Directed Evolution of P450 BM3 towards Functionalization of Aromatic O-Heterocycles

 ,
,
Abstract
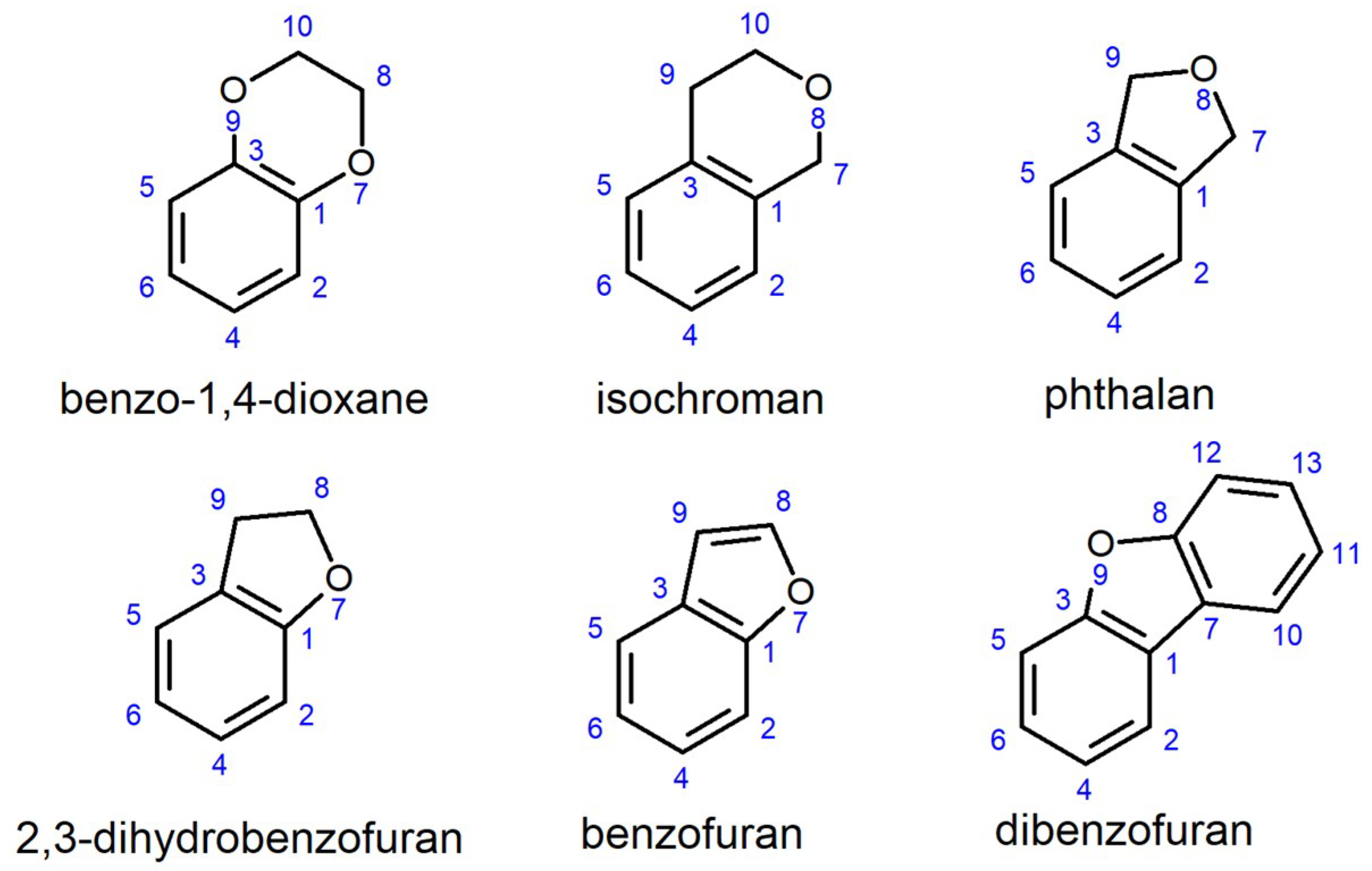
:1. Introduction
2. Results and Discussion
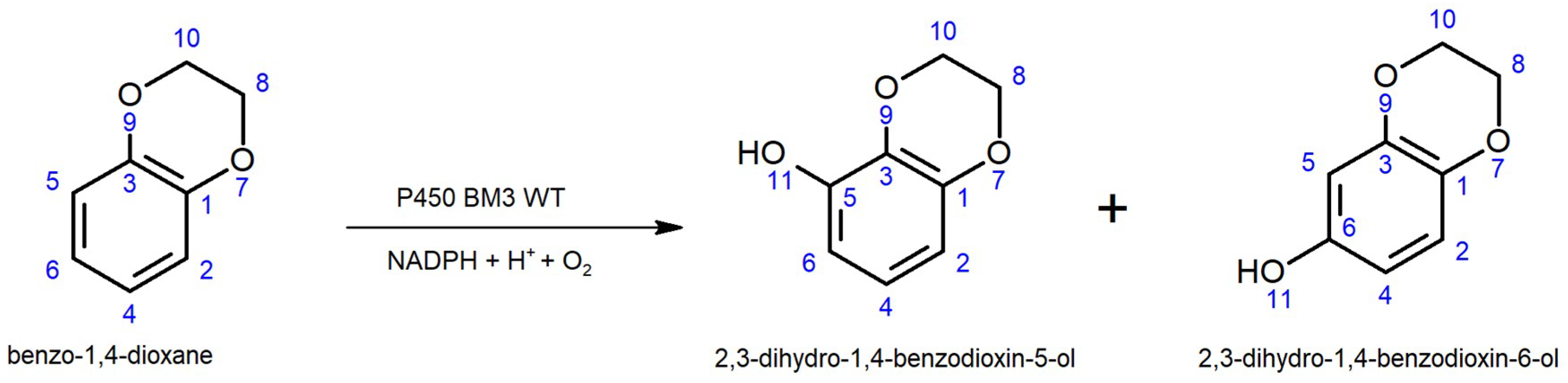
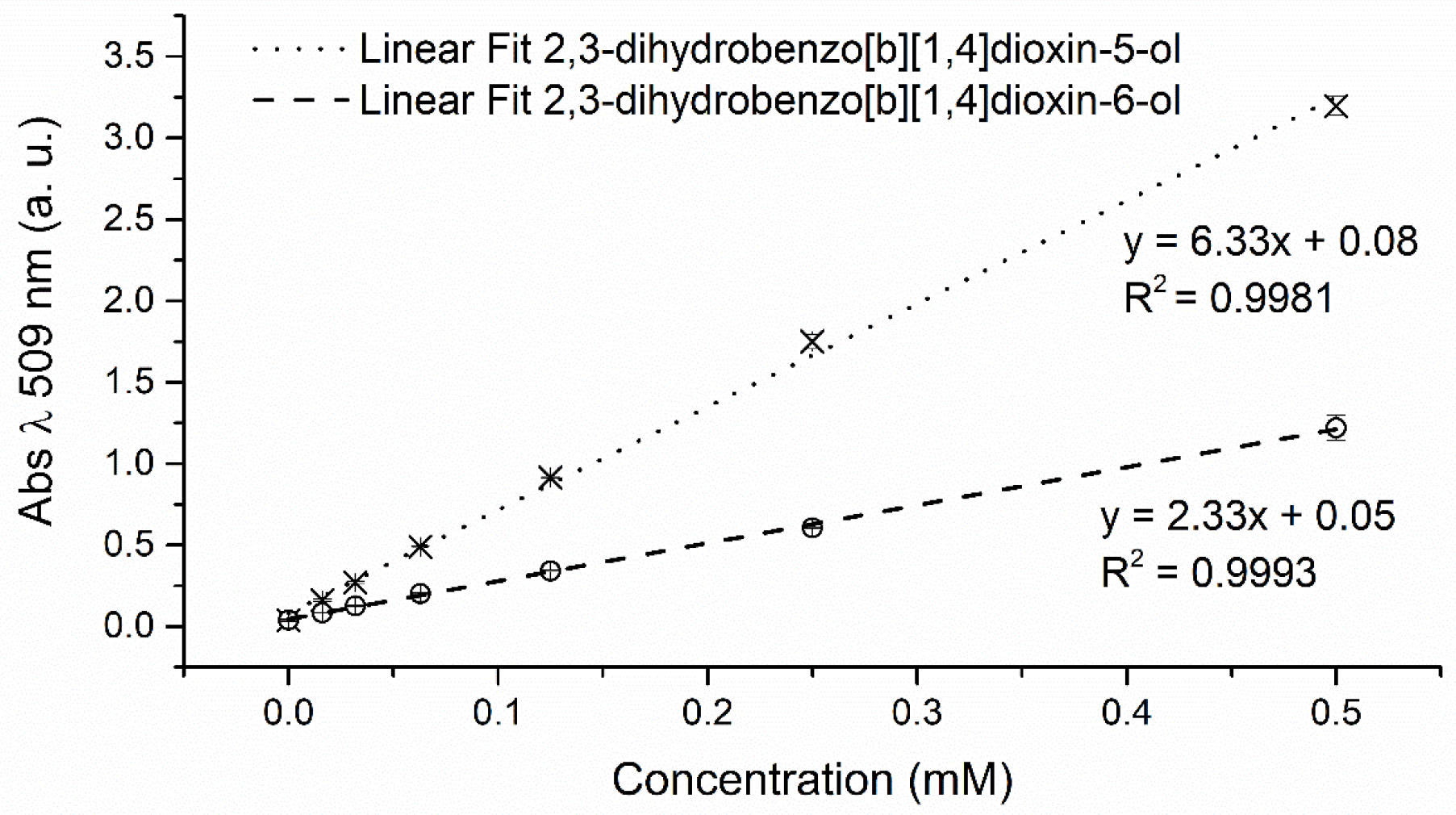
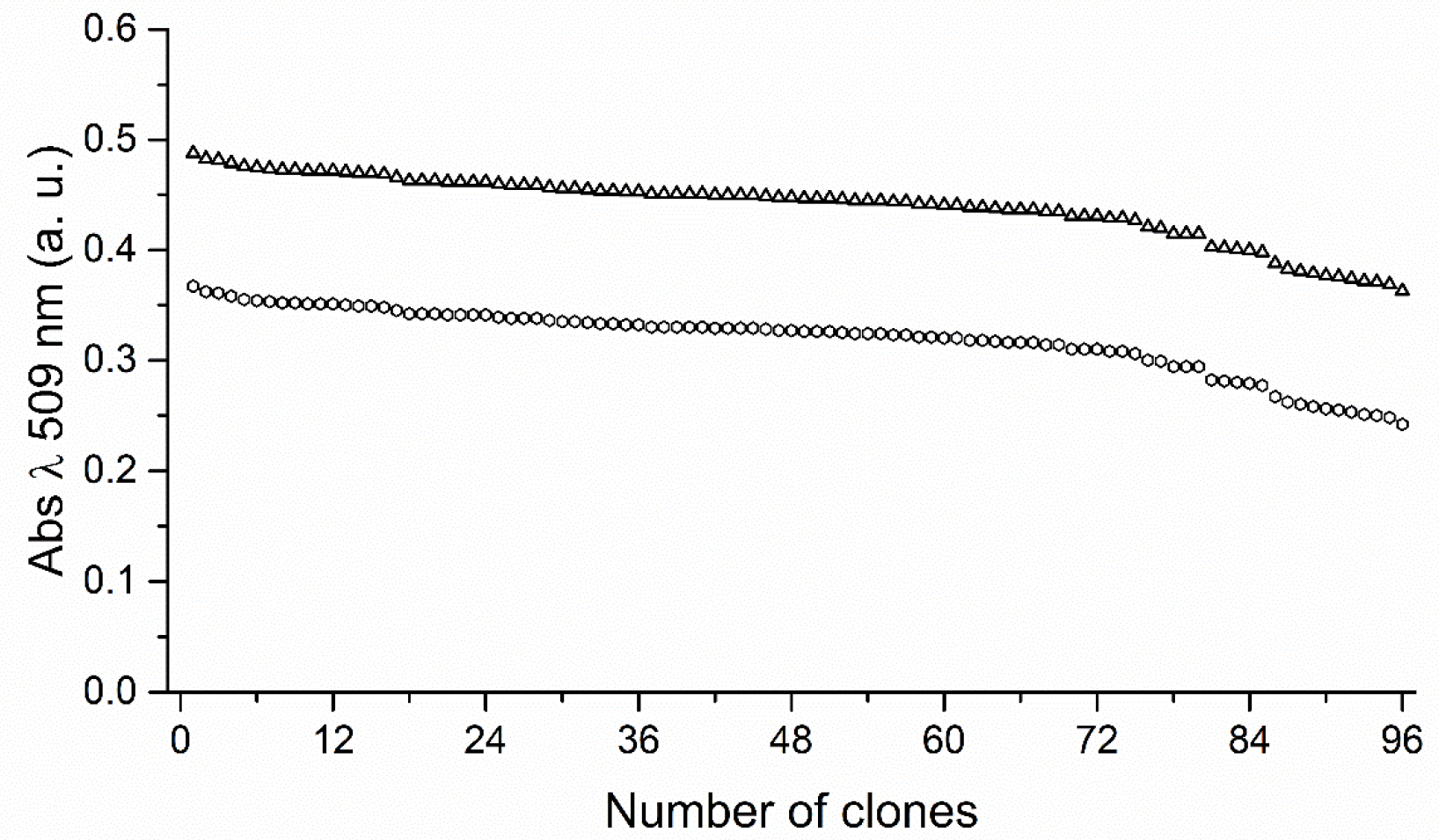
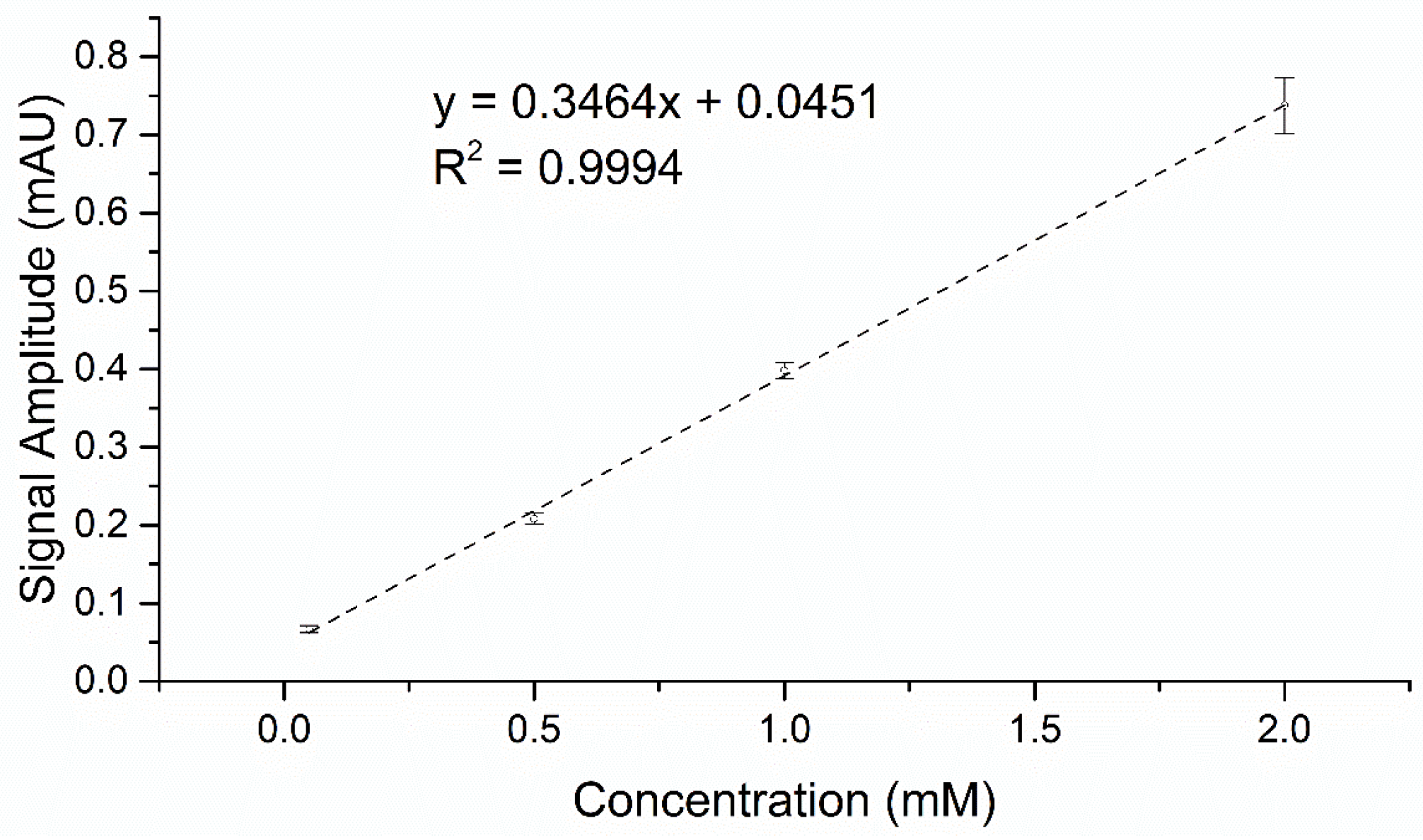
2.1. Development of 4-AAP and CE Screening Systems for Product-Based Quantification of 2,3-Dihydro-1,4-Benzodioxin-5-ol and 2,3 Dihydro-1,4-Benzodioxin-6-ol
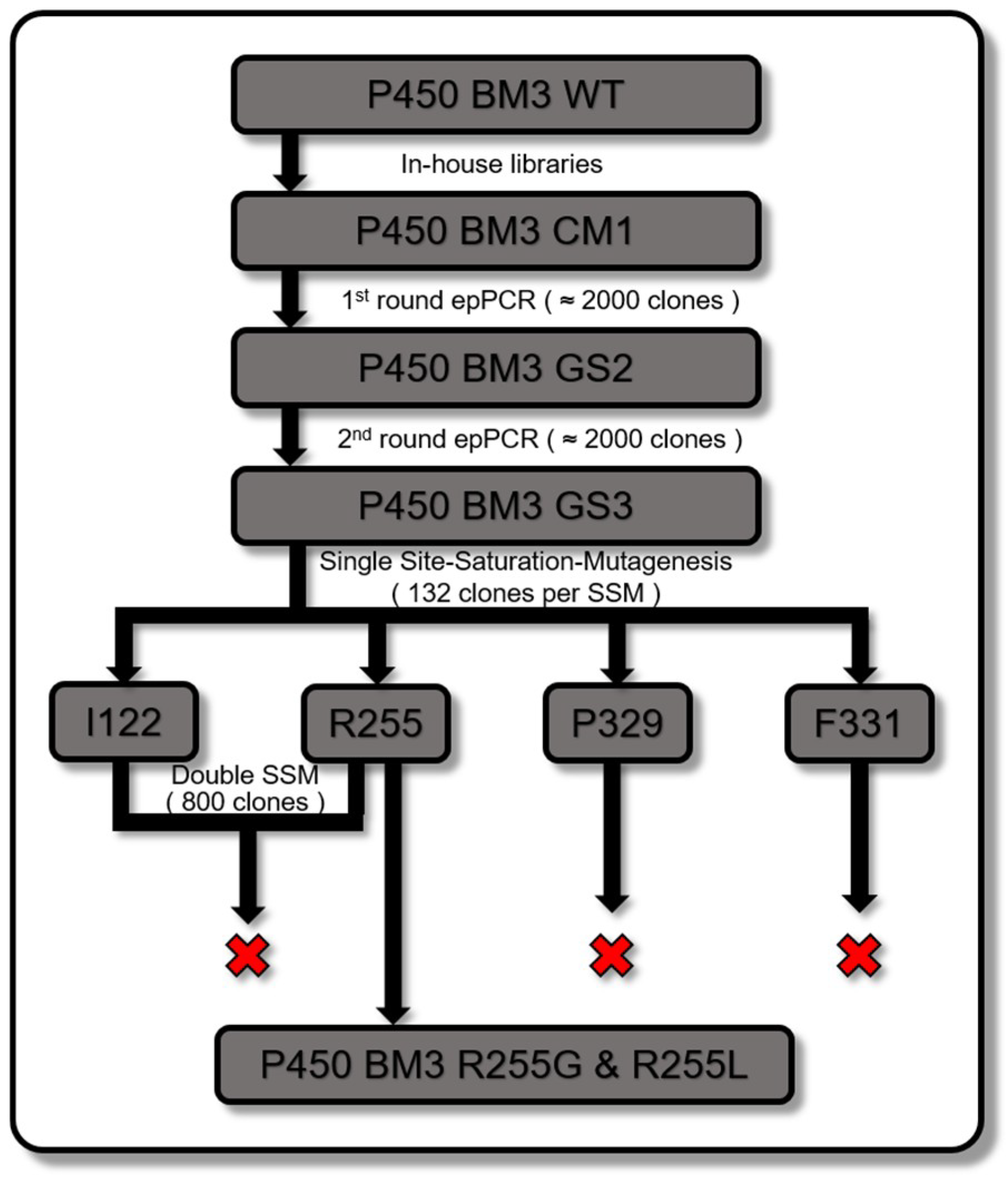
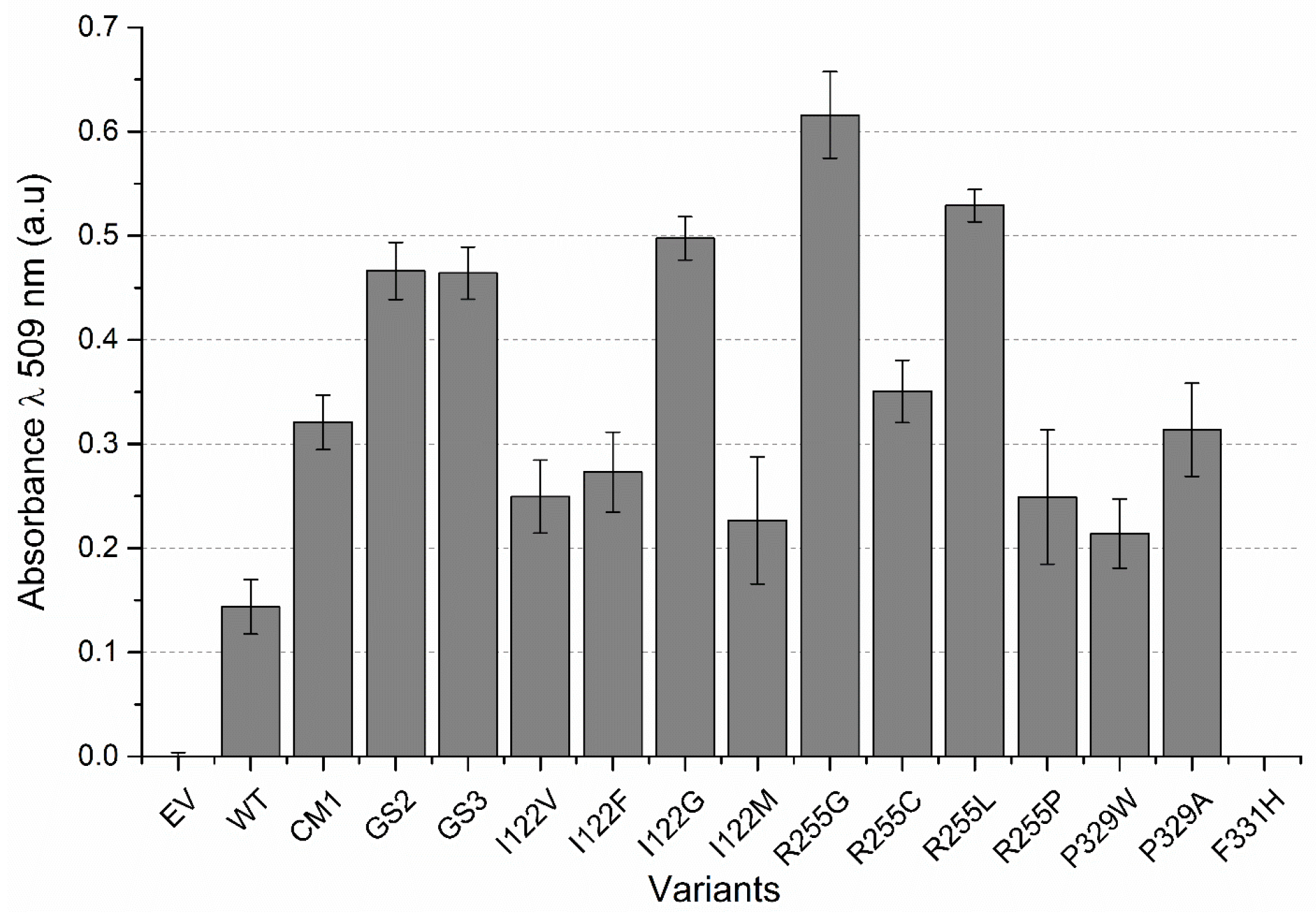
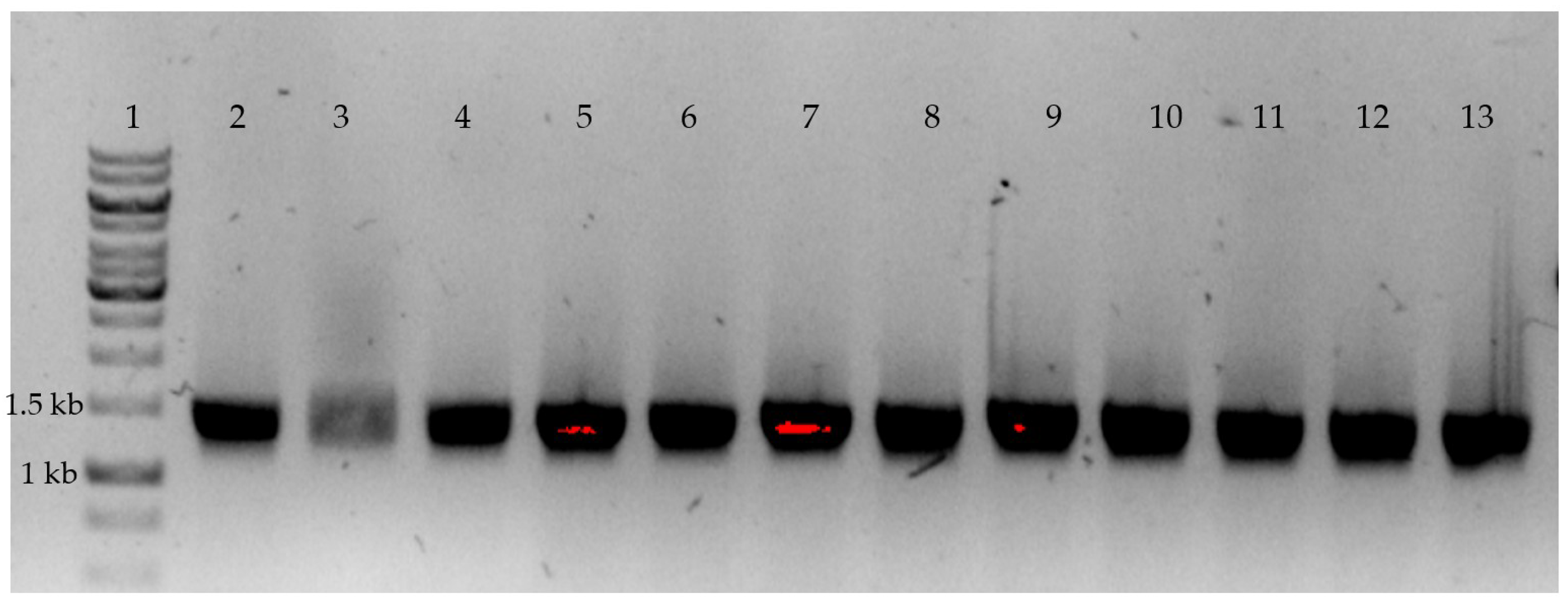
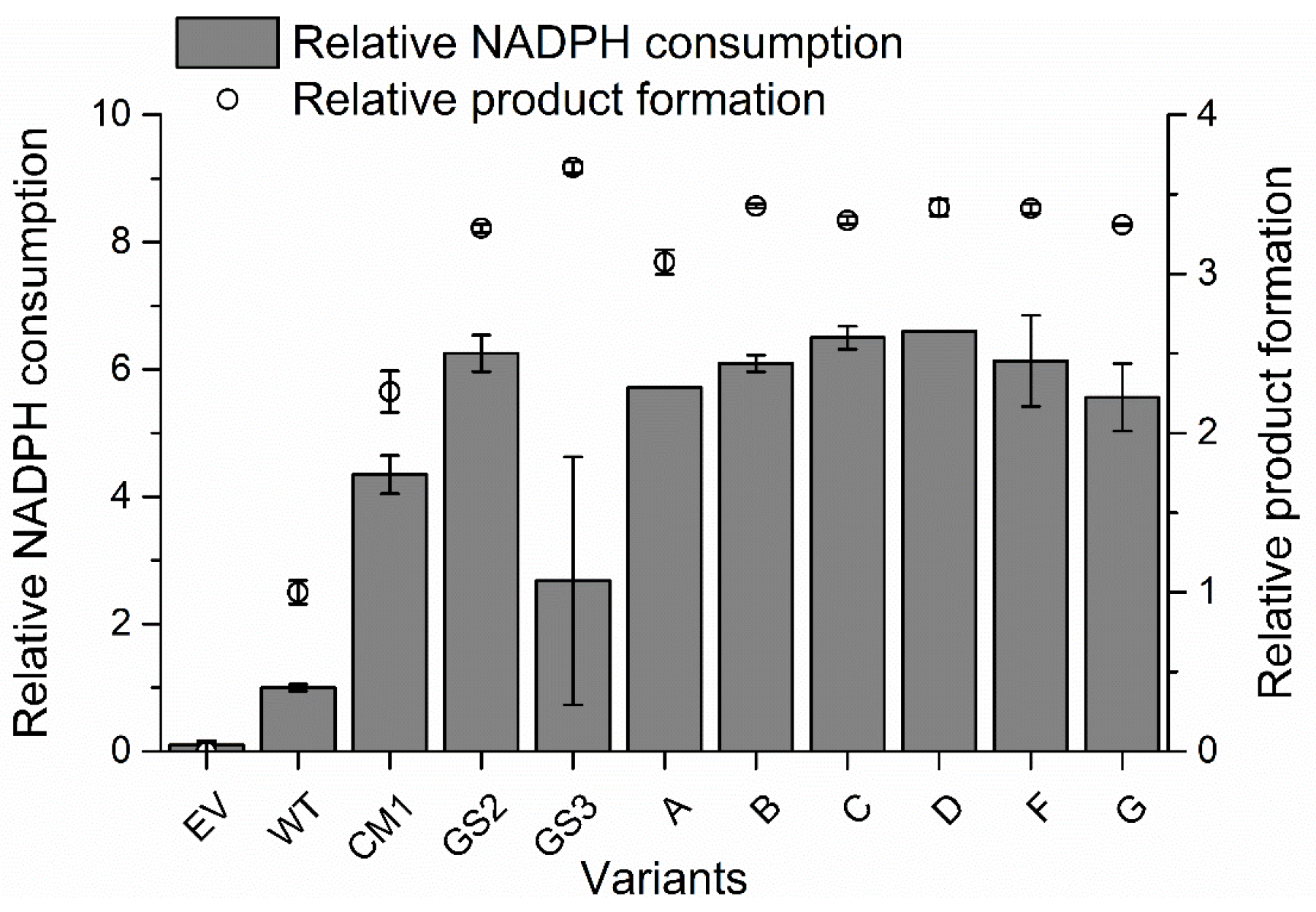
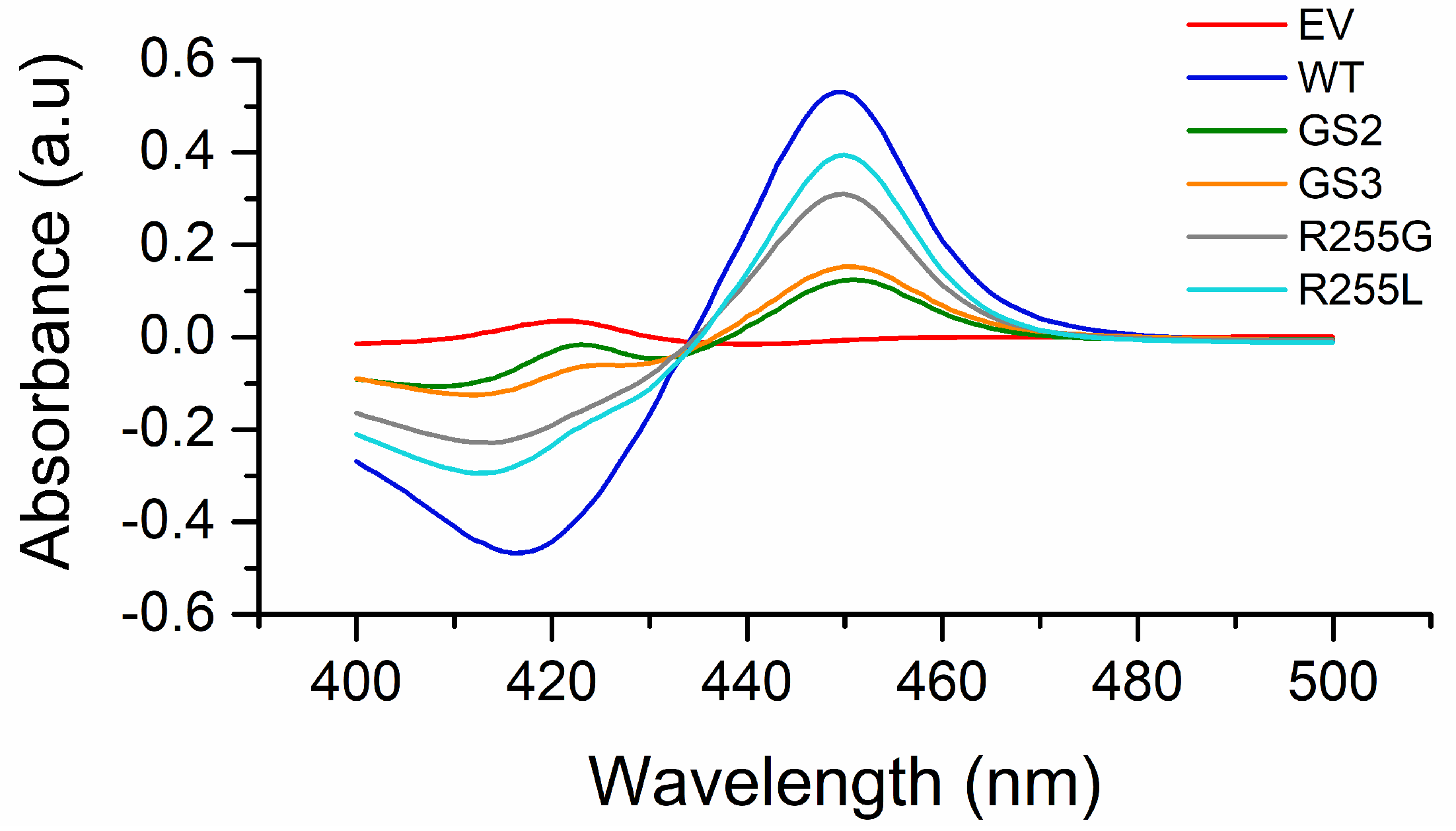
2.2. P450 BM3 Library Generation and Screening
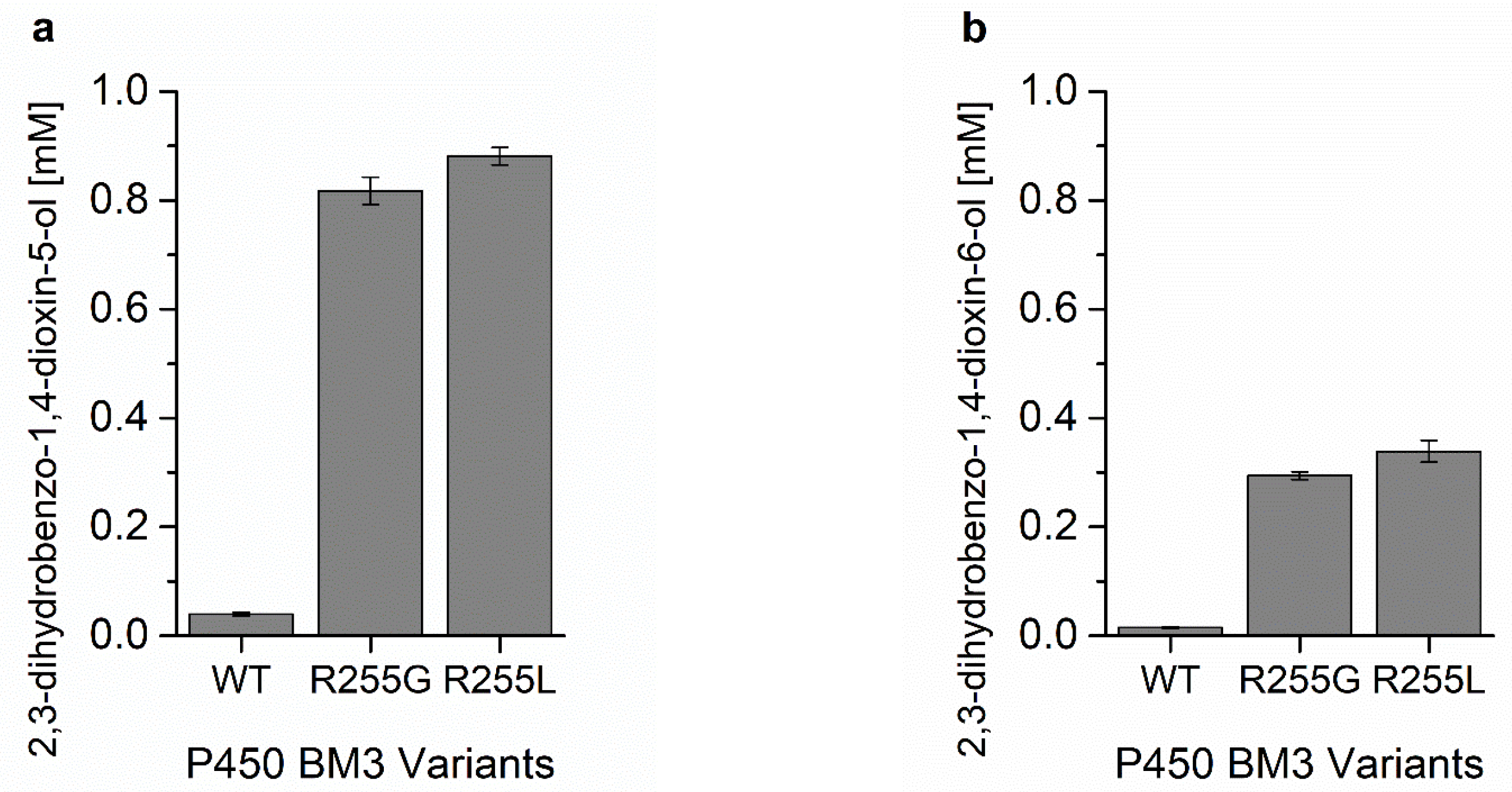
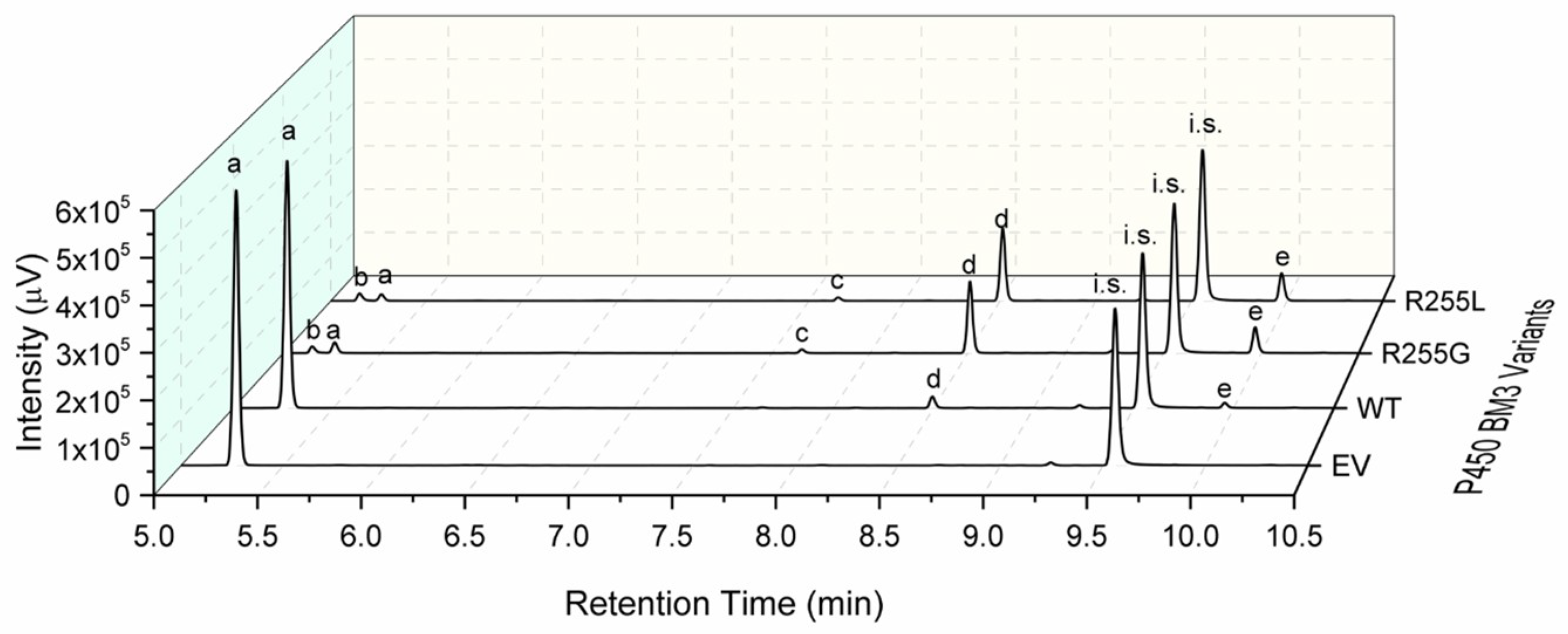
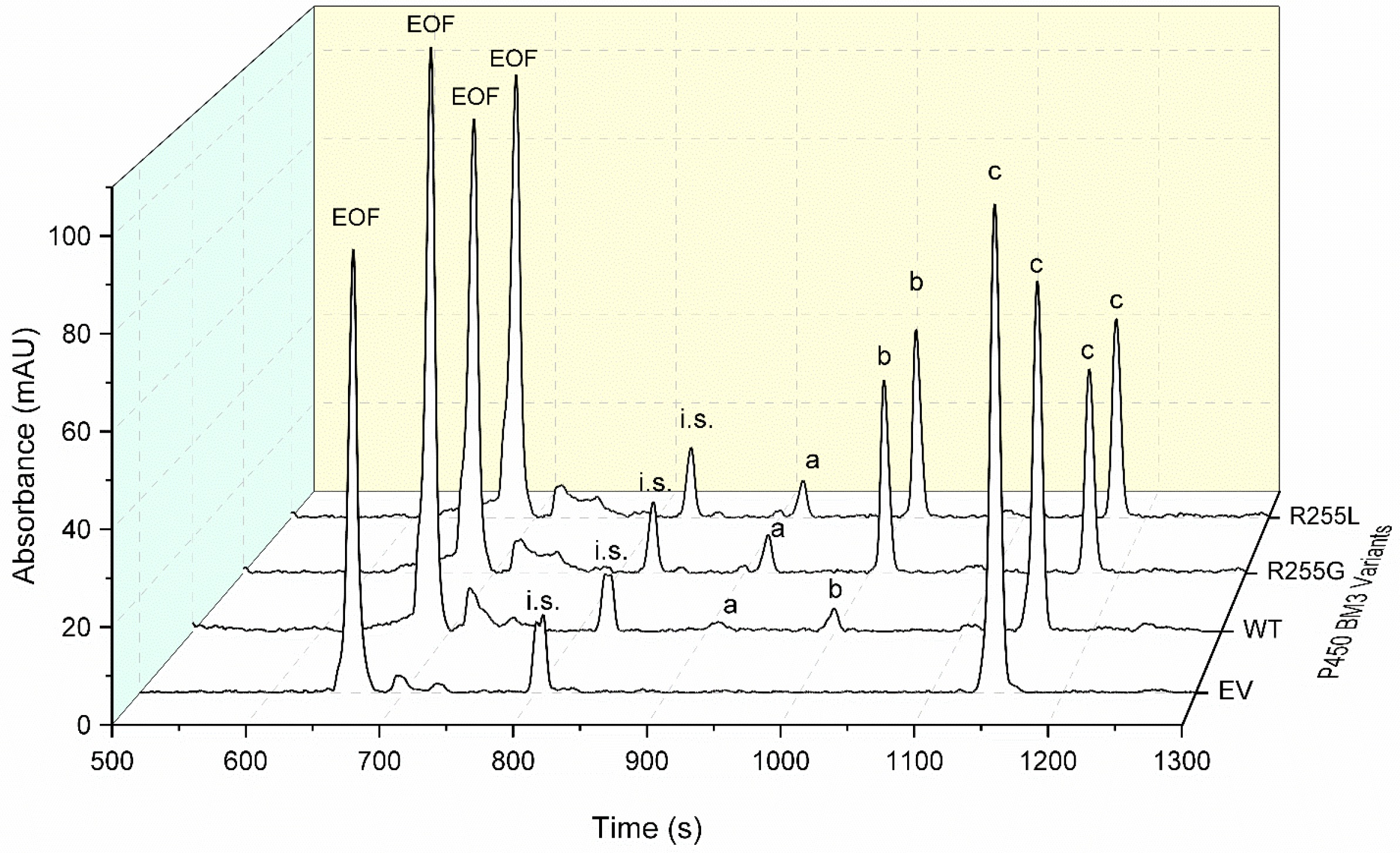
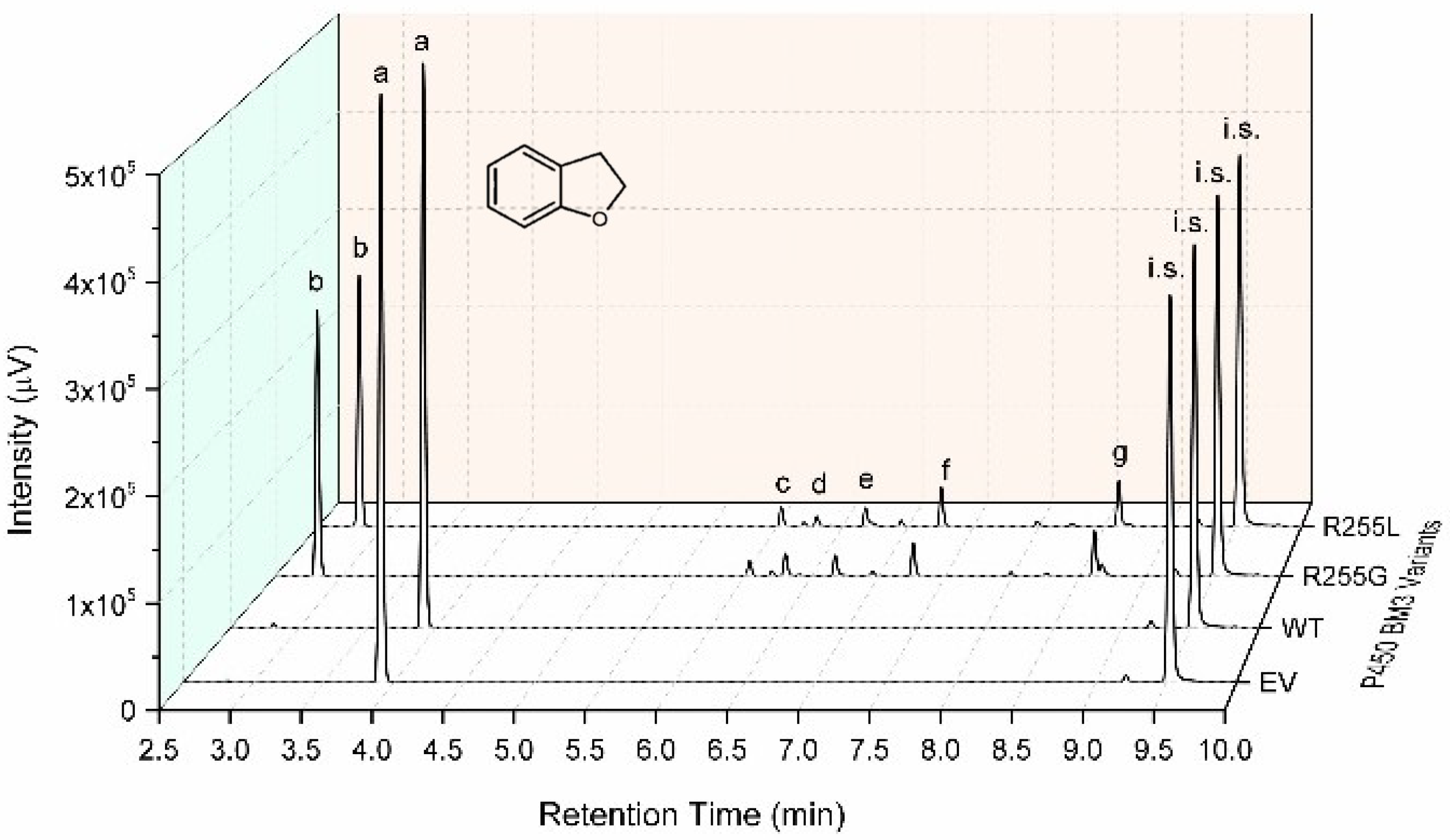
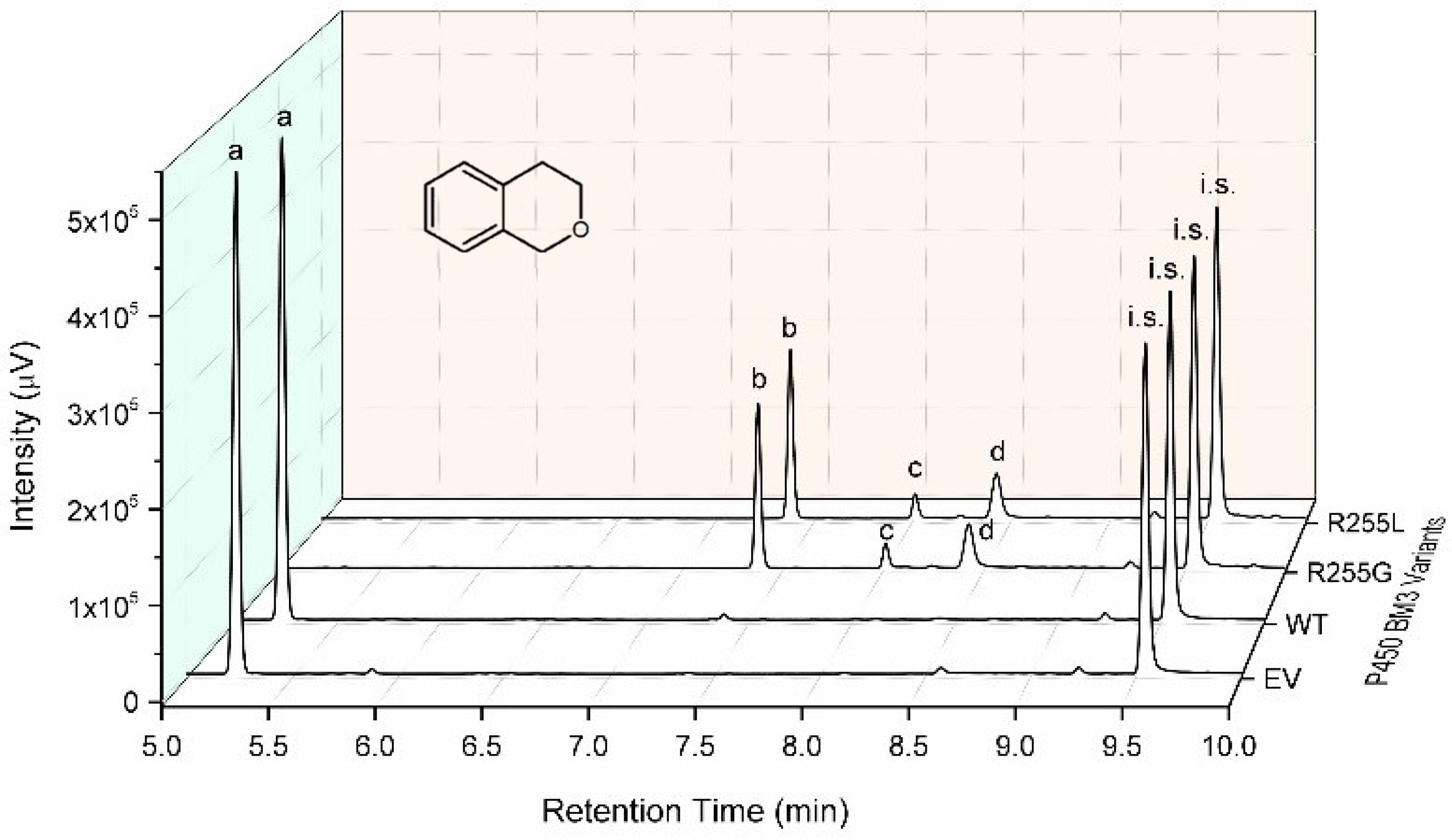
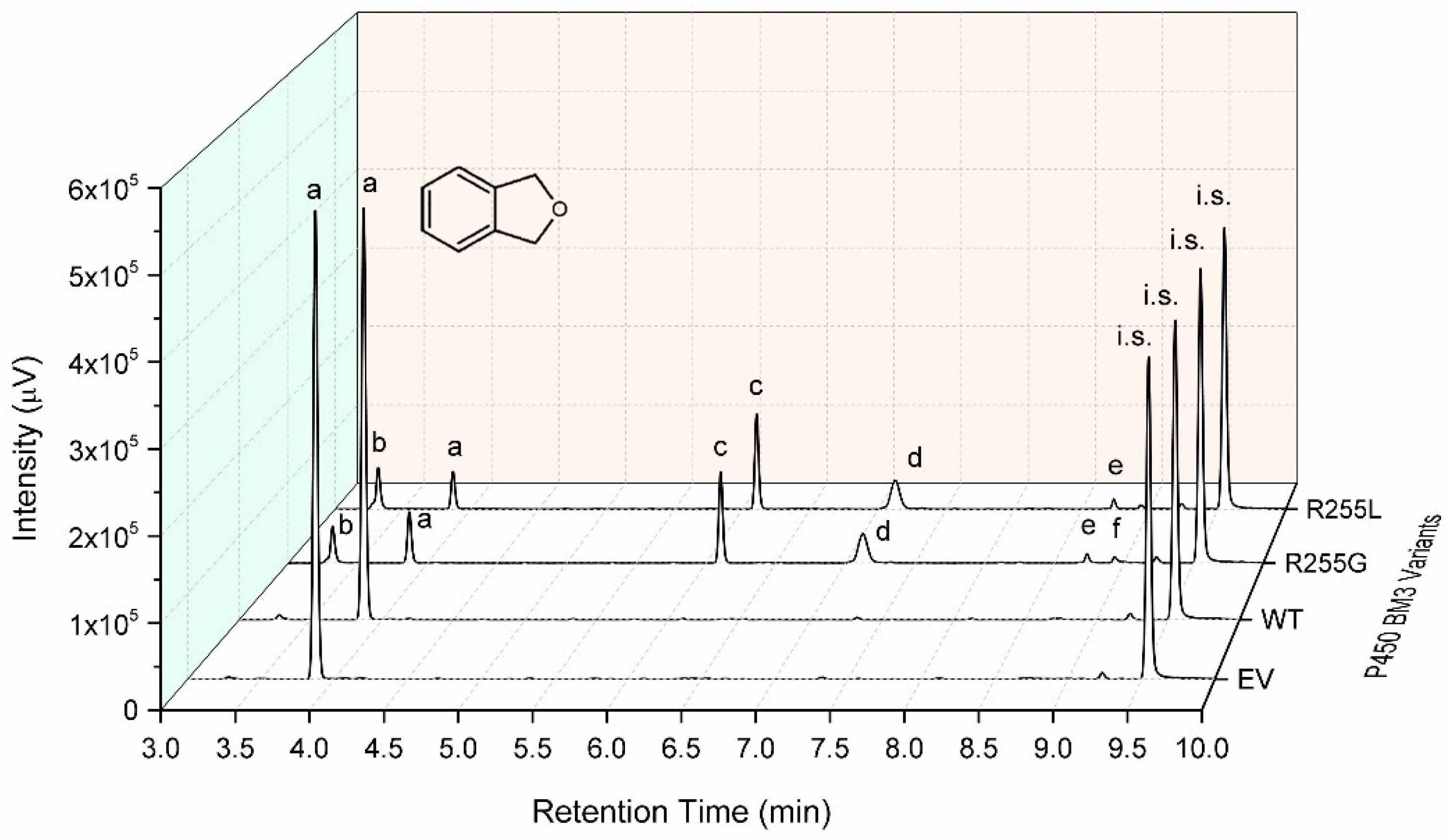
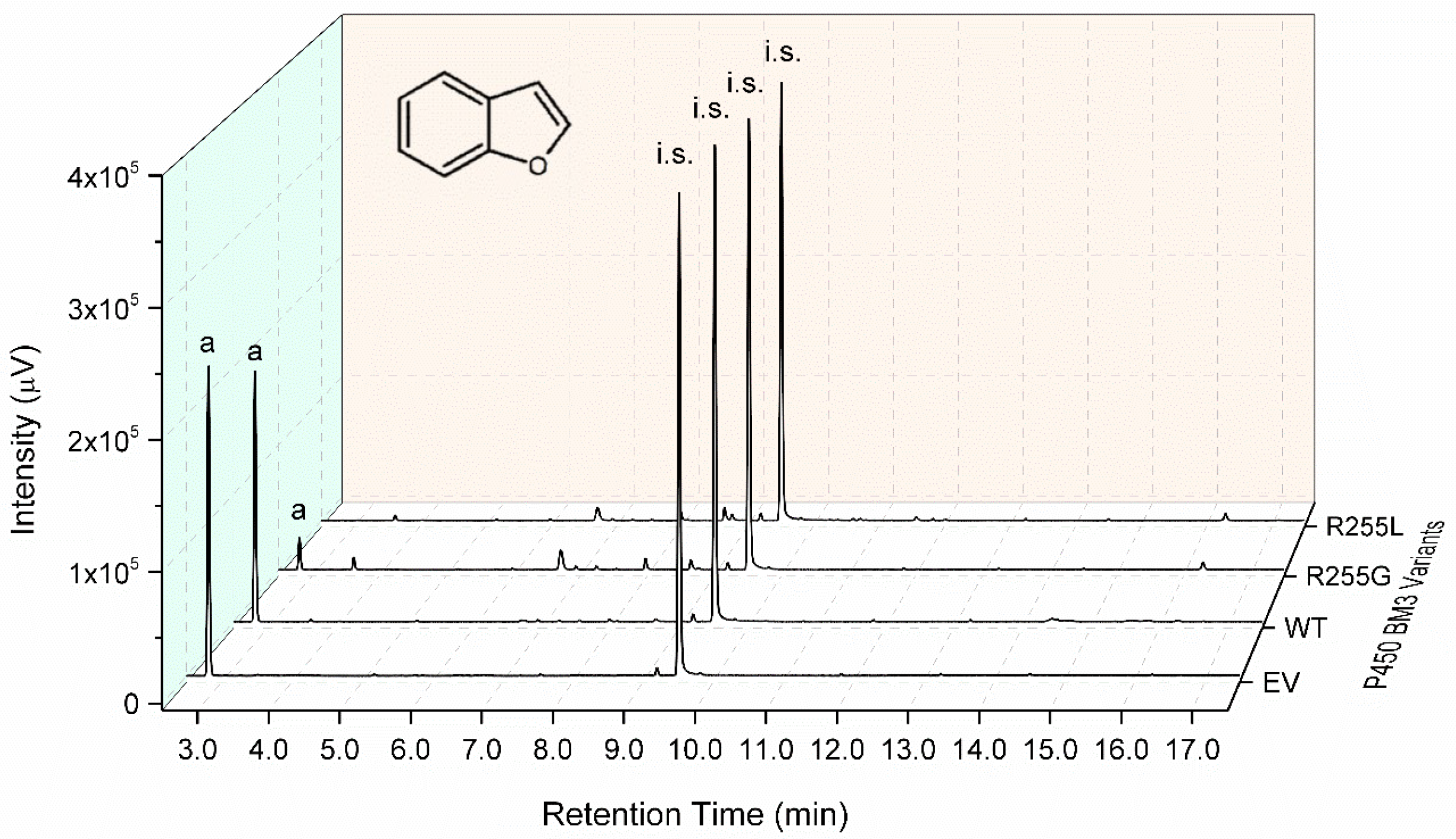
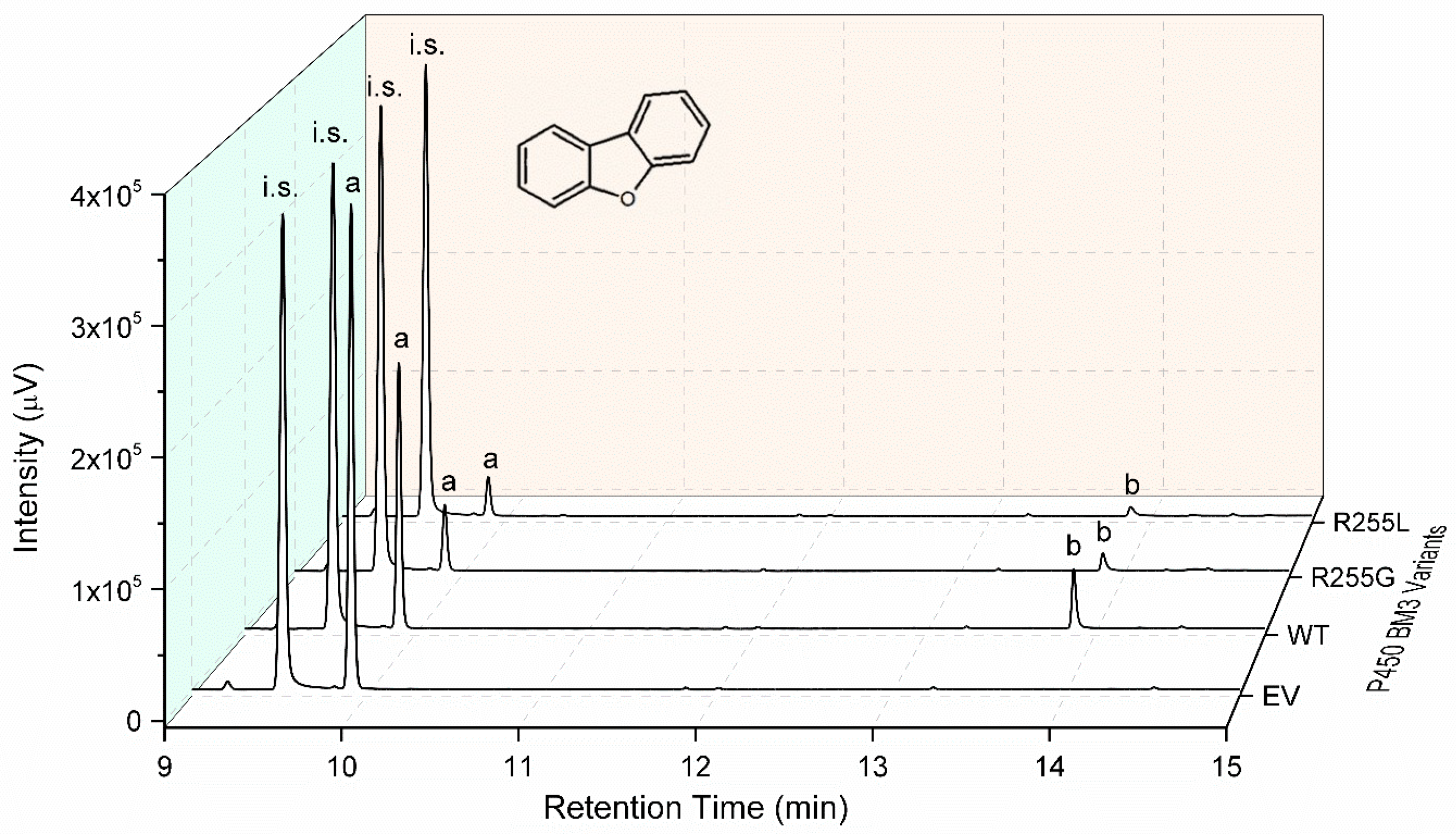
2.3. Characterization of P450 BM3 WT and Variants R255G and R255L in Respect to Hydroxylation of the Six Selected O-Heterocycles
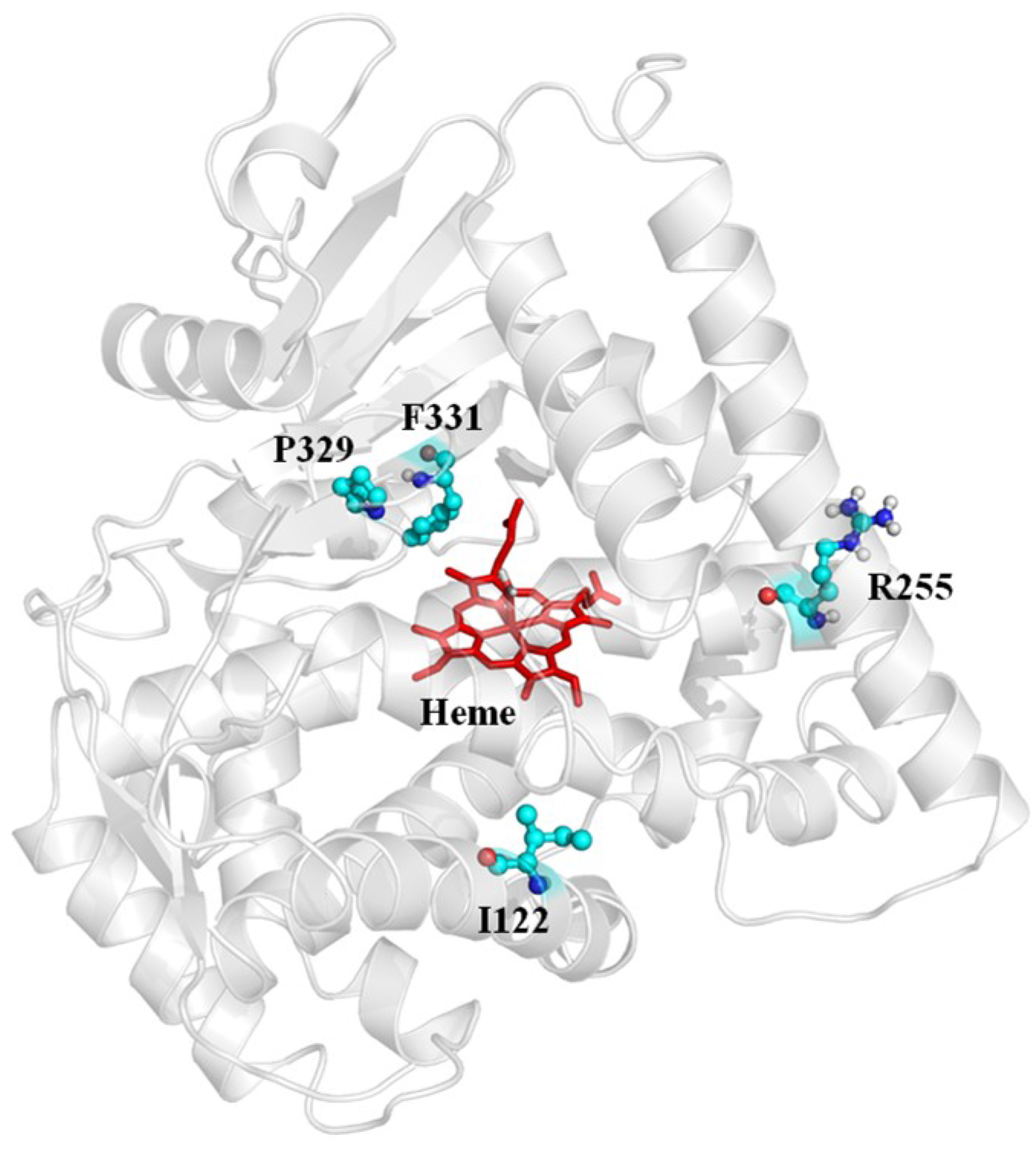
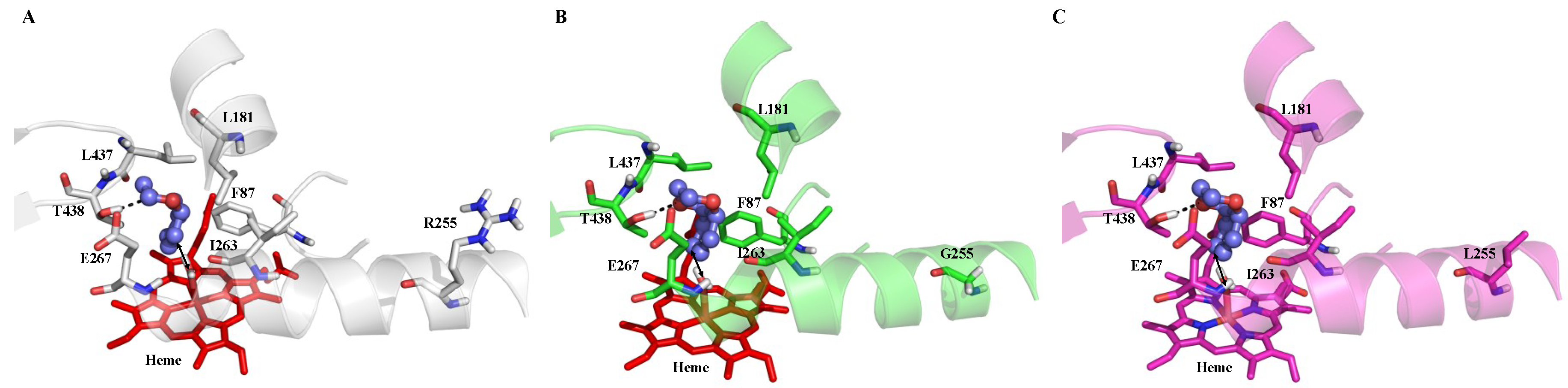
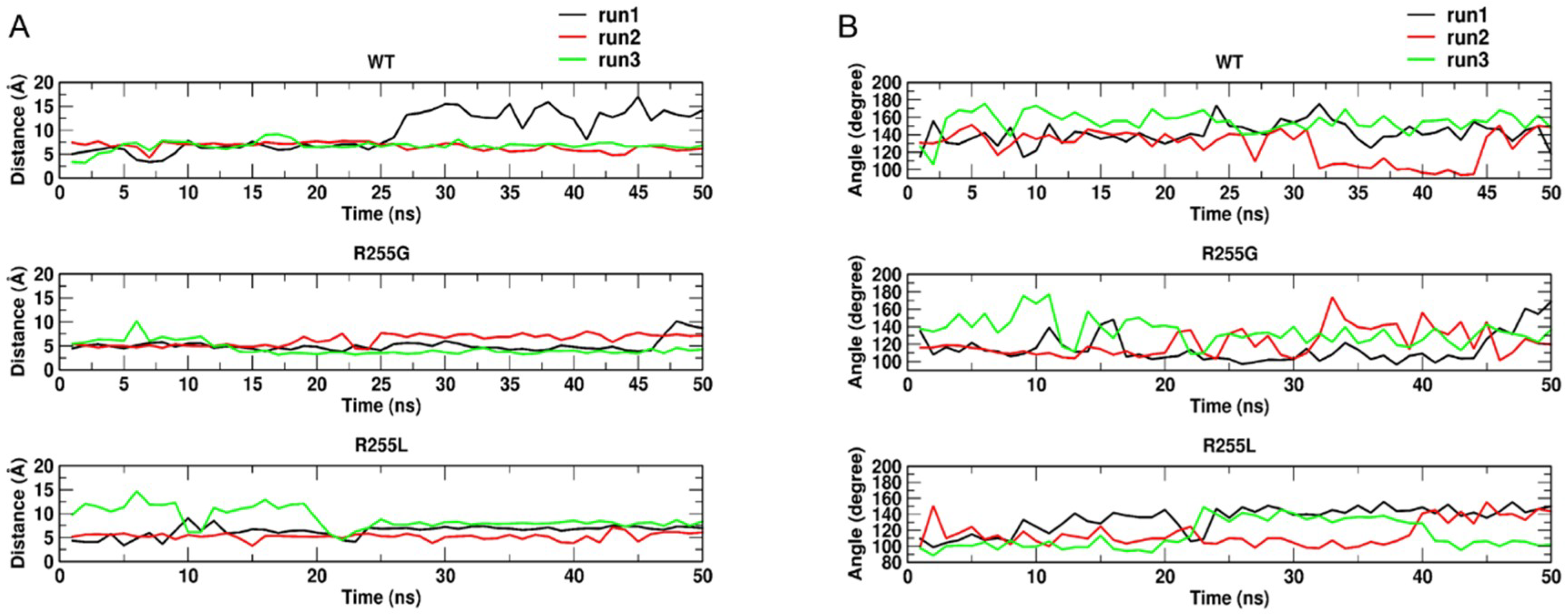
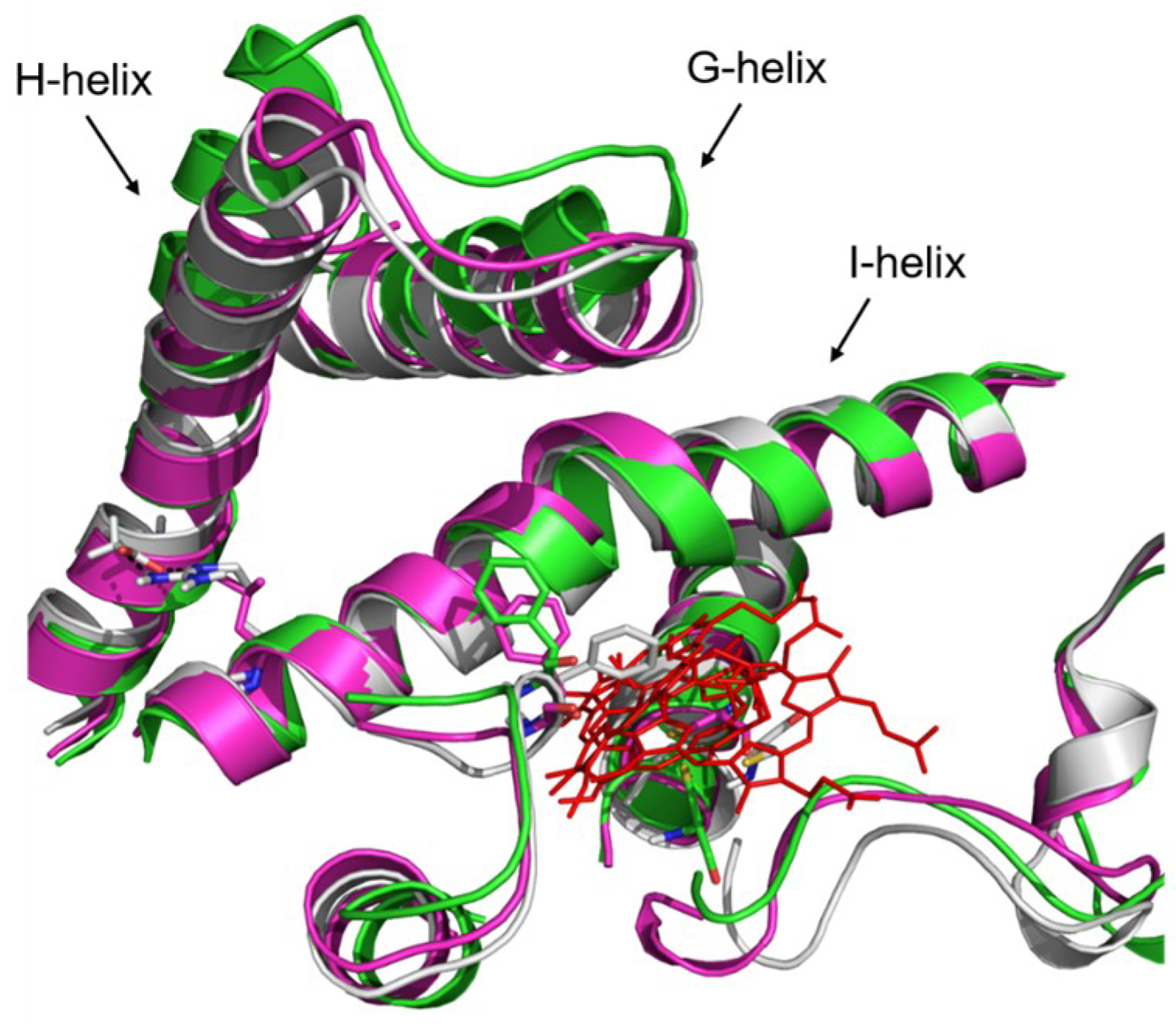
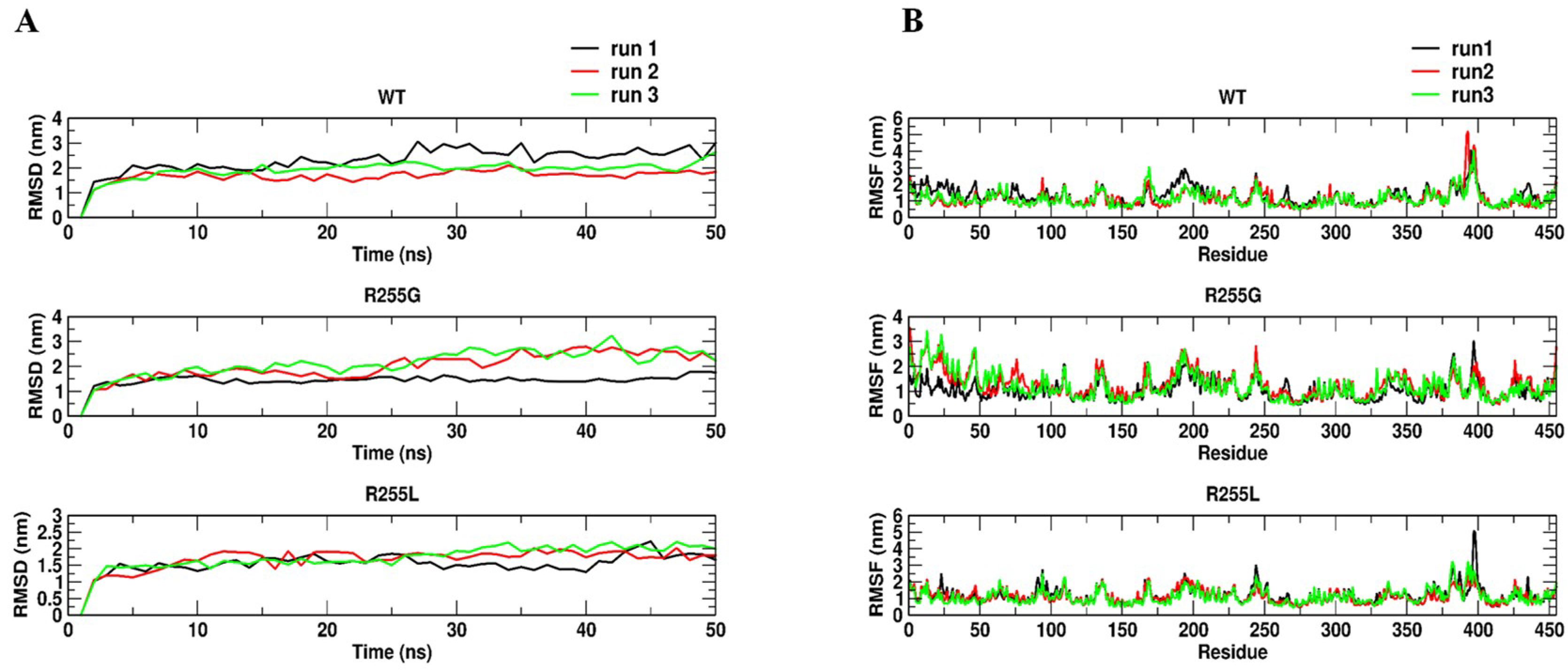
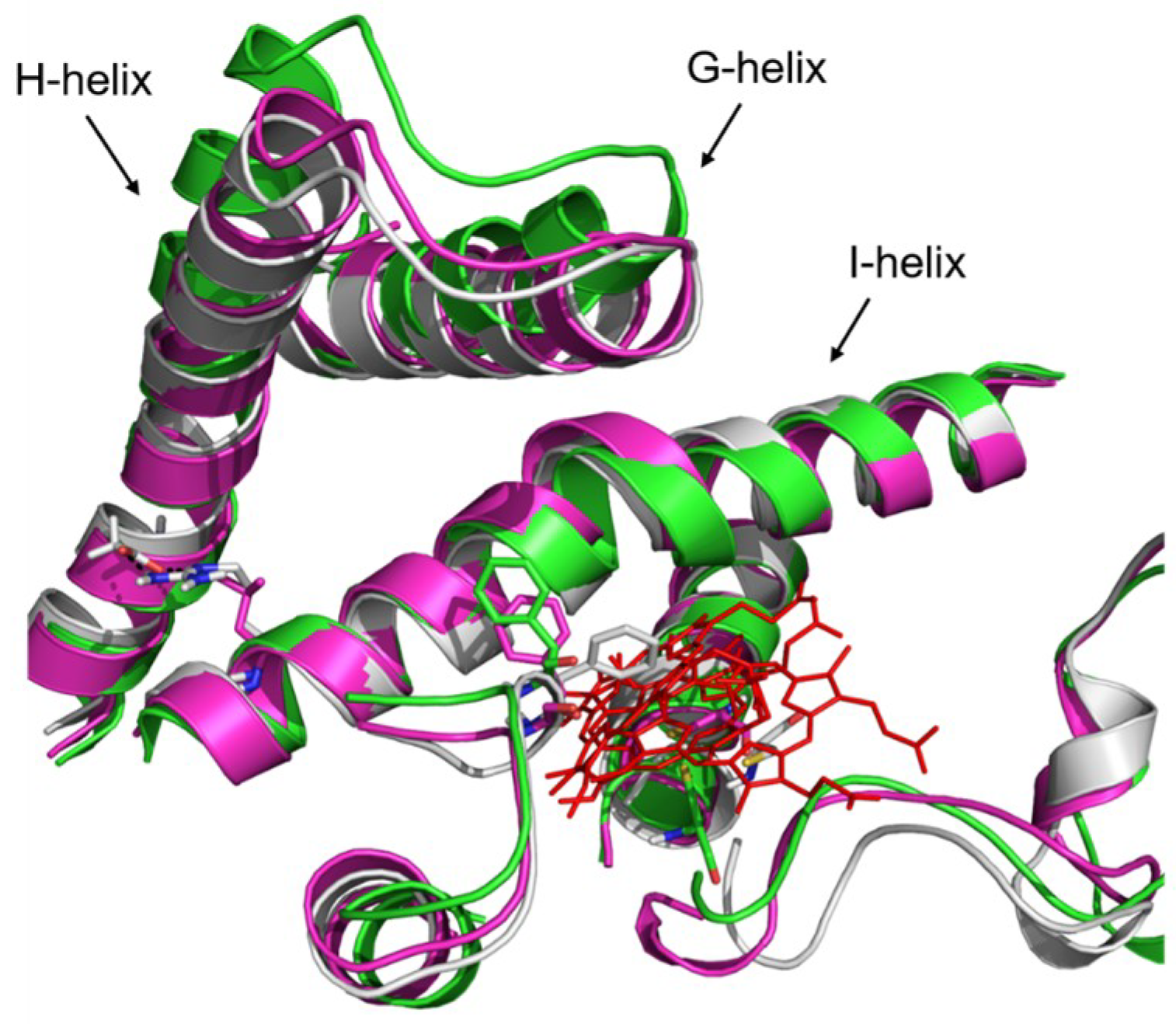
2.4. Rationale behind the Activity Improvement of R255G and R255L Variants over the WT
3. Materials and Methods
3.1. Strains, Plasmids, and Target Gene
3.2. Error-Prone PCR
3.3. Site Saturation Mutagenesis
3.4. Cultivation of P450 BM3 in 96-Deep-Well Plates
3.5. Screening for Improved P450 BM3 Variants
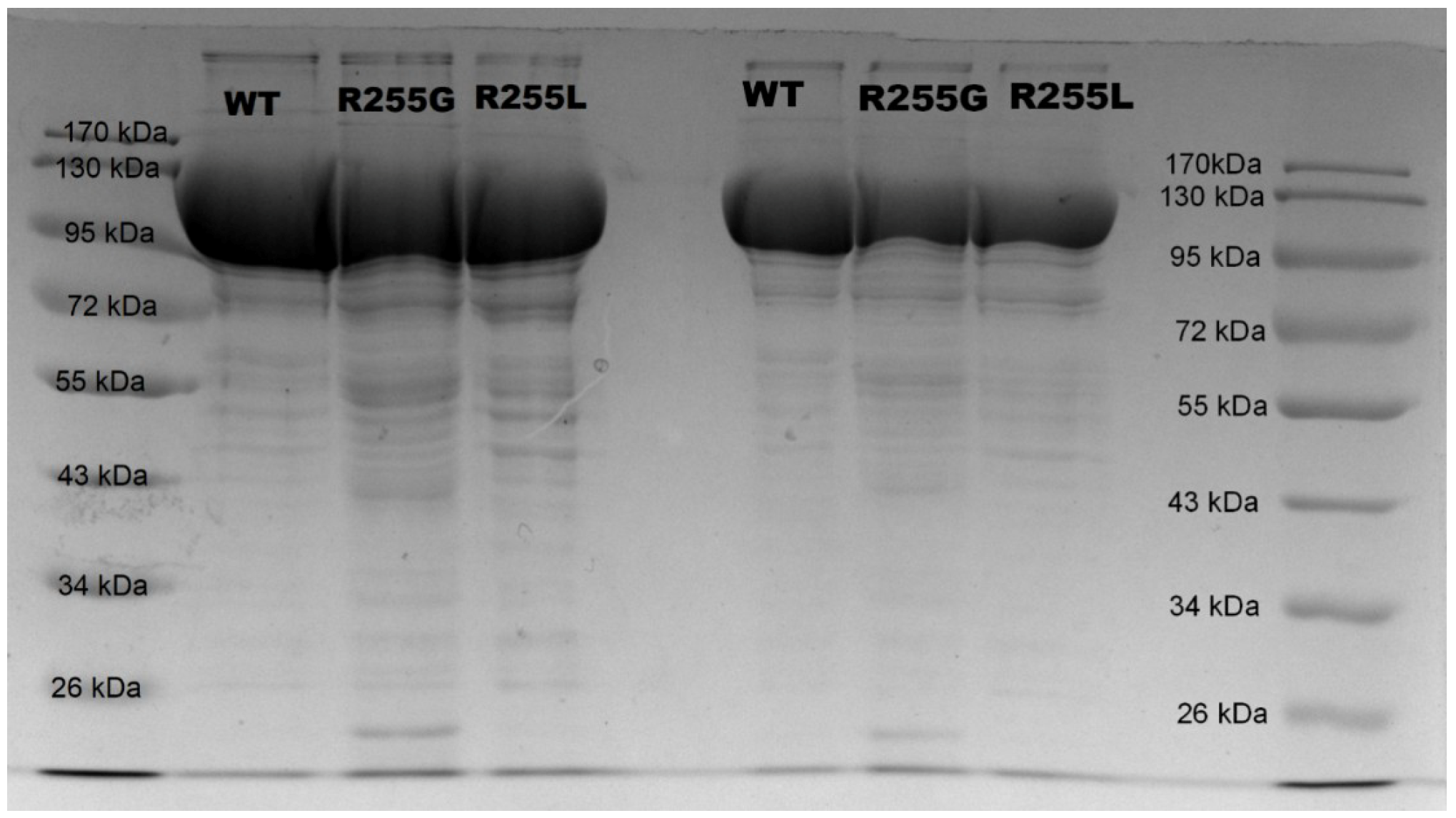
3.6. Expression and Purification of P450 BM3 Variants
3.7. Substrate Conversion and Kinetic Characterization of P450 BM3 Variants
3.8. Molecular Modeling
3.8.1. Molecular Docking
3.8.2. Molecular Dynamics Simulations
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAP | Aminoantipyrine |
| BM3 | Bacillus megaterium 3 |
| CE | Capillary electrophoresis |
| CO | Carbon monoxide |
| EV | Empty vector |
| FAD | Flavin adenine dinucleotide |
| FID | Flame ionization detector |
| FMN | Flavin mononucleotide |
| HPSF | High purity salt free |
| GC | Gas chromatography |
| GDH | Glucose dehydrogenase |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| KPi | Phosphate buffer |
| MD | Molecular dynamics |
| MS | Mass spectroscopy |
| MTBE | Methyl tert-butyl ether |
| MTP | Microtiter plate |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NaPi | Sodium phosphate buffer |
| P450 | Cytochrome P450 monooxygenase |
| PCR | Polymerase chain reaction |
| RESP | Restrained Electrostatic Potential |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| SSM | Site saturation mutagenesis |
| TTN | Total turnover number |
| WT | Wild type |
Appendix A
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Target | Sequence (5′–3′) |
|---|---|---|
| P450_BM3_FWD | P450 BM3 Heme | catgggcatGACAATTAAAGAAATGCCTCA- |
| P450_BM3_REV | P450 BM3 Heme | gcgtattatgaGCGTTTTCTGCCTTTTTGC |
| pALX_FWD | pALXtreme-1a with P450 BM3 reductase | ctcataatacGCCGCTGCTTGTGCTATACG |
| pALX_REV | pALXtreme-1a with P450 BM3 reductase | gcgtattatgAGCGTTTTCTGCCTTTTTGC |
| 122.FWD | Position 122 | ATGATGGTCGATNNKGCCGTGCAGCTT |
| 122.REV | Position 122 | AAGCTGCACGGCMNNATCGACCATCAT |
| 255.FWD | Position 255 | GACGAGAACATTNNKTATCAAATTATT |
| 255.REV | Position 255 | AATAATTTGATAMNNAATGTTCTCGTC |
| 329.FWD | Position 329 | TGGCCAACTGCTNNKGCGTTTTCCCTA |
| 329.REV | Position 329 | TAGGGAAAACGCMNNAGCAGTTGGCCA |
| 331.FWD | Position 331 | ACTGCTCCTGCGNNKTCCCTATATGCA |
| 331.REV | Position 331 | TGCATATAGGGAMNNCGCAGGAGCAGT |

References
- Shimamura, T.; Shiroishi, M.; Weyand, S.; Tsujimoto, H.; Winter, G.; Katritch, V.; Abagyan, R.; Cherezov, V.; Liu, W.; Han, G.W.; et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Griffith, E.C.; Su, Z.; Turk, B.E.; Chen, S.; Chang, Y.-H.; Wu, Z.; Biemann, K.; Liu, J.O. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem. Biol. 1997, 4, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Garrett, R.H.; Grisham, C.M. Biochemistry, 4th ed.; Mary Finch: Boston, MA, USA, 2010; ISBN 978-0-495-10935-8. [Google Scholar]
- Pilkington, L.I.; Barker, D. Asymmetric synthesis and CD investigation of the 1,4-benzodioxane lignans eusiderins A, B, C, G, L, and M. J. Org. Chem. 2012, 77, 8156–8166. [Google Scholar] [CrossRef]
- Pilkington, L.I.; Barker, D. Synthesis and biology of 1,4-benzodioxane lignan natural products. Nat. Prod. Rep. 2015, 32, 1369–1388. [Google Scholar] [CrossRef] [Green Version]
- Merlini, L.; Zanarotti, A. A biogenetically patterned synthesis of (+)-eusiderin. Tetrahedron Lett. 1975, 16, 3621–3622. [Google Scholar] [CrossRef]
- Arnone, A.; Merlin, L.; Zanarotti, A. Constituents of Silybum marianum. Structure of isosilybin and stereochemistry of silybin. J. Chem. Soc. Chem. Commun. 1979, 696–697. [Google Scholar] [CrossRef]
- Debenedetti, S.L.; Nadinic, E.L.; Coussio, J.D.; De Kimpe, N.; Feneau-Dupont, J.; Declerc, J.P. Purpurenol, a Highly Oxygenated Coumarin from Pterocaulon purpurascens. Phytochemistry 1991, 30, 2757–2758. [Google Scholar] [CrossRef]
- Bosseray, P.; Guillaumet, G.; Coudert, G.; Wassermann, H. Synthesis of Polyether Carboxylic Acid with a Benyodioxinic Subunit. Tetrahedron Lett. 1989, 30, 1387–1390. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Kato, H.; Hirota, H.; Fusetani, N. 3,4-Dihydroxystyrene dimers, inducers of larval metamorphosis in ascidians, from a marine sponge Jaspis sp. Tetrahedron 1994, 50, 13583–13592. [Google Scholar] [CrossRef]
- Singh, P.K.; Silakari, O. The Current Status of O-Heterocycles: A Synthetic and Medicinal Overview. ChemMedChem 2018, 13, 1071–1087. [Google Scholar] [CrossRef]
- Campbell, S.F.; Davey, M.J.; Hardstone, J.D.; Lewis, B.N.; Palmer, M.J. 2,4-Diamino-6,7-dimethoxy quinazolines. 1. 2-[4-(1,4-Berizodioxan-2-ylcarbonyl)piperazin-1-yl] Derivatives as α1-Adrenoceptor Antagonists and Antihypertensive Agents. J. Med. Chem. 1987, 30, 49–57. [Google Scholar] [CrossRef]
- Guthrie, R.M.; Siegel, R.L. A multicenter, community-based study of doxazosin in the treatment of concomitant hypertension and symptomatic benign prostatic hyperplasia: The hypertension and BPH intervention trial (HABIT). Clin. Ther. 1999, 21, 1732–1748. [Google Scholar] [CrossRef]
- Vincent, J.; Elliott, H.L.; Meredith, P.A.; Reid, J.L. Effect Relationships in Man. Blood Press. 1983, 719–725. [Google Scholar]
- Tomiyama, T.; Wakabayashi, S.; Yokota, M. Synthesis and Biological Activity of Novel Carbacyclins Having Bicyclic Substituents on the w-Chain. J. Med. Chem. 1989, 32, 1988–1996. [Google Scholar] [CrossRef]
- Ertan, R.; Goker, H. Studies on some new flavone derivatives possessing spasmolytic activity. FABAD J. Pharm. Sci. 1987, 12, 152–157. [Google Scholar]
- Hibert, M.F.; Gittos, M.W.; Middlemiss, D.N.; Mir, A.K.; Fozard, J.R. Graphics Computer-Aided Receptor Mapping as a Predictive Tool for Drug Design: Development of Potent, Selective, and Stereospecific Ligands for the 5-HT1A Receptor. J. Med. Chem. 1988, 31, 1087–1093. [Google Scholar] [CrossRef]
- Mir, A.K.; Hibert, M.; Tricklebank, M.D.; Middlemiss, D.N.; Kidd, E.J.; Fozard, J.R. MDL 72832: A potent and stereoselective ligand at central and peripheral 5-HT1A receptors. Eur. J. Pharmacol. 1988, 149, 107–120. [Google Scholar] [CrossRef]
- Vogel, G.; Trost, W.; Braatz, R. Pharmacodynamics, and site and mechanism of action of silymarin, the antihepatotoxic component of Silybum marianum (L.) Gaertn. II. Special studies on site and mechanism of action (also in organs other than the liver). Arzneimittel-Forschung/Drug Res. 1975, 25, 179–188. (In German) [Google Scholar]
- Giardina, D.; Bertini, R.; Brancia, E.; Brasili, L.; Melchiorre, C. Structure-Activity Relationships for Prazosin and WB 4101 Analogues as arAdrenoreceptor Antagonists. J. Med. Chem. 1985, 28, 1354–1357. [Google Scholar] [CrossRef]
- Welbourn, A.P.; Chapleo, C.B.; Lane, A.C.; Myers, P.L.; Roach, A.G.; Smith, C.F.; Stillings, M.R.; Tulloch, I.F. A-Adrenoreceptor Reagents. 4. Resolution of Some Potent Selective Prejunctional α2-Adrenoreceptor Antagonists. J. Med. Chem. 1986, 29, 2000–2003. [Google Scholar] [CrossRef]
- Su, B.; Cao, Z.-C.; Shi, Z.-J. Exploration of Earth-Abundant Transition Metals (Fe, Co, and Ni) as Catalysts in Unreactive Chemical Bond Activations. Acc. Chem. Res. 2015, 48, 886–896. [Google Scholar] [CrossRef]
- Fürstner, A. Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion. ACS Cent. Sci. 2016, 2, 778–789. [Google Scholar] [CrossRef]
- Hollmann, F.; Arends, I.W.C.E.; Buehler, K.; Schallmey, A.; Bühler, B. Enzyme-mediated oxidations for the chemist. Green Chem. 2011, 13, 226–265. [Google Scholar] [CrossRef]
- Fessner, N.D. P450 Monooxygenases Enable Rapid Late-Stage Diversification of Natural Products via C-H Bond Activation. ChemCatChem 2019, 11, 2226–2242. [Google Scholar] [CrossRef]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 Monooxygenases in Biotechnology and Synthetic Biology. Trends Biotechnol. 2019. [Google Scholar] [CrossRef]
- Julsing, M.K.; Cornelissen, S.; Bühler, B.; Schmid, A. Heme-iron oxygenases: Powerful industrial biocatalysts? Curr. Opin. Chem. Biol. 2008, 12, 177–186. [Google Scholar] [CrossRef]
- Gribble, G.W.; Joule, J.J. Progress in Heterocyclic Chemistry; Elsevier: Amsterdam, The Netherlands, 2009; Volume 21, ISBN 9780080965161. [Google Scholar]
- Knochel, P.; Schade, M.A.; Bernhardt, S.; Manolikakes, G.; Metzger, A.; Piller, F.M.; Rohbogner, C.J.; Mosrin, M. Functionalization of heterocyclic compounds using polyfunctional magnesium and zinc reagents. Beilstein J. Org. Chem. 2011, 7, 1261–1277. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.J.; Peck, N.E.; Renata, H.; Arnold, F.H. Cytochrome P450-catalyzed insertion of carbenoids into N–H bonds. Chem. Sci. 2014, 5, 598–601. [Google Scholar] [CrossRef]
- Singh, R.; Kolev, J.N.; Sutera, P.A.; Fasan, R. Enzymatic C(sp3)-H Amination: P450-Catalyzed Conversion of Carbonazidates into Oxazolidinones. ACS Catal. 2015, 5, 1685–1691. [Google Scholar] [CrossRef]
- Coelho, P.S.; Brustad, E.M.; Kannan, A.; Arnold, F.H. Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Coelho, P.S.; Wang, Z.J.; Ener, M.E.; Baril, S.A.; Kannan, A.; Arnold, F.H.; Brustad, E.M. A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat. Chem. Biol. 2013, 9, 485–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farwell, C.C.; McIntosh, J.A.; Hyster, T.K.; Wang, Z.J.; Arnold, F.H. Enantioselective Imidation of Sulfides via Enzyme-Catalyzed Intermolecular Nitrogen-Atom Transfer. J. Am. Chem. Soc. 2014, 136, 8766–8771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nardo, G.; Gilardi, G.; Di Nardo, G.; Gilardi, G. Optimization of the Bacterial Cytochrome P450 BM3 System for the Production of Human Drug Metabolites. Int. J. Mol. Sci. 2012, 13, 15901–15924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ost, T.W.; Miles, C.S.; Murdoch, J.; Cheung, Y.-F.; Reid, G.A.; Chapman, S.K.; Munro, A.W. Rational re-design of the substrate binding site of flavocytochrome P450 BM3. FEBS Lett. 2000, 486, 173–177. [Google Scholar] [CrossRef]
- Sawayama, A.M.; Chen, M.M.Y.; Kulanthaivel, P.; Kuo, M.-S.; Hemmerle, H.; Arnold, F.H. A Panel of Cytochrome P450 BM3 Variants to Produce Drug Metabolites and Diversify Lead Compounds. Chem. Eur. J. 2009, 15, 11723–11729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmichael, A.B.; Wong, L.-L. Protein engineering of Bacillus megaterium CYP102. Eur. J. Biochem. 2001, 268, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Kille, S.; Zilly, F.E.; Acevedo, J.P.; Reetz, M.T. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 2011, 3, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.F.; Peet, C.; Mason, A.E.; Voice, M.W.; Leys, D.; Munro, A.W. Key mutations alter the cytochrome P450 BM3 conformational landscape and remove inherent substrate bias. J. Biol. Chem. 2013, 288, 25387–25399. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Westlake, A.C.G.; Maréchal, J.D.; Joyce, M.G.; Moody, P.C.E.; Roberts, G.C.K. Filling a Hole in Cytochrome P450 BM3 Improves Substrate Binding and Catalytic Efficiency. J. Mol. Biol. 2007, 373, 633–651. [Google Scholar] [CrossRef] [Green Version]
- Kaluzna, I.; Schmitges, T.; Straatman, H.; Van Tegelen, D.; Müller, M.; Schürmann, M.; Mink, D. Enabling Selective and Sustainable P450 Oxygenation Technology. Production of 4-Hydroxy-α-isophorone on Kilogram Scale. Org. Process Res. Dev. 2016, 20, 814–819. [Google Scholar] [CrossRef]
- Wong, T.S.; Wu, N.; Roccatano, D.; Zacharias, M.; Schwaneberg, U. Sensitive assay for laboratory evolution of hydroxylases toward aromatic and heterocyclic compounds. J. Biomol. Screen. 2005, 10, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Weingartner, A.M.; Sauer, D.F.; Dhoke, G.V.; Davari, M.D.; Ruff, A.J.; Schwaneberg, U. A hydroquinone-specific screening system for directed P450 evolution. Appl. Microbiol. Biotechnol. 2018, 102, 9657–9667. [Google Scholar] [CrossRef] [Green Version]
- Tee, K.L.; Schwaneberg, U. A Screening System for the Directed Evolution of Epoxygenases: Importance of Position 184 in P450 BM3 for Stereoselective Styrene Epoxidation. Angew. Chem. Int. Ed. 2006, 45, 5380–5383. [Google Scholar] [CrossRef] [PubMed]
- Virus, C.; Bernhardt, R. Molecular Evolution of a Steroid Hydroxylating Cytochrome P450 Using a Versatile Steroid Detection System for Screening. Lipids 2008, 43, 1133–1141. [Google Scholar] [CrossRef]
- Gärtner, A.; Ruff, A.J.; Schwaneberg, U. A 96-multiplex capillary electrophoresis screening platform for product based evolution of P450 BM3. Sci. Rep. 2019. under review. [Google Scholar]
- Lauer, H.H.; Rozing, G.P. High Performance Capillary Electrophoresis; Agilent Technologies: Santa Clara, CA, USA, 2018; ISBN 5990-3777EN. [Google Scholar]
- Petersen, J.R.; Okorodudu, A.O.; Mohammad, A.; Payne, D.A. Capillary electrophoresis and its application in the clinical laboratory. Clin. Chim. Acta 2003, 330, 1–30. [Google Scholar] [CrossRef]
- Emerson, E. The Condensation of Aminoantipyrine. A new color test for phenolic compounds. J. Org. Chem. 1943, 8, 417–428. [Google Scholar] [CrossRef]
- Glieder, A.; Meinhold, P. High-Throughput Screens Based on NAD(P)H Depletion. In Directed Enzyme Evolution: Screening and Selection Methods; Arnold, F.H., Georgiou, G., Eds.; Humana Press: Totowa, NJ, USA, 2003; Volume 230, pp. 157–170. ISBN 978-1-59259-396-5. [Google Scholar]
- Cheng, F.; Zhu, L.; Schwaneberg, U. Directed evolution 2.0: Improving and deciphering enzyme properties. Chem. Commun. 2015, 51, 9760–9772. [Google Scholar] [CrossRef]
- Dennig, A.; Marienhagen, J.; Ruff, A.J.; Guddat, L.; Schwaneberg, U. Directed Evolution of P450 BM3 into a p-Xylene Hydroxylase. ChemCatChem 2012, 4, 771–773. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Li, H.; Zhang, H.; Peterson, J.A.; Poulos, T.L. Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. USA 1999, 96, 1863–1868. [Google Scholar] [CrossRef]
- Pochapsky, T.C.; Kazanis, S.; Dang, M. Conformational Plasticity and Structure/Function Relationships in Cytochromes P450. Antioxid. Redox Signal. 2010, 13, 1273–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavanti, M.; Parmeggiani, F.; Castellanos, J.R.G.; Mattevi, A.; Turner, N.J. One-Pot Biocatalytic Double Oxidation of α-Isophorone for the Synthesis of Ketoisophorone. ChemCatChem 2017, 9, 3338–3348. [Google Scholar] [CrossRef]
- Blanusa, M.; Schenk, A.; Sadeghi, H.; Marienhagen, J.; Schwaneberg, U. Phosphorothioate-based ligase-independent gene cloning (PLICing): An enzyme-free and sequence-independent cloning method. Anal. Biochem. 2010, 406, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, R.C.; Joyce, G.F. Mutagenic PCR. Genome Res. 1994, 3, S136–S140. [Google Scholar] [CrossRef]
- Nazor, J.; Dannenmann, S.; Adjei, R.O.; Fordjour, Y.B.; Ghampson, I.T.; Blanusa, M.; Roccatano, D.; Schwaneberg, U. Laboratory evolution of P450 BM3 for mediated electron transfer yielding an activity-improved and reductase-independent variant. Protein Eng. Des. Sel. 2007, 21, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaneberg, U.; Sprauer, A.; Schmidt-Dannert, C.; Schmid, R.D. P450 monooxygenase in biotechnology. I. Single-step, large-scale purification method for cytochrome P450 BM-3 by anion-exchange chromatography. J. Chromatogr. A 1999, 848, 149–159. [Google Scholar] [CrossRef]
- Omura, T.; Sato, R. The Carbon Monoxide-binding Pigment of Liver Microsomes. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Wang, Q.; Canutescu, A.A.; Dunbrack, R.L. SCWRL and MolIDE: Computer programs for side-chain conformation prediction and homology modeling. Nat. Protoc. 2008, 3, 1832–1847. [Google Scholar] [CrossRef]
- Wang, J.M.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Shahrokh, K.; Orendt, A.; Yost, G.S.; Cheatham, T.E. Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012, 33, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian Inc.: Wallingford, UK, 2016. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA, 2010.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]





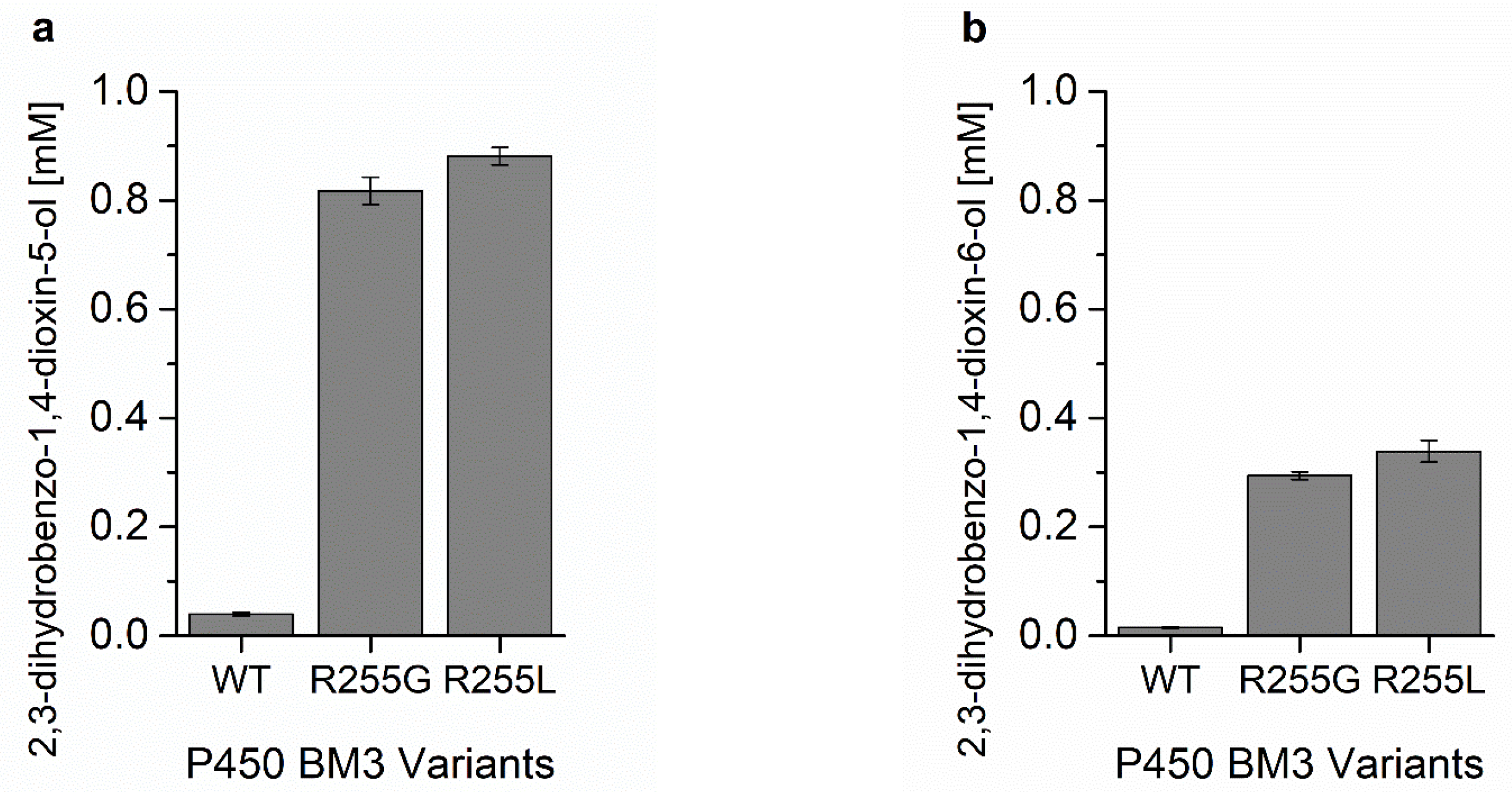



| Variant | NADPH Oxidation Rate [min−1] | Coupling EFFICIENCY [%] | TTN |
|---|---|---|---|
| WT | 8.3 ± 1.3 | 8.8 ± 0.1 | 40 ± 3 |
| R255G | 1719 ± 231 | 23.7 ± 0.5 | 798 ± 24 |
| R255L | 1168 ± 163 | 25.7 ± 1.0 | 860 ± 15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, G.d.A.; Dhoke, G.V.; Davari, M.D.; Ruff, A.J.; Schwaneberg, U. Directed Evolution of P450 BM3 towards Functionalization of Aromatic O-Heterocycles. Int. J. Mol. Sci. 2019, 20, 3353. https://doi.org/10.3390/ijms20133353
Santos GdA, Dhoke GV, Davari MD, Ruff AJ, Schwaneberg U. Directed Evolution of P450 BM3 towards Functionalization of Aromatic O-Heterocycles. International Journal of Molecular Sciences. 2019; 20(13):3353. https://doi.org/10.3390/ijms20133353
Chicago/Turabian StyleSantos, Gustavo de Almeida, Gaurao V. Dhoke, Mehdi D. Davari, Anna Joëlle Ruff, and Ulrich Schwaneberg. 2019. "Directed Evolution of P450 BM3 towards Functionalization of Aromatic O-Heterocycles" International Journal of Molecular Sciences 20, no. 13: 3353. https://doi.org/10.3390/ijms20133353
APA StyleSantos, G. d. A., Dhoke, G. V., Davari, M. D., Ruff, A. J., & Schwaneberg, U. (2019). Directed Evolution of P450 BM3 towards Functionalization of Aromatic O-Heterocycles. International Journal of Molecular Sciences, 20(13), 3353. https://doi.org/10.3390/ijms20133353