Quantifying the Potential for Future Gene Therapy to Lower Lifetime Risk of Polygenic Late-Onset Diseases


Abstract
:1. Introduction
2. Results
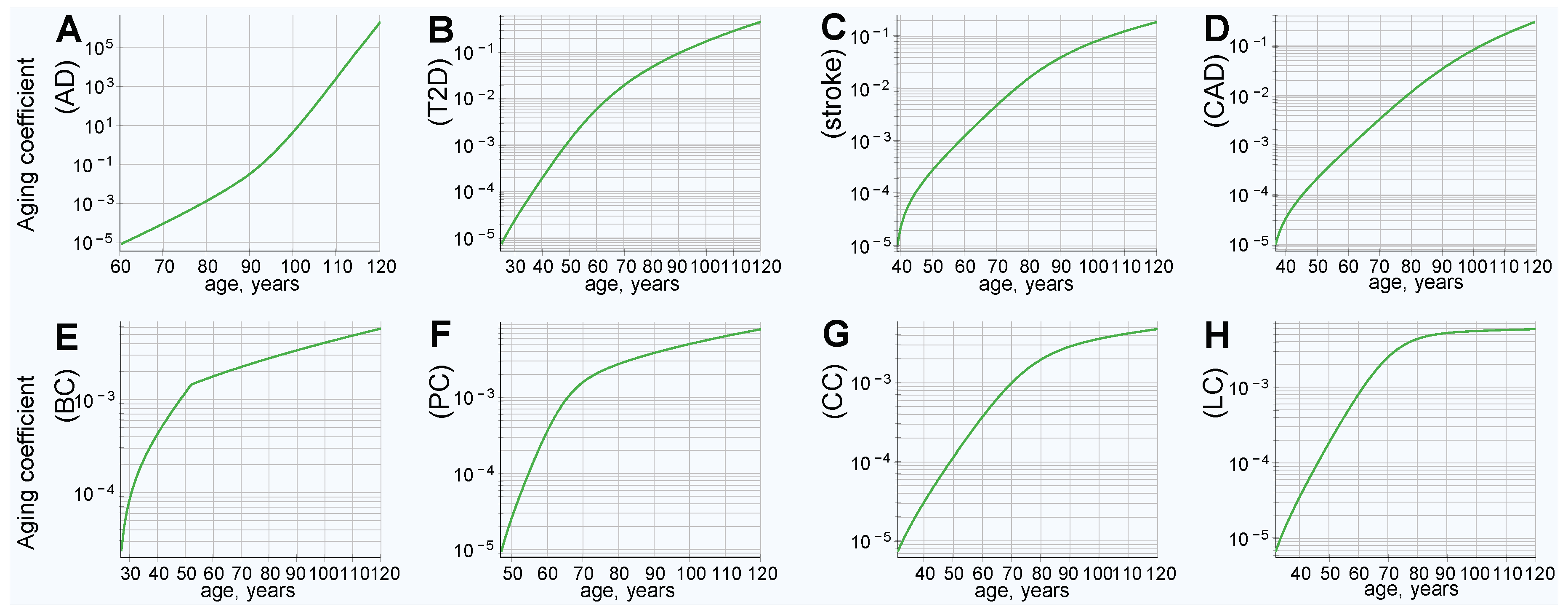
2.1. Characteristics of the Aging Coefficient
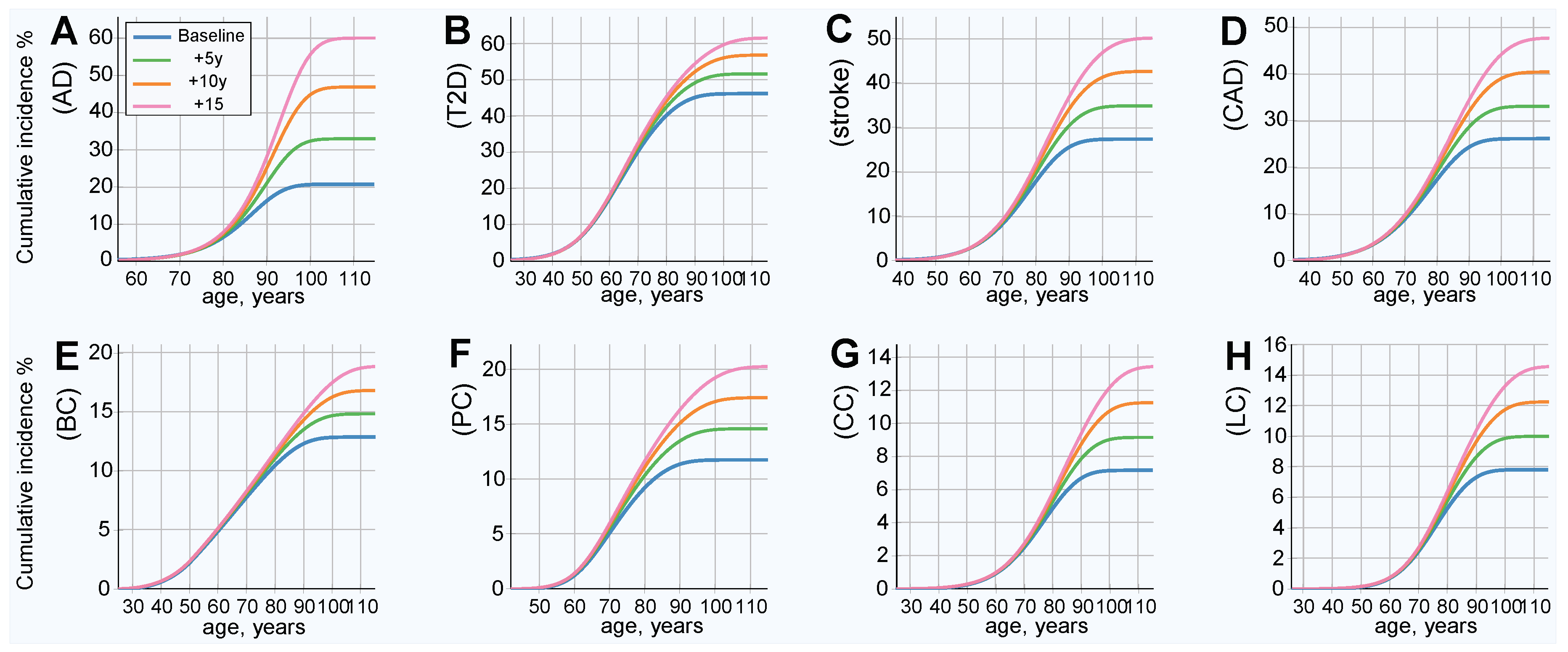
2.2. Longer Life Expectancy Corresponds to Increasing Lifetime Risk
2.3. Lifetime Risk Estimates for Discrete Hazard Ratio Multiples
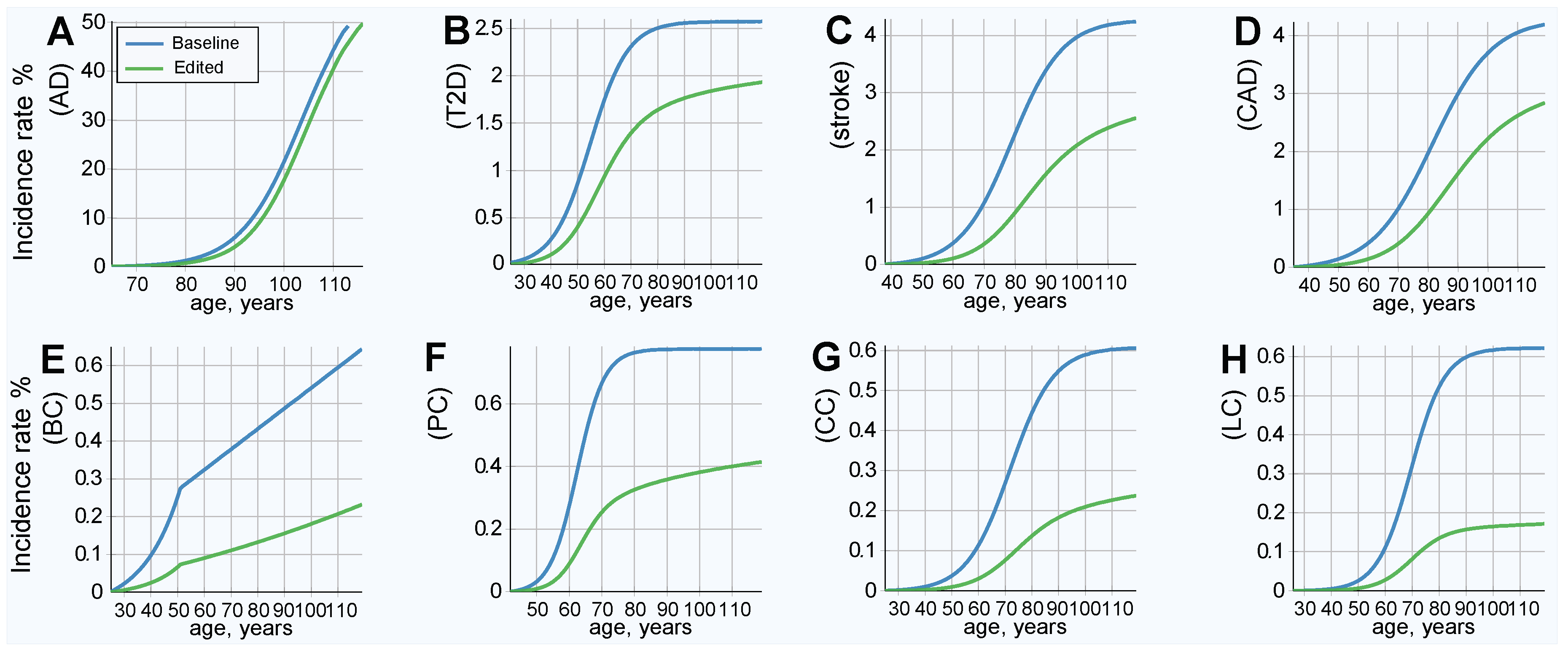
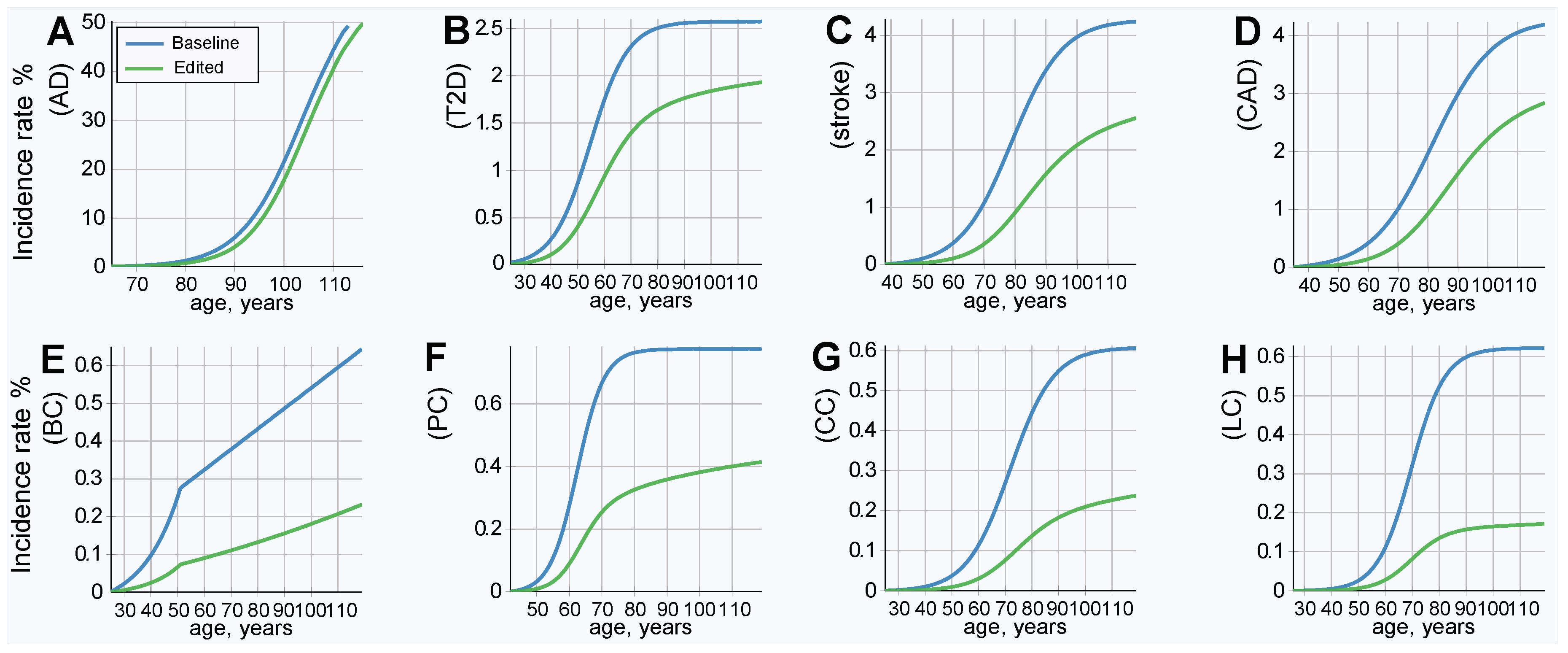
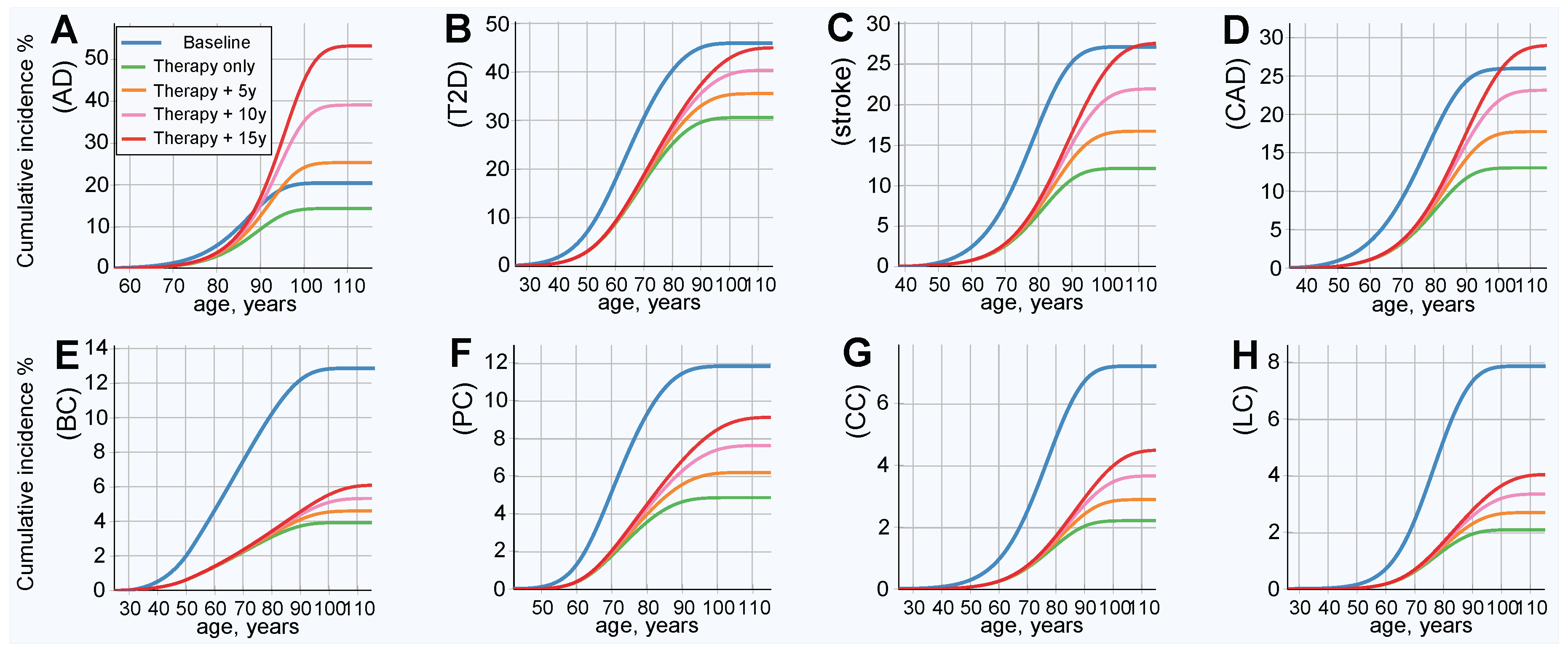
2.4. Results of Simulated Gene Therapy Lowering Population PRS
3. Discussion
4. Methods
4.1. Conceptual Summary
4.1.1. Cox’s Proportional Hazards Model
4.1.2. Allele Distribution Models
4.1.3. LOD Incidence Rate Functional Approximation
4.2. The Aging Coefficient: Mapping PRS to Age-Dependent Probability of LODs
4.3. Data Sources, Programming, and Equipment
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| CAD | coronary artery disease |
| GWAS | genome-wide association study |
| HR | hazard ratio |
| LOD | late-onset disease |
| MAF | minor allele frequency; customarily implying the ’effect allele frequency’ |
| OR | odds ratio |
| PRS | polygenic risk score |
| SNP | single nucleotide polymorphism; in context of this study used synonymously with the term ’allele’ |
| T2D | type 2 diabetes |
References
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [PubMed]
- OMIM. 2019. Available online: http://omim.org/statistics/geneMap (accessed on 2 June 2019).
- Steck, A.K.; Rewers, M.J. Genetics of type 1 diabetes. Clin. Chem. 2011, 57, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Ghani, M.; Reitz, C.; St George-Hyslop, P.; Rogaeva, E. Genetic Complexity of Early-Onset Alzheimer’s Disease. In Neurodegenerative Diseases; Springer International Publishing: Cham, Switzerland, 2018; pp. 29–50. [Google Scholar]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [PubMed] [Green Version]
- Sobrin, L.; Ripke, S.; Yu, Y.; Fagerness, J.; Bhangale, T.R.; Tan, P.L.; Souied, E.H.; Buitendijk, G.H.; Merriam, J.E.; Richardson, A.J. Heritability and genome-wide association study to assess genetic differences between advanced age-related macular degeneration subtypes. Ophthalmology 2012, 119, 1874–1885. [Google Scholar] [PubMed]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [PubMed]
- Eyre-Walker, A. Genetic architecture of a complex trait and its implications for fitness and genome-wide association studies. Proc. Natl. Acad. Sci. USA 2010, 107, 1752–1756. [Google Scholar]
- Yang, J.; Ferreira, T.; Morris, A.P.; Medland, S.E.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; Weedon, M.N.; Loos, R.J. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012, 44, 369–375. [Google Scholar] [CrossRef]
- Ribezzo, F.; Shiloh, Y.; Schumacher, B. Systemic DNA damage responses in aging and diseases. In Seminars in Cancer Biology; Elsevier: Atlanta, GA, USA, 2016; Volume 37, pp. 26–35. [Google Scholar]
- Nelson, P.; Masel, J. Intercellular competition and the inevitability of multicellular aging. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef]
- Fedarko, N.S. Theories and Mechanisms of Aging. In Geriatric Anesthesiology; Springer International Publishing: Cham, Switzerland, 2018; pp. 19–25. [Google Scholar]
- Franceschi, C.; Garagnani, P.G.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The continuum of aging and age-related diseases: Common mechanisms but different rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Lupski, J.R.; Gibbs, R.A. Human genome sequencing in health and disease. Annu. Rev. Med. 2012, 63, 35–61. [Google Scholar] [CrossRef] [PubMed]
- Yashin, A.I.; Arbeev, K.G.; Wu, D.; Arbeeva, L.; Kulminski, A.; Kulminskaya, I.; Akushevich, I.; Ukraintseva, S.V. How Genes Modulate Patterns of Aging-Related Changes on the Way to 100: Biodemographic Models and Methods in Genetic Analyses of Longitudinal Data. N. Am. Actuar J. 2016, 20, 201–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzhetsky, A.; Wajngurt, D.; Park, N.; Zheng, T. Probing genetic overlap among complex human phenotypes. Proc. Natl. Acad. Sci. USA 2007, 104, 11694–11699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.A.; Soranzo, N.; Zeggini, E.; Barrett, J.C. Synthetic associations are unlikely to account for many common disease genome-wide association signals. PLoS Biol. 2011, 9, e1000580. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bakshi, A.; Zhu, Z.; Hemani, G.; Vinkhuyzen, A.A.; Lee, S.H.; Robinson, M.R.; Perry, J.R.; Nolte, I.M.; van Vliet-Ostaptchouk, J.V. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 2015, 47, 1114. [Google Scholar] [CrossRef]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Fan, I.; Tang, J.; Li, S.; Zhang, S.; Shaw, P.A. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: A kin–cohort study in Ontario, Canada. J. Natl. Cancer Inst. 2006, 98, 1694–1706. [Google Scholar] [CrossRef]
- Haley, B. Hereditary Breast Cancer: The Basics of BRCA and Beyond. Internal Medicine Grand Rounds; UT Southwestern Medical Center; 2016; Available online: https://utswmed-ir.tdl.org/handle/2152.5/3095 (accessed on 7 July 2019).
- Wu, X.; Gu, J. Heritability of prostate cancer: A tale of rare variants and common single nucleotide polymorphisms. Ann. Transl. Med. 2016, 4, 206. [Google Scholar] [CrossRef]
- Mancuso, N.; Rohland, N.; Rand, K.A.; Tandon, A.; Allen, A.; Quinque, D.; Mallick, S.; Li, H.; Stram, A.; Sheng, X. The contribution of rare variation to prostate cancer heritability. Nat. Genet. 2016, 48, 30. [Google Scholar] [CrossRef]
- Walsh, P.C. The search for the missing heritability of prostate cancer. Eur. Urol. 2017, 72, 657–659. [Google Scholar] [CrossRef]
- Lecarpentier, J.; Silvestri, V.; Kuchenbaecker, K.B.; Barrowdale, D.; Dennis, J.; McGuffog, L.; Soucy, P.; Leslie, G.; Rizzolo, P.; Navazio, A.S. Prediction of breast and prostate cancer risks in male BRCA1 and BRCA2 mutation carriers using polygenic risk scores. J. Clin. Oncol. 2017, 35, 2240–2250. [Google Scholar] [CrossRef]
- De Voer, R.M.; Hahn, M.M.; Weren, R.D.; Mensenkamp, A.R.; Gilissen, C.; van Zelst-Stams, W.A.; Spruijt, L.; Kets, C.M.; Zhang, J.; Venselaar, H. Identification of novel candidate genes for early-onset colorectal cancer susceptibility. PLoS Genet. 2016, 12, e1005880. [Google Scholar] [CrossRef] [PubMed]
- Graff, R.E.; Möller, S.; Passarelli, M.N.; Witte, J.S.; Skytthe, A.; Christensen, K.; Tan, Q.; Adami, H.O.; Czene, K.; Harris, J.R. Familial risk and heritability of colorectal cancer in the nordic twin study of cancer. Clin. Gastroenterol. Hepatol. 2017, 15, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Weissfeld, J.L.; Lin, Y.; Lin, H.M.; Kurland, B.F.; Wilson, D.O.; Fuhrman, C.R.; Pennathur, A.; Romkes, M.; Nukui, T.; Yuan, J.M. Lung cancer risk prediction using common SNPs located in GWAS-identified susceptibility regions. J. Thorac. Oncol. 2015, 10, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.C.; Wang, X. Tomorrow’s genome medicine in lung cancer. In Seminars in Cancer Biology; Elsevier: Shanghai, China, 2017; Volume 42, pp. 39–43. [Google Scholar]
- Kanwal, M.; Ding, X.J.; Cao, Y. Familial risk for lung cancer. Oncol. Lett. 2017, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R. The Biology of Cancer; Garland Science: New York, NY, USA, 2013. [Google Scholar]
- Alzheimer’s Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. So! What’s aging? Is cardiovascular aging a disease? J. Mol. Cell. Cardiol. 2015, 83, 1–13. [Google Scholar] [CrossRef]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell 2018, 173, 611–623. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. Human Genome Editing: Science, Ethics, and Governance; National Academies Press: Washington, DC, USA, 2017. [Google Scholar]
- Nuffield Council on Bioethics. Genome Editing and Human Reproduction: Social and Ethical Issues; Nuffield Council on Bioethics: London, UK, 2018. [Google Scholar]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3. 0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Beck, T.; Hastings, R.K.; Gollapudi, S.; Free, R.C.; Brookes, A.J. GWAS Central: A comprehensive resource for the comparison and interrogation of genome-wide association studies. Eur. J. Hum. Genet. 2014, 22, 949. [Google Scholar] [CrossRef]
- Wright, A.V.; Nuñez, J.K.; Doudna, J.A. Biology and applications of CRISPR systems: Harnessing nature’s toolbox for genome engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Castanon, O.; Said, K.; Volf, V.; Khoshakhlagh, P.; Hornick, A.; Ferreira, R.; Wu, C.T.; Güell, M.; Garg, S.; et al. Enabling large-scale genome editing by reducing DNA nicking. bioRxiv 2019. [Google Scholar] [CrossRef]
- Thompson, D.; Aboulhouda, S.; Hysolli, E.; Smith, C.; Wang, S.; Castanon, O.; Church, G. The future of multiplexed eukaryotic genome engineering. ACS Chem. Biol. 2017, 13, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Kohman, R.E.; Kunjapur, A.M.; Hysolli, E.; Wang, Y.; Church, G.M. From Designing the Molecules of Life to Designing Life: Future Applications Derived from Advances in DNA Technologies. Angew. Chem. 2018, 57, 4313–4328. [Google Scholar] [CrossRef] [PubMed]
- Makeham, W.M. On the law of mortality and construction of annuity tables. J. Inst. Actuar. 1860, 8, 301–310. [Google Scholar] [CrossRef]
- Missov, T.I.; Lenart, A. Gompertz–Makeham life expectancies: Expressions and applications. Theor. Popul. Biol. 2013, 90, 29–35. [Google Scholar] [CrossRef]
- Stallard, E. Compression of morbidity and mortality: New perspectives. N. Am. Actuar. J. 2016, 20, 341–354. [Google Scholar] [CrossRef]
- Geddes, L. Human age limit claim sparks debate. Nat. News 2016. [Google Scholar] [CrossRef]
- Barbi, E.; Lagona, F.; Marsili, M.; Vaupel, J.W.; Wachter, K.W. The plateau of human mortality: Demography of longevity pioneers. Science 2018, 360, 1459–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliynyk, R.T. Age-related late-onset disease heritability patterns and implications for genome-wide association studies. PeerJ 2019, 7, e7168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stearns, F.W. One hundred years of pleiotropy: A retrospective. Genetics 2010, 186, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Paaby, A.B.; Rockman, M.V. The many faces of pleiotropy. Trends Genet. 2013, 29, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Beiser, A.S.; Breteler, M.M.; Fratiglioni, L.; Helmer, C.; Hendrie, H.C.; Honda, H.; Ikram, M.A.; Langa, K.M.; Lobo, A. The changing prevalence and incidence of dementia over time - current evidence. Nat. Rev. Neurol. 2017, 13, 327. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, R.B., Sr.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care. Circulation 2008. [Google Scholar] [CrossRef] [PubMed]
- Boehme, M.W.; Buechele, G.; Frankenhauser-Mannuss, J.; Mueller, J.; Lump, D.; Boehm, B.O.; Rothenbacher, D. Prevalence, incidence and concomitant co-morbidities of type 2 diabetes mellitus in South Western Germany—A retrospective cohort and case control study in claims data of a large statutory health insurance. BMC Public Health 2015, 15, 855. [Google Scholar] [CrossRef]
- Cox, D. Regression Models and Life-Tables. J. R. Stat. Soc. Ser. B (Methodol.) 1972, 34, 187–220. [Google Scholar] [CrossRef]
- Chatterjee, N.; Shi, J.; García-Closas, M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 2016, 17, 392. [Google Scholar] [CrossRef]
- Belsky, D.W.; Moffitt, T.E.; Baker, T.B.; Biddle, A.K.; Evans, J.P.; Harrington, H.; Houts, R.; Meier, M.; Sugden, K.; Williams, B.; et al. Polygenic risk and the developmental progression to heavy, persistent smoking and nicotine dependence: Evidence from a 4-decade longitudinal study. JAMA Psychiatry 2013, 70, 534–542. [Google Scholar] [CrossRef]
- Hjelmborg, J.; Korhonen, T.; Holst, K.; Skytthe, A.; Pukkala, E.; Kutschke, J.; Harris, J.R.; Mucci, L.A.; Christensen, K.; Czene, K.; et al. Lung cancer, genetic predisposition and smoking: The Nordic Twin Study of Cancer. Thorax 2017, 72, 1021–2027. [Google Scholar] [CrossRef] [PubMed]
- Brookmeyer, R.; Gray, S.; Kawas, C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am. J. Public Health 1998, 88, 1337–1342. [Google Scholar] [PubMed]
- Noh, M.; Yip, B.; Lee, Y.; Pawitan, Y. Multicomponent variance estimation for binary traits in family-based studies. Genet. Epidemiol. 2006, 30, 37–47. [Google Scholar] [PubMed]
- Pawitan, Y.; Seng, K.C.; Magnusson, P.K. How many genetic variants remain to be discovered? PLoS ONE 2009, 4, e7969. [Google Scholar]
- Social Security Administration (US). 2014. Available online: https://www.ssa.gov/oact/STATS/table4c6.html (accessed on 2 June 2019).
- Edland, S.D.; Rocca, W.A.; Petersen, R.C.; Cha, R.H.; Kokmen, E. Dementia and Alzheimer disease incidence rates do not vary by sex in Rochester, Minn. Arch. Neurol. 2002, 59, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Kokmen, E.; Chandra, V.; Schoenberg, B.S. Trends in incidence of dementing illness in Rochester, Minnesota, in three quinquennial periods, 1960–1974. Neurology 1988, 38, 975–980. [Google Scholar] [PubMed]
- Hebert, L.E.; Scherr, P.A.; Beckett, L.A.; Albert, M.S.; Pilgrim, D.M.; Chown, M.J.; Funkenstein, H.H.; Evans, D.A. Age-specific incidence of Alzheimer’s disease in a community population. JAMA 1995, 273, 1354–1359. [Google Scholar] [PubMed]
- Rothwell, P.; Coull, A.; Silver, L.; Fairhead, J.; Giles, M.; Lovelock, C.; Redgrave, J.; Bull, L.; Welch, S.; Cuthbertson, F. Population-based study of event-rate, incidence, case fatality, and mortality for all acute vascular events in all arterial territories (Oxford Vascular Study). Lancet 2005, 366, 1773–1783. [Google Scholar]
- Cancer Research UK. 2018. Available online: http://www.cancerresearchuk.org/health-professional/cancer-statistics-for-the-uk (accessed on 10 November 2018).
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017, 317, 2402–2416. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Highly Prevalent LODs | Cancers | |||||||
|---|---|---|---|---|---|---|---|---|
| AD | T2D | Stroke | CAD | Breast | Prostate | Colorectal | Lung | |
| 1.0 | 43.3 | 78.7 | 26.8 | 21.1 | 8.27 | 3.94 | 3.33 | 7.12 |
| 0.5 | 31.9 | 39.9 | 13.5 | 10.6 | 4.14 | 1.97 | 1.66 | 3.56 |
| 0.25 | 22.0 | 20.0 | 6.75 | 5.30 | 2.07 | 0.984 | 0.832 | 1.78 |
| Highly Prevalent LODs | Cancers | |||||||
|---|---|---|---|---|---|---|---|---|
| AD | T2D | Stroke | CAD | Breast | Prostate | Colorectal | Lung | |
| 16.0 → 4.0 | 4 | 7 | 10 | 10 | 21 | 17 | 13 | 10 |
| 4.0 → 1.0 | 3 | 12 | 13 | 14 | 31 | 16 | 16 | 20 |
| 1.0 → 0.25 | 4 | 13 | 11 | 16 | 37 | 21 | 19 | 16 |
| Highly Prevalent LODs | Cancers | |||||||
|---|---|---|---|---|---|---|---|---|
| AD | T2D | Stroke | CAD | Breast | Prostate | Colorectal | Lung | |
| 16.0 → 4.0 | 1 | 0 | 2 | 3 | >40 | 19 | 24 | 21 |
| 4.0 → 1.0 | 2 | 9 | 14 | 12 | >40 | 28 | 34 | >40 |
| 1.0 → 0.25 | 3 | 20 | 17 | 14 | >40 | 29 | 35 | >40 |
| Highly Prevalent LODs | Cancers | |||||||
|---|---|---|---|---|---|---|---|---|
| AD | T2D | Stroke | CAD | Breast | Prostate | Colorectal | Lung | |
| Literature and clinical data: | ||||||||
| Heritability | 0.795 | 0.69 | 0.55 | 0.41 | 0.57 | 0.40 | 0.31 | 0.10 |
| Max yearly incidence rate | >20% | 2.5% | 4.4% | 3.6% | <0.5% | <0.8% | <0.6% | <0.6% |
| Genetic model SNP count | 3575 | 2125 | 1175 | 625 | 1250 | 600 | 400 | 100 |
| Lifetime risk, baseline + longer life: | ||||||||
| +5 years life expectancy | 160% | 112% | 128% | 127% | 115% | 123% | 127% | 128% |
| +10 years life expectancy | 228% | 123% | 156% | 155% | 130% | 147% | 156% | 156% |
| +15 years life expectancy | 293% | 134% | 184% | 184% | 146% | 172% | 187% | 186% |
| Lifetime risk, odds ratio (OR) 0.25 therapy versus baseline: | ||||||||
| Therapy, unchanged life expectancy | 70% | 67% | 44% | 50% | 30% | 41% | 31% | 27% |
| Therapy, +5 years life expectancy | 124% | 77% | 61% | 69% | 36% | 52% | 40% | 34% |
| Therapy, +10 years life expectancy | 191% | 88% | 81% | 89% | 41% | 65% | 51% | 43% |
| Therapy, +15 years life expectancy | 260% | 98% | 101% | 112% | 47% | 77% | 62% | 52% |
| Scenario | MAF Range | OR Range | MAF Values | Allele OR Values |
|---|---|---|---|---|
| A. Common low | 0.073–0.499 | 1.05–1.15 | 0.073, 0.18, 0.286, 0.393, 0.5 | 1.05, 1.075, 1.1, 1.125, 1.15 |
| B. Rare medium | 0.0146–0.0998 | 1.28–2.01 | 0.0146, 0.036, 0.0572, 0.0785, 0.0998 | 1.28, 1.463, 1.645, 1.828, 2.01 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliynyk, R.T. Quantifying the Potential for Future Gene Therapy to Lower Lifetime Risk of Polygenic Late-Onset Diseases. Int. J. Mol. Sci. 2019, 20, 3352. https://doi.org/10.3390/ijms20133352
Oliynyk RT. Quantifying the Potential for Future Gene Therapy to Lower Lifetime Risk of Polygenic Late-Onset Diseases. International Journal of Molecular Sciences. 2019; 20(13):3352. https://doi.org/10.3390/ijms20133352
Chicago/Turabian StyleOliynyk, Roman Teo. 2019. "Quantifying the Potential for Future Gene Therapy to Lower Lifetime Risk of Polygenic Late-Onset Diseases" International Journal of Molecular Sciences 20, no. 13: 3352. https://doi.org/10.3390/ijms20133352
APA StyleOliynyk, R. T. (2019). Quantifying the Potential for Future Gene Therapy to Lower Lifetime Risk of Polygenic Late-Onset Diseases. International Journal of Molecular Sciences, 20(13), 3352. https://doi.org/10.3390/ijms20133352