Z.; Zhaowei, Z.; Xinqun, H.; Chang, S. HMGB1: A critical mediator for oxidized-low density lipoproteins induced atherosclerosis. Int. J. Cardiol. 2016, 202, 956–957. [Google Scholar]
- Tian, K.; Ogura, S.; Little, P.J.; Xu, S.W.; Sawamura, T. Targeting LOX-1 in atherosclerosis and vasculopathy: Current knowledge and future perspectives. Ann. N.Y. Acad. Sci. 2018, 1443, 34–53. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Khaidakov, M.; Dai, Y.; Deng, X.; Fan, Y.; Xiang, D.; Mehta, J.L. Lectin-like ox-LDL receptor-1 (LOX-1)-Toll-like receptor 4 (TLR4) interaction and autophagy in CATH. a differentiated cells exposed to angiotensin II. Mol. Neurobiol. 2015, 51, 623–632. [Google Scholar] [CrossRef]
- Balzan, S.; Lubrano, V. LOX-1 receptor: A potential link in atherosclerosis and cancer. Life Sci. 2018, 198, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Ragusa, S.; Sacco, I.; Muscoli, C.; Sculco, F.; Visalli, V.; Palma, E.; Muscoli, S.; Mondello, L.; Dugo, P.; et al. The protective effect of bergamot oil extract on lecitine-like oxyLDL receptor-1 expression in balloon injury-related neointima formation. J. Cardiovasc Pharm. 2008, 3, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Gliozzi, M.; Musolino, V.; Carresi, C.; Muscoli, S.; Mollace, R.; Tavernese, A.M.; Gratteri, S.; Palma, E.; Morabito, C.; et al. Oxidized LDL attenuates protective autophagy and induces apoptotic cell death of endothelial cells: Role of oxidative stress and LOX-1 receptor expression. Int. J. Cardiol. 2015, 184, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Muscoli, C.; Sacco, I.; Alecce, W.; Palma, E.; Nisticò, R.; Costa, N.; Clementi, F.; Rotiroti, D.; Romeo, F.; Salvemini, D.; et al. The protective effect of superoxide dismutase mimetic M40401 on balloon injury-related neointima formation: Role of the lectin-like oxidized low-density lipoprotein receptor-1. J. Pharm. Exp. 2004, 311, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Cong, S. LOX-1 and atherosclerotic-related diseases. Clin. Chim. Acta. 2019, 491, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Jono, T.; Miyazaki, A.; Nagai, R.; Sawamura, T.; Kitamura, T.; Horiuchi, S. Lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) serves as an endothelial receptor for advanced glycation end products (AGE). Febs. Lett. 2002, 511, 170–174. [Google Scholar] [CrossRef]
- Otsuki, T.; Maeda, S.; Mukai, J.; Ohki, M.; Nakanishi, M.; Yoshikawa, T. Association between plasma sLOX-1 concentration and arterial stiffness in middle-aged and older individuals. Clin. Biochem. Nutr. 2015, 57, 151–155. [Google Scholar] [CrossRef]
- Yavuzer, S.; Yavuzer, H.; Cengiz, M.; Erman, H.; Altıparmak, M.R.; Korkmazer, B.; Balci, H.; Simsek, G.; Yaldıran, A.L.; Karter, Y.; et al. Endothelial damage in white coat hypertension: Role of lectin-like oxidized low-density lipoprotein-1. J. Hum. Hypertens 2015, 29, 92–98. [Google Scholar] [CrossRef]
- Chen, M.; Nagase, M.; Fujita, T.; Narumiya, S.; Masaki, T.; Sawamura, T. Diabetes enhances lectin-like oxidized LDL receptor-1 (LOX-1) expression in the vascular endothelium: Possible role of LOX-1 ligand and AGE. Biochem. Biophys Res. Commun. 2001, 287, 962–968. [Google Scholar] [CrossRef]
- Chen, H.; Li, D.; Sawamura, T.; Inoue, K.; Mehta, J.L. Upregulation of LOX-1 expression in aorta of hypercholesterolemic rabbits: Modulation by losartan. Biochem Biophys Res. Commun. 2000, 276, 1100–1104. [Google Scholar] [CrossRef]
- Nagase, M.; Hirose, S.; Sawamura, T.; Masaki, T.; Fujita, T. Enhanced expression of endothelial oxidized low-density lipoprotein receptor (LOX-1) in hypertensive rats. Biochem. Biophys Res. Commun. 1997, 237, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Aluganti Narasimhulu, C.; Burge, K.Y.; Doomra, M.; Riad, A.; Parthasarathy, S. Primary prevention of atherosclerosis by pretreatment of low-density lipoprotein receptor knockout mice with sesame oil and its aqueous components. Sci Rep. 2018, 8, 12270. [Google Scholar] [CrossRef] [PubMed]
- Grell, A.S.; Frederiksen, S.D.; Edvinsson, L.; Ansar, S. Cerebrovascular gene expression in spontaneously hypertensive rats. PLoS ONE 2017, 12, e0184233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, C.; Wang, H.; Lu, M.; Li, Y.; Feng, H.; Lin, J.; Yuan, Z.; Wang, X. Ox-LDL promotes migration and adhesion of bone marrow-derived mesenchymal stem cells via regulation of MCP-1 expression. Mediat. Inflamm. 2013, 2013, 691023. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.W.; Yang, E.; Yoo, K.H.; Choi, I.H. Macrophage Differentiation from Monocytes Is Influenced by the Lipid Oxidation Degree of Low Density Lipoprotein. Mediat. Inflamm. 2015, 2015, 235797. [Google Scholar] [CrossRef]
- Chernyavskiy, I.; Veeranki, S.; Sen, U.; Tyagi, S.C. Atherogenesis: Hyperhomocysteinemia interactions with LDL, macrophage function, paraoxonase 1, and exercise. Ann. N.Y. Acad. Sci. 2016, 1363, 138–154. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Aw, T.Y.; Kvietys, P.R.; Granger, D.N. Oxidized LDL-induced microvascular dysfunction. Dependence on oxidation procedure. Arter. Thromb Vasc. Biol. 1995, 15, 2305–2311. [Google Scholar] [CrossRef]
- Ou, H.C.; Song, T.Y.; Yeh, Y.C.; Huang, C.Y.; Yang, S.F.; Chiu, T.H.; Tsai, K.L.; Chen, K.L.; Wu, Y.J.; Tsai, C.S.; et al. EGCG protects against oxidized LDL induced endothelial dysfunction by inhibiting LOX-1-mediated signalling. J. Appl. Physiol. 2010, 108, 1745–1756. [Google Scholar] [CrossRef]
- Kume, N.; Kita, T. Apoptosis of vascular cells by oxidized LDL: Involvement of caspases and LOX-1 and its implication in atherosclerotic plaque rupture. Circ. Res. 2004, 94, 269–270. [Google Scholar] [CrossRef]
- Salvemini, D.; Kim, S.F.; Mollace, V. Reciprocal regulation of the nitric oxide and cyclooxygenase pathway in pathophysiology: Relevance and clinical implications. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, 473–487. [Google Scholar] [CrossRef]
- Akinwumi, B.C.; Bordun, K.A.M.; Anderson, H.D. Biological Activities of Stilbenoids. Int. J. Mol. Sci. 2018, 19, 792. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Lu, J.; Walterscheid, J.P.; Chen, H.H.; Engler, D.A.; Sawamura, T.; Chang, P.Y.; Safi, H.J.; Yang, C.Y.; Chen, C.H. Electronegative LDL circulating in smokers impairs endothelial progenitor cell differentiation by inhibiting Akt phosphorylation via LOX-1. J. Lipid Res. 2008, 49, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Janda, E.; Lascala, A.; Carresi, C.; Parafati, M.; Aprigliano, S.; Russo, V.; Savoia, C.; Ziviani, E.; Musolino, V.; Morani, F.; et al. Parkinsonian toxin-induced oxidative stress inhibits basal autophagy in astrocytes via NQO2/quinone oxidoreductase 2: Implications for neuroprotection. Autophagy 2015, 11, 1063–1080. [Google Scholar] [CrossRef]
- Gliozzi, M.; Walker, R.; Muscoli, S.; Vitale, C.; Gratteri, S.; Carresi, C.; Musolino, V.; Russo, V.; Janda, E.; Ragusa, S.; et al. Bergamot polyphenolic fraction enhances rosuvastatin-induced effect on LDL-cholesterol, LOX-1 expression and protein kinase B phosphorylation in patients with hyperlipidemia. Int. J. Cardiol. 2013, 170, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Roumeliotis, N.; Sawamura, T.; Renier, G. C-reactive protein enhances LOX-1 expression in human aortic endothelial cells: Relevance of LOX-1 to C-reactive protein-induced endothelial dysfunction. Circ. Res. 2004, 95, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Stancel, N.; Chen, C.C.; Ke, L.Y.; Chu, C.S.; Lu, J.; Sawamura, T.; Chen, C.H. Interplay between CRP, Atherogenic LDL, and LOX-1 and Its Potential Role in the Pathogenesis of Atherosclerosis. Clin. Chem. 2016, 62, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Eagle, K.A.; Ginsburg, G.S.; Musunuru, K.; Aird, W.C.; Balaban, R.S.; Bennett, S.K.; Blumenthal, R.S.; Coughlin, S.R.; Davidson, K.W.; Frohlich, E.D.; et al. Identifying patients at high risk of a cardiovascular event in the near future: Current status and future directions: Report of a national heart, lung, and blood institute working group. Circulation 2010, 121, 1447–1454. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef]
- Kita, T.; Kume, N.; Minami, M.; Hayashida, K.; Murayama, T.; Sano, H.; Moriwaki, H.; Kataoka, H.; Nishi, E.; Horiuchi, H.; et al. Role of oxidized LDL in atherosclerosis. Ann. N.Y. Acad. Sci. 2001, 947, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.L. The role of LOX-1, a novel lectin-like receptor for oxidized low density Dec;lipoprotein, in atherosclerosis. Can. J. Cardiol. 2004, 20 (Suppl. B), 32–36. [Google Scholar]
- Li, D.; Mehta, J.L. Intracellular signaling of LOX-1 in endothelial cell apoptosis. Circ. Res. 2009, 104, 566–568. [Google Scholar] [CrossRef]
- Lee, A.S.; Xi, Y.; Lai, C.H.; Chen, W.Y.; Peng, H.Y.; Chan, H.C.; Chen, C.H.; Chang, K.C. Human electronegative low-density lipoprotein modulates cardiac repolarization via LOX-1-mediated alteration of sarcolemmal ion channels. Sci. Rep. 2017, 7, 10889. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Carresi, C.; Musolino, V.; Gliozzi, M.; Maiuolo, J.; Mollace, R.; Nucera, S.; Maretta, A.; Sergi, D.; Muscoli, S.; Gratteri, S.; et al. Anti-oxidant effect of bergamot polyphenolic fraction counteracts doxorubicin-induced cardiomyopathy: Role of autophagy and ckitposCD45negCD31neg cardiac stem cell activation. J. Mol. Cell Cardiol. 2018, 119, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Park, H.A.; Broman, K.; Stumpf, A.; Kazyak, S.; Jonas, E.A. Nutritional Regulators of Bcl-xL in the Brain. Molecules 2018, 23, 3019. [Google Scholar] [CrossRef] [PubMed]
- Pihán, P.; Carreras-Sureda, A.; Hetz1, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 9, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Q.; Lin, J.L.; Wang, Y.; Zhang, R.X.; Hou, J.B.; Yu, B. Recombinant Recombinant Human Thioredoxin-1 Protects Macrophages from Oxidized Low-Density Lipoprotein-Induced Foam Cell Formation and Cell Apoptosis. Biomology 2018, 26, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Akyol, S.; Lu, J.; Akyol, O.; Akcay, F.; Armutcu, F.; Ke, L.Y.; Chen, C.H. The role of electronegative low-density lipoprotein in cardiovascular diseases and its therapeutic implications. Trends Cardiovasc. Med. 2017, 27, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Melino, G.; Knight, R.A. Cell death pathology: Cross-talk with autophagy and its clinical implications. Biochem. Biophys Res. Commun. 2011, 414, 277–281. [Google Scholar] [CrossRef]
- Parafati, M.; Lascala, A.; Morittu, V.M.; Trimboli, F.; Rizzuto, A.; Brunelli, E.; Coscarelli, F.; Costa, N.; Britti, D.; Ehrlich, J.; et al. Bergamot polyphenol fraction prevents nonalcoholic fatty liver disease via stimulation of lipophagy in cafeteria diet-induced rat model of metabolic syndrome. J. Nutr. Biochem. 2015, 26, 938–948. [Google Scholar] [CrossRef]
- Tang, D.Y.; Ellis, R.A.; Lovat, P.E. Prognostic Impact of Autophagy Biomarkers for Cutaneous Melanoma. Front. Oncol. 2016, 6, 236. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NOS Isoform | Expression | Function |
|---|---|---|
| eNOS | endothelial | Under basal conditions, the release of NO from eNOS is pulsed and tightly dependent on the rise of Ca2+ intracellular levels. This, in turn, leads to strong binding of calmodulin to the enzyme, which generates nM concentration of NO, which regulates:
|
| nNOS | neuronal | It is involved in neurotransmission in astrocytes and neurons [31,32]. |
| iNOS | inducible | It is overexpressed in response to different inflammatory stimuli, such as endogenous cytokines and bacterial lipopolysaccharide endotoxin (LPS) and causes a delayed, but persistent, synthesis of a large amount of NO. Leads to endothelial dysfunction and, in the late stages, to the development of atherothrombosis. NO production by iNOS causes the generation of high levels of peroxynitrite, which has been correlated with endothelial cell (EC) death via apoptosis [24,25,28]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. https://doi.org/10.3390/ijms20133294
Gliozzi M, Scicchitano M, Bosco F, Musolino V, Carresi C, Scarano F, Maiuolo J, Nucera S, Maretta A, Paone S, et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. International Journal of Molecular Sciences. 2019; 20(13):3294. https://doi.org/10.3390/ijms20133294
Chicago/Turabian StyleGliozzi, Micaela, Miriam Scicchitano, Francesca Bosco, Vincenzo Musolino, Cristina Carresi, Federica Scarano, Jessica Maiuolo, Saverio Nucera, Alessia Maretta, Sara Paone, and et al. 2019. "Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development" International Journal of Molecular Sciences 20, no. 13: 3294. https://doi.org/10.3390/ijms20133294
APA StyleGliozzi, M., Scicchitano, M., Bosco, F., Musolino, V., Carresi, C., Scarano, F., Maiuolo, J., Nucera, S., Maretta, A., Paone, S., Mollace, R., Ruga, S., Zito, M. C., Macrì, R., Oppedisano, F., Palma, E., Salvemini, D., Muscoli, C., & Mollace, V. (2019). Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. International Journal of Molecular Sciences, 20(13), 3294. https://doi.org/10.3390/ijms20133294