A Unique Panel of Patient-Derived Cutaneous Squamous Cell Carcinoma Cell Lines Provides a Preclinical Pathway for Therapeutic Testing

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
2.1. Case Histories and Histopathology
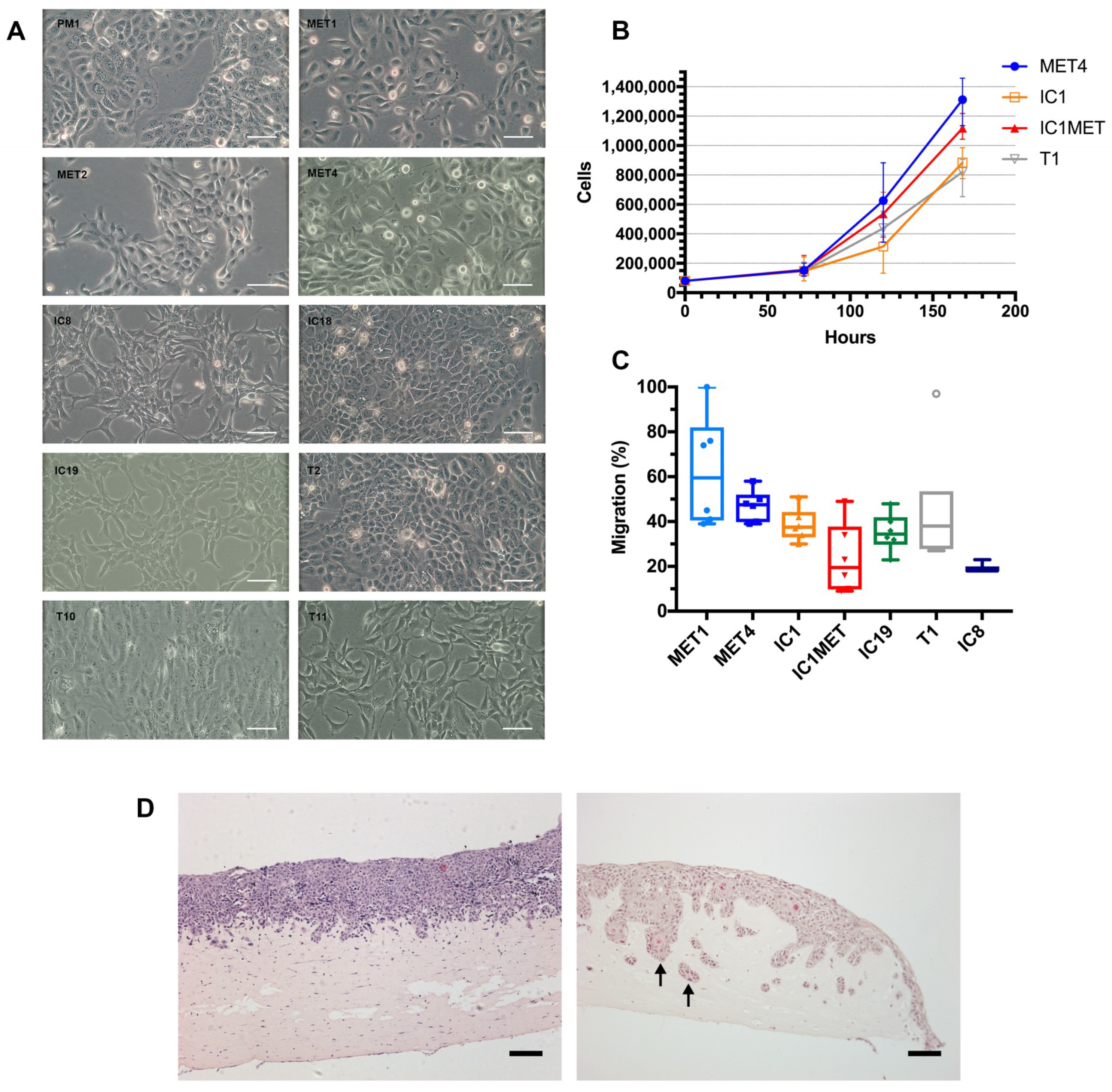
2.2. In Vitro Characteristics: Cell Morphology, Growth Curves, Transwell Migration and Organotypic Invasion
2.3. In Vivo Tumorigenicity
2.4. Genetic Analyses
2.4.1. Short Tandem Repeat (STR) DNA Profiling
2.4.2. The Genomic Landscape of Cell Lines
2.4.3. Phylogenetic Analysis of Isogenic Lines
2.4.4. Genome-Wide Methylation Profiling of cSCC Cell Lines
3. Discussion
3.1. Existing cSCC Cell Lines
3.2. Tumour Microenvironment
3.3. Characterisation of cSCC Cell Lines for Future Experimentation
3.4. Methylation Profiling
3.5. Application of cSCC Cell Lines in Drug Screening
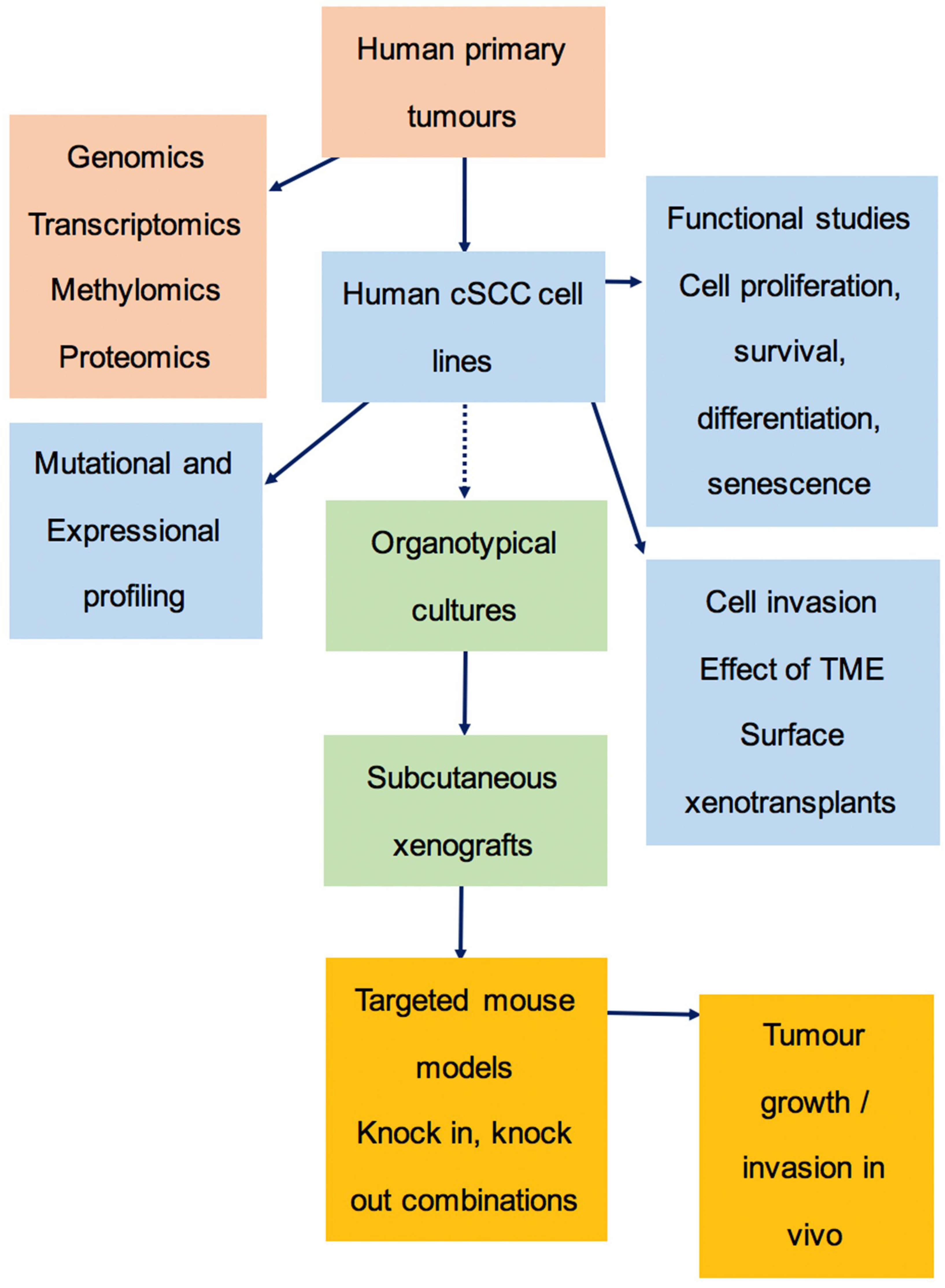
3.6. The Preclinical Pipeline
3.7. Models for Metastasis
4. Materials and Methods
4.1. Patient Samples
4.2. Establishment of cSCC Cell Lines
4.3. 3-D Organotypic Cultures
4.4. Growth Curve and Cell Migration Assay
4.5. In Vivo Xenograft Experiments
4.6. DNA Extraction and STR Profiling Analysis
4.7. Whole Exome Sequencing (WES) Data Processing and Analysis
4.8. Copy Number Analysis
4.9. Phylogenetic Analysis
4.10. Genome-Wide Methylation Profiling
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cSCC | cutaneous squamous cell carcinoma |
| OTR | organ transplant recipients |
| SCID | severe combined immune deficiency |
| STR | short tandem repeat |
| HNSCC | head and neck squamous cell carcinoma |
| ATCC | American Type Culture Collection |
| WES | whole exome sequencing |
| CNA | copy number alterations |
| DAVID | Database for Annotation, Visualization and Integrated Discovery |
| FDR | False discovery rate |
| NHK | normal human keratinocyte |
| ECACC | European Collection of Authenticated Cell Cultures |
| DSMZ | German Collection of Microorganisms and Cell Cultures GmbH |
| HPV | human papillomavirus |
| CAFs | cancer-associated fibroblasts |
| RDEB | recessive dystrophic epidermolysis bullosa |
| TME | tumour microenvironment |
| MEK | mitogen-activated protein kinase kinase |
| EGFR | epidermal growth factor receptor |
| SSUV | solar-simulated ultraviolet radiation |
| EDTA | ethylenediaminetetraacetic acid |
| EGF | epidermal growth factor |
| H&E | hematoxylin and eosin |
| BSA | bovine serum albumin |
| SDS | sodium dodecyl sulfate |
| NHF | normal human fibroblasts |
| SNP | single nucleotide polymorphism |
| LOH | loss of heterozygosity |
| CBS | circular binary segmentation |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Cell Lines Utilised (Nomenclature Changes) [N.B. SCC Prefix Sometimes Included] | Experimental Overview |
|---|---|---|
| [7] | PM1, MET1, MET2, MET4 | PM1 and additional isogenic premalignant cells retain features of normal keratinocytes compared to MET1-4 cells, which displayed reduced growth requirements, abnormal differentiation, aberrant K18 expression and tumorigenicity in vivo. HPV is not necessary for immortal phenotype maintenance. |
| [43] | MET1, MET2, MET3, MET4 | Genetic analysis of MET series. |
| [51] | PM1, MET1, MET4 | The oncogenic receptor tyrosine kinase Axl plays a role in driving cSCC; it is upregulated in MET1 and MET4 compared to PM1. |
| [52] | MET1, SCCIC1 | Type VII collagen knockdown increases cell migration and invasion in MET1 and SCCIC1 cells. Knockdown also leads to the disorganisation of epithelial differentiation and the promotion of epithelial to mesenchymal transition. |
| [53] | MET1, MET4 | The flavonoid luteolin inhibited UVB-induced inflammation and apoptosis in normal human keratinocytes. Exposure to luteolin did not increase cSCC line UVB resistance. |
| [54] | PM1, MET1, MET4 | Sensitisation of cSCC cells to cisplatin-induced cell death is achieved by the induction of apoptosis after Akt inhibition and can be increased with an Akt- and autophagy-inhibitor combination. |
| [55] | PM1, MET1 | The activated DNA damage response protein kinase ATM (pATM) is located in the perinucleus and, focally, in the cytoplasm of PM1 and MET1 cSCC cells. UVB-irradiation resulted in a degree of nuclear localisation in premalignant PM1 but not MET1 cells. DNA damage response proteins are upregulated in early carcinogenesis. |
| [56] | MET1, SCCIC1 | Axl knockdown in MET1 sensitises cells to UV-induced apoptosis via increased activation of pro-apoptotic protein Bad, altered conformation of pro-apoptotic proteins Bax and Bak, cytosolic cytochrome c release and caspase activation. Expression levels upon Axl knockdown were confirmed in SCCIC1 cells. UV-induced apoptosis was via the mitochondrial-mediated pathway. Axl confers cSCC resistance to apoptosis. |
| [57] | SCCT1, SCCT2, SCCT8, SCCIC1, SCCIC8, SCCIC12 | NOTCH1 and NOTCH2 mutations found in ~75% of cSCCs lines illustrating central roles in the disruption of microenvironmental communication in cSCC progression. |
| [58] | SCCT1, SCCT2, SCCT8, SCCIC1 | The overexpressed cSCC tumour cell-survival genes, PLK1 and C20orf20, when inhibited by siRNA-mediated knockdown, induced apoptosis in vitro and tumour growth reduction in vivo. |
| [59] | MET1, MET4 | The retinoid signalling molecule TRIM16 is reduced during progression from normal to actinic keratosis and cSCC. In cSCC TRIM16, cytoplasmic and nuclear expression is lower than normal lines due to reduced TRIM16 stability. In cSCC TRIM16, bound and downregulated E2F1 are required for replication. In normal cells, retinoids increase nuclear TRIM16 but not in retinoid-resistant cSCC. Overexpression of TRIM16 in cSCC reduced migration. TRIM16 plays a role in cSCC development and retinoid sensitivity. |
| [60] | MET1, MET4 | The flavonoid luteolin induced caspase-dependent cell death in cSCC. No cytotoxicity occurred in normal cell lines. Luteolin-induced apoptosis was accompanied by Akt signalling inhibition. Sensitivity decreased with tumour progression, with MET1 more sensitive to luteolin than MET4. Luteolin stimulates autophagy in MET4 cells. In blocking autophagy, luteolin-induced apoptosis in MET4 was enhanced. |
| [32] | SCCIC1 | Expression of full-length type VII collagen in RDEB fibroblasts retards SCCIC1 growth and invasion compared to growth and invasion facilitated by RDEB fibroblasts expressing empty vectors. Matrix composition in RDEB patients provides a tumour permissive environment, with type VII collagen directly regulating cancer-associated fibroblast protein secretion and cSCC progression. |
| [61] | PM1, MET1, MET4 | Sensitivity to apoptosis induced by UVB or oxidative stress differs between isogenic cell lines compared to cisplatin-induced apoptosis. MET1 was most sensitive to UVB- and oxidative stress-mediated apoptosis, with PM1 being the most resistant to apoptosis induction. |
| [62] | MET1, SCCIC1 | Axl knockdown in MET1 cells leads to ALDH1 cancer stem marker downregulation, CTGF, TWIST and VIM EMT marker downregulation, and E-cadherin upregulation. Subpopulations of MET1 cells expressing higher levels of cancer stem marker CD44 have correspondingly higher levels of Axl expression. Morphological and cell–cell interaction changes occur with Axl knockdown. Chemotherapeutic drug-induced cell death was inhibited by Axl expression. |
| [63] | MET1, MET2, MET4, SCCT8, SCCIC8, SCCIC18, SCCIC19 | The actin regulatory scaffold protein Eps8 is elevated in cSCC compared to normal skin. There were no differences in Eps8 levels based on cSCC progression or immune status. Eps8 in cSCC forms as a complex with focal adhesion kinase (FAK) at focal adhesions, promoting actin-associated cancer phenotypes, including direction sensing and invasive migration. In the absence of FAK, Eps8 is co-recruited with active Src. This provides a role for Eps8 as a mediator of intracellular kinase fate in cSCC. |
| [64] | SCCT1, SCCT2, SCCT8, SCCIC1, MET1, SCCIC1MET, MET4 | The SLCO1B3 gene, encoding the anion transporter frequently-mutated in cancer, OATP1B3, is upregulated in RDEB cSCC and UV-induced cSCC compared to normal keratinocytes. With the exception of SCCT8, all cSCC cell lines showed lower expression of SLCO1B3 compared to RDEB cSCC keratinocytes. Compared to normal keratinocytes, OATP1B3 is expressed in cSCC keratinocytes; however, membrane-localised expression is only readily detected in 3D cultures, mouse xenografts or in OATP1B3-overexpressing 2D cultures. COL7A1 knockdown increases SLCO1B3 expression in SCCT8 cells. SLCO1B3 expression was also regulated by COL7A1 and ELMO2 in cSCC cells. |
| [65] | MET1, MET2, MET4 | Flightless I is elevated in cSCC lines compared to HaCaT cell line controls. There was no significant difference in expression between isogenic lines MET1, MET2 and MET4. |
| [66] | SCCIC1 | Type VII collagen knockdown in SCCIC1 cells reduces cell–cell adhesion, increases blood vessel formation by increased vascular endothelial growth factor (VEGF) secretion, increases invasion, and reduces differentiation in cellular, organotypic and xenograft studies. |
| [41] | SCCIC1, SCCIC8, SCCIC12, SCCIC18, SCCIC19, SCCT1, SCCT2, SCCT8, PM1, MET1, MET4, SCCT9, SCCT10, SCCT11 | Identification of frequent TGFBR1/2 mutations in human vemurafenib-induced skin lesions and in sporadic cSCC. LGR5+ stem cells may act as origins for cSCC, driven by RAS/RAF/MAPK pathway hyperactivation or TP53 mutation (coupled with reduction in TGFb signalling). The TGFBR2-mutant cell lines SCCIC8 and SCCIC12 failed to respond to exogenous TGFb stimulation. |
| [27] | SCCT2, SCCT8, SCCIC1 | MEK inhibition results in senescence, but not apoptosis. Sensitivity to MEK inhibition was seen across all lines at high concentration, low concentration-response between lines was heterogenous. MEK inhibition may be a basis for molecularly targeted cSCC chemoprevention and therapy. |
| [67] | SCCIC18 | Activated SMAD2/3 was significantly reduced in perilesional cSCC and, to a greater extent, lesional cSCC tissue. Increased tumour thickness inversely correlates with phospho-SMAD presence. Lines were used for IHC antibody validation. |
| [37] | MET1 (SCCT), MET4 (SCCTMet), SCCIC1, SCCIC1Met | Continuous treatment with the ubiquitin E1 inhibitor MLN7243 selectively killed MET1 and SCC IC1MET cells. The latter cells were resistant to the inhibitor bortezomib. SCCIC1 cells were susceptible to bortezomib pulses but highly resistant to MLN7243. Bortezomib-resistant cells can be sensitive to MLN7243. Generally, pulse proteasome inhibitor exposure kills cSCC cells more effectively than extended exposure. C-MYC-dependent pro-apoptotic protein NOXA upregulation confers inhibitor susceptibility for rapid BAK-dependent death. |
| [68] | T1, T8, MET1, MET4 | Early stage T1 demonstrates the greatest proliferative response to IL-22 treatment. cSCC proliferation was inhibited by JAK1/2 inhibitor ruxolitinib in vitro with tumour size reduction in vivo; however, this was demonstrated in the A431 cSCC epidermoid carcinoma line only and not in T1, T8, MET1 or MET4 lines. |
| [3] | All lines (nomenclature consistent) | Whole exome sequencing analysis confirmed COSMIC mutation signature 7 was present in all lines. Signature 32 was present in eight lines (four patients), all immunosuppressed and receiving azathioprine. PM1, MET1/2/4 and T9 (all isogenic lines) showed similar mutational signatures. MET4 had additional signature 26 associated with defective DNA mismatch repair. Altered mismatch repair genes are found in all cSCC cell lines, but none correlate with signature 26. |
| [38] | MET1 (SCCT), MET4 (SCCTMet), SCCT1, SCCT2, SCCT8, SCCT10, SCCT11, SCCIC1, SCCIC1Met, SCCIC8, SCCIC12, SCCIC18 | Spliceosome suppression has potential as a cSCC treatment. Endogenous C-MYC expression in cSCC cells plays a role in conferring spliceosome inhibition-sensitivity and can act as a determinant of cSCC sensitivity to spliceosome targeting. Mutant p53 depletion does not attenuate spliceosome-inhibition-induced cell death in cSCC cells. |
| [69] | MET1, IC1 | Dual-action lysine specific demethylase 1- and histone deacetylase- inhibitor, coined Corin, exhibits anti-proliferative activity in cSCC lines, compared to mono-functional inhibitors alone. |
| [70] | MET1, MET4, IC8 | The HDAC-inhibitor MS-275 potently inhibits the proliferation of MET4, MET1 and IC8 cells. MS-275 also induced expression of p21 in cSCC cells. |
References
- Venables, Z.C.; Autier, P.; Nijsten, T.; Wong, K.F.; Langan, S.M.; Rous, B.; Broggio, J.; Harwood, C.; Henson, K.; Proby, C.M.; et al. Nationwide Incidence of Metastatic Cutaneous Squamous Cell Carcinoma in England. JAMA Dermatol. 2019, 155, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Eng. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef] [PubMed]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.Y.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 Mutations Occur Early during Cutaneous Squamous Cell Carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef] [Green Version]
- Harwood, C.A.; Proby, C.M.; Inman, G.J.; Leigh, I.M. The Promise of Genomics and the Development of Targeted Therapies for Cutaneous Squamous Cell Carcinoma. Acta Derm. Venereol. 2016, 96, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Sano, D.; Pickering, C.R.; Jasser, S.A.; Henderson, Y.C.; Clayman, G.L.; Sturgis, E.M.; Ow, T.J.; Lotan, R.; Carey, T.E.; et al. Assembly and Initial Characterization of a Panel of 85 Genomically Validated Cell Lines from Diverse Head and Neck Tumor Sites. Clin. Cancer Res. 2011, 17. [Google Scholar] [CrossRef]
- Proby, C.M.; Purdie, K.J.; Sexton, C.J.; Purkis, P.; Navsaria, H.A.; Stables, J.N.; Leigh, I.M. Spontaneous keratinocyte cell lines representing early and advanced stages of malignant transformation of the epidermis. Exp. Dermatol. 2000, 9, 104–117. [Google Scholar] [CrossRef]
- Giard, D.J.; Aaronson, S.A.; Parks, W.P.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H. In Vitro Cultivation of Human Tumors: Establishment of Cell Lines Derived From a Series of Solid Tumors2. JNCI J. Natl.Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef]
- Hunter, T.; Cooper, J.A. Epidermal growth factor induces rapid tyrosine phosphorylation of proteins in A431 human tumor cells. Cell 1981, 24, 741–752. [Google Scholar] [CrossRef]
- Rosdy, M.; Bernard, B.A.; Schmidt, R.; Darmon, M. Incomplete epidermal differentiation of A431 epidermoid carcinoma cells. In Vitro Cell. Dev. Biol. 1986, 22, 295–300. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Beckett, M.A. Defective terminal differentiation in culture as a consistent and selectable character of malignant human keratinocytes. Cell 1980, 22, 629–632. [Google Scholar] [CrossRef]
- Hu, L.; Crowe, D.L.; Rheinwald, J.G.; Chambon, P.; Gudas, L.J. Abnormal Expression of Retinoic Acid Receptors and Keratin 19 by Human Oral and Epidermal Squamous Cell Carcinoma Cell Lines. Cancer Res. 1991, 51, 3972. [Google Scholar] [CrossRef]
- Junttila, M.R.; Ala-aho, R.; Jokilehto, T.; Peltonen, J.; Kallajoki, M.; Grenman, R.; Jaakkola, P.; Westermarck, J.; Kähäri, V.M. p38α and p38δ mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene 2007, 26, 5267. [Google Scholar] [CrossRef] [PubMed]
- Farshchian, M.; Kivisaari, A.; Ala-Aho, R.; Riihilä, P.; Kallajoki, M.; Grénman, R.; Peltonen, J.; Pihlajaniemi, T.; Heljasvaara, R.; Kähäri, V.-M. Serpin peptidase inhibitor clade A member 1 (SerpinA1) is a novel biomarker for progression of cutaneous squamous cell carcinoma. Am. J. Pathol. 2011, 179, 1110–1119. [Google Scholar] [CrossRef]
- Moilanen, J.M.; Löffek, S.; Kokkonen, N.; Salo, S.; Väyrynen, J.P.; Hurskainen, T.; Manninen, A.; Riihilä, P.; Heljasvaara, R.; Franzke, C.-W.; et al. Significant Role of Collagen XVII And Integrin β4 in Migration and Invasion of The Less Aggressive Squamous Cell Carcinoma Cells. Sci. Rep. 2017, 7, 45057. [Google Scholar] [CrossRef]
- Hail, N.; Lotan, R. Mitochondrial Respiration Is Uniquely Associated with the Prooxidant and Apoptotic Effects ofN-(4-Hydroxyphenyl)retinamide. J. Biol. Chem. 2001, 276, 45614–45621. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Villanueva, J.; McDonnell, T.J. Induction of apoptotic cell death in non-melanoma skin cancer by interferon-α. Int. J. Cancer 1995, 61, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.E.; Merrick, S.B.; Woods, L.K.; Arabasz, N.M. A Human Squamous Cell Carcinoma Cell Line. Cancer Res. 1975, 35, 2684. [Google Scholar]
- Liu, Z.-L.; Li, H.; Liu, J.; Wu, M.-L.; Chen, X.-Y.; Liu, L.-H.; Wang, Q. Inactivated Wnt signaling in resveratrol-treated epidermal squamous cancer cells and its biological implication. Oncol. Lett. 2017, 14, 2239–2243. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.-N.; Li, Y.; Wu, M.-L.; Liu, Z.-L.; Fu, Y.-S.; Kong, Q.-Y.; Chen, X.-Y.; Li, H.; Liu, J. CRABP-II- and FABP5-independent all-trans retinoic acid resistance in COLO 16 human cutaneous squamous cancer cells. Exp. Dermatol. 2012, 21, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.W.; Younes, M.N.; Jasser, S.A.; Yigitbasi, O.G.; Zhou, G.; Bucana, C.D.; Bekele, B.N.; Myers, J.N. AEE788, a Dual Tyrosine Kinase Receptor Inhibitor, Induces Endothelial Cell Apoptosis in Human Cutaneous Squamous Cell Carcinoma Xenografts in Nude Mice. Clin. Cancer Res. 2005, 11, 1963. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.H.C.; Olsen, S.L.; Rankin, W.A.; Ho, P.W.M.; Martin, T.J.; Gillespie, M.T.; Moseley, J.M. PTHrP and cell division: Expression and localization of PTHrP in a keratinocyte cell line (HaCaT) during the cell cycle. J. Cell. Physiol. 1997, 173, 433–446. [Google Scholar] [CrossRef]
- Sonavane, K.; Phillips, J.; Ekshyyan, O.; Moore-Medlin, T.; Roberts Gill, J.; Rong, X.; Lakshmaiah, R.R.; Abreo, F.; Boudreaux, D.; Clifford, J.L.; et al. Topical curcumin-based cream is equivalent to dietary curcumin in a skin cancer model. J. Skin Cancer 2012, 2012, 147863. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.M.; Clark, C.; Herman-Ferdinandez, L.; Moore-Medlin, T.; Rong, X.; Gill, J.R.; Clifford, J.L.; Abreo, F.; Nathan, C.-A.O. Curcumin Inhibits Skin Squamous Cell Carcinoma Tumor Growth In Vivo. Otolaryngol. Head Neck Surg. 2011, 145, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kleiner-Hancock, H.E.; Shi, R.; Remeika, A.; Robbins, D.; Prince, M.; Gill, J.N.; Syed, Z.; Adegboyega, P.; Mathis, J.M.; Clifford, J.L. Effects of ATRA combined with citrus and ginger-derived compounds in human SCC xenografts. BMC Cancer 2010, 10, 394. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Glisson, B.S.; Khuri, F.R.; Clifford, J.L.; Clayman, G.; Benner, S.E.; Forastiere, A.A.; Ginsberg, L.; Liu, D.; Lee, J.J.; et al. Phase II and Biologic Study of Interferon Alfa, Retinoic Acid, and Cisplatin in Advanced Squamous Skin Cancer. J. Clin. Oncol. 2002, 20, 364–370. [Google Scholar] [CrossRef]
- Adelmann, C.H.; Truong, K.A.; Liang, R.J.; Bansal, V.; Gandee, L.; Saporito, R.C.; Lee, W.; Du, L.; Nicholas, C.; Napoli, M.; et al. MEK Is a Therapeutic and Chemopreventative Target in Squamous Cell Carcinoma. J. Investig. Dermatol. 2016, 136, 1920–1924. [Google Scholar] [CrossRef] [Green Version]
- Ridky, T.W.; Chow, J.M.; Wong, D.J.; Khavari, P.A. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat. Med. 2010, 16, 1450–1455. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, M.B.; Ramirez, R.D.; Andrews, C.M.; Wright, W.E.; Shay, J.W. H-Ras Expression in Immortalized Keratinocytes Produces an Invasive Epithelium in Cultured Skin Equivalents. PLoS ONE 2009, 4, e7908. [Google Scholar] [CrossRef]
- Fusenig, N.E.; Boukamp, P. Multiple stages and genetic alterations in immortalization, malignant transformation, and tumor progression of human skin keratinocytes. Mol. Carcinog. 1998, 23, 144–158. [Google Scholar] [CrossRef]
- Mueller, M.M.; Peter, W.; Mappes, M.; Huelsen, A.; Steinbauer, H.; Boukamp, P.; Vaccariello, M.; Garlick, J.; Fusenig, N.E. Tumor progression of skin carcinoma cells in vivo promoted by clonal selection, mutagenesis, and autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor. Am. J. Pathol. 2001, 159, 1567–1579. [Google Scholar] [CrossRef]
- Ng, Y.-Z.; Pourreyron, C.; Salas-Alanis, J.C.; Dayal, J.H.S.; Cepeda-Valdes, R.; Yan, W.; Wright, S.; Chen, M.; Fine, J.-D.; Hogg, F.J.; et al. Fibroblast-Derived Dermal Matrix Drives Development of Aggressive Cutaneous Squamous Cell Carcinoma in Patients with Recessive Dystrophic Epidermolysis Bullosa. Cancer Res. 2012, 72, 3522. [Google Scholar] [CrossRef]
- Scheitz, C.J.F.; Tumbar, T. New insights into the role of Runx1 in epithelial stem cell biology and pathology. J. Cell. Biochem. 2013, 114, 985–993. [Google Scholar] [CrossRef]
- Pourreyron, C.; Reilly, L.; Proby, C.; Panteleyev, A.; Fleming, C.; McLean, K.; South, A.P.; Foerster, J. Wnt5a Is Strongly Expressed at the Leading Edge in Non-Melanoma Skin Cancer, Forming Active Gradients, while Canonical Wnt Signalling Is Repressed. PLoS ONE 2012, 7, e31827. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603. [Google Scholar] [CrossRef]
- McHugh, A.; Fernandes, K.; South, A.P.; Mellerio, J.E.; Salas-Alanís, J.C.; Proby, C.M.; Leigh, I.M.; Saville, M.K. Preclinical comparison of proteasome and ubiquitin E1 enzyme inhibitors in cutaneous squamous cell carcinoma: The identification of mechanisms of differential sensitivity. Oncotarget 2018, 9, 20265–20281. [Google Scholar] [CrossRef]
- Hepburn, L.A.; McHugh, A.; Fernandes, K.; Boag, G.; Proby, C.M.; Leigh, I.M.; Saville, M.K. Targeting the spliceosome for cutaneous squamous cell carcinoma therapy: A role for c-MYC and wild-type p53 in determining the degree of tumour selectivity. Oncotarget 2018, 9, 23029–23046. [Google Scholar] [CrossRef]
- Nassar, D.; Latil, M.; Boeckx, B.; Lambrechts, D.; Blanpain, C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat. Med. 2015, 21, 946. [Google Scholar] [CrossRef]
- Knatko, E.V.; Praslicka, B.; Higgins, M.; Evans, A.; Purdie, K.J.; Harwood, C.A.; Proby, C.M.; Ooi, A.; Dinkova-Kostova, A.T. Whole-Exome Sequencing Validates a Preclinical Mouse Model for the Prevention and Treatment of Cutaneous Squamous Cell Carcinoma. Cancer Prev. Res. 2017, 10, 67. [Google Scholar] [CrossRef]
- Cammareri, P.; Rose, A.M.; Vincent, D.F.; Wang, J.; Nagano, A.; Libertini, S.; Ridgway, R.A.; Athineos, D.; Coates, P.J.; McHugh, A.; et al. Inactivation of TGFβ receptors in stem cells drives cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12493. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.A.; Mesher, D.; McGregor, J.M.; Mitchell, L.; Leedham-Green, M.; Raftery, M.; Cerio, R.; Leigh, I.M.; Sasieni, P.; Proby, C.M. A Surveillance Model for Skin Cancer in Organ Transplant Recipients: A 22-Year Prospective Study in an Ethnically Diverse Population. Am. J. Transplant. 2013, 13, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Popp, S.; Waltering, S.; Boukamp, P.; Holtgreve-Grez, H.; Jauch, A.; Proby, C.; Leigh, I.M. Genetic Characterization of a Human Skin Carcinoma Progression Model: From Primary Tumor to Metastasis. J. Investig. Dermatol. 2000, 115, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Purdie, K.J.; Pourreyron, C.; South, A.P. Isolation and Culture of Squamous Cell Carcinoma Lines. In Cancer Cell Culture: Methods and Protocols; Cree, I.A., Ed.; Humana Press: Totowa, NJ, USA, 2011; 14p. [Google Scholar]
- Van Loo, P.; Nordgard, S.H.; Lingjærde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910. [Google Scholar] [CrossRef] [PubMed]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, H.; Simpson, K.; Bullard, J.; Hansen, K. aroma. affymetrix: A Generic Framework in R for Analyzing Small to Very Large Affymetrix Data Sets in Bounded Memory; Technical Report; Department of Epidemiology and Biostatistics, University of California San Francisco: San Francisco, CA, USA, 2008; pp. 1–9. [Google Scholar]
- Bengtsson, H.; Neuvial, P.; Speed, T.P. TumorBoost: Normalization of allele-specific tumor copy numbers from a single pair of tumor-normal genotyping microarrays. BMC Bioinform. 2010, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Olshen, A.B.; Bengtsson, H.; Neuvial, P.; Spellman, P.T.; Olshen, R.A.; Seshan, V.E. Parent-specific copy number in paired tumor-normal studies using circular binary segmentation. Bioinformatics 2011, 27, 2038–2046. [Google Scholar] [CrossRef]
- Nei, M.; Saitou, N. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evolut. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Green, J.; Ikram, M.; Vyas, J.; Patel, N.; Proby, C.M.; Ghali, L.; Leigh, I.M.; O’Toole, E.A.; Storey, A. Overexpression of the Axl tyrosine kinase receptor in cutaneous SCC-derived cell lines and tumours. Br. J. Cancer 2006, 94, 1446–1451. [Google Scholar] [CrossRef] [Green Version]
- Martins, V.L.; Vyas, J.J.; Chen, M.; Purdie, K.; Mein, C.A.; South, A.P.; Storey, A.; McGrath, J.A.; O’Toole, E.A. Increased invasive behaviour in cutaneous squamous cell carcinoma with loss of basement-membrane type VII collagen. J. Cell Sci. 2009, 122, 1788–1799. [Google Scholar] [CrossRef] [Green Version]
- Verschooten, L.; Smaers, K.; Van Kelst, S.; Proby, C.; Maes, D.; Declercq, L.; Agostinis, P.; Garmyn, M. The Flavonoid Luteolin Increases the Resistance of Normal, but Not Malignant Keratinocytes, Against UVB-Induced Apoptosis. J. Investig. Dermatol. 2010, 130, 2277–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claerhout, S.; Verschooten, L.; Van Kelst, S.; De Vos, R.; Proby, C.; Agostinis, P.; Garmyn, M. Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int. J. Cancer 2010, 127, 2790–2803. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.; Ikram, M.; Purdie, K.; Harwood, C.; Leigh, I.; Storey, A. Cutaneous Squamous Cell Carcinoma (SCC) and the DNA Damage Response: pATM Expression Patterns in Pre-Malignant and Malignant Keratinocyte Skin Lesions. PLoS ONE 2011, 6, e21271. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, E.S.; Cichoń, M.A.; Vyas, J.J.; Patel, N.; Ghali, L.; Cerio, R.; Storey, A.; O’Toole, E.A. Axl Promotes Cutaneous Squamous Cell Carcinoma Survival through Negative Regulation of Pro-Apoptotic Bcl-2 Family Members. J. Investig. Dermatol. 2011, 131, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761. [Google Scholar] [CrossRef] [PubMed]
- Watt, S.A.; Pourreyron, C.; Purdie, K.; Hogan, C.; Cole, C.L.; Foster, N.; Pratt, N.; Bourdon, J.C.; Appleyard, V.; Murray, K.; et al. Integrative mRNA profiling comparing cultured primary cells with clinical samples reveals PLK1 and C20orf20 as therapeutic targets in cutaneous squamous cell carcinoma. Oncogene 2011, 30, 4666–4677. [Google Scholar] [CrossRef]
- Cheung, B.B.; Koach, J.; Tan, O.; Kim, P.; Bell, J.L.; D’Andreti, C.; Sutton, S.; Malyukova, A.; Sekyere, E.; Norris, M.; et al. The retinoid signalling molecule, TRIM16, is repressed during squamous cell carcinoma skin carcinogenesis in vivo and reduces skin cancer cell migration in vitro. J. Pathol. 2012, 226, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Verschooten, L.; Barrette, K.; Van Kelst, S.; Rubio Romero, N.; Proby, C.; De Vos, R.; Agostinis, P.; Garmyn, M. Autophagy inhibitor chloroquine enhanced the cell death inducing effect of the flavonoid luteolin in metastatic squamous cell carcinoma cells. PLoS ONE 2012, 7, e48264. [Google Scholar] [CrossRef] [PubMed]
- Barrette, K.; Zutterman, N.; Van Kelst, S.; Proby, C.; Garmyn, M. Pattern of sensitivity of progressive cutaneous squamous cell carcinoma cells to UVB and oxidative stress-induced cell death. Photochem. Photobiol. Sci. 2013, 12, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Cichoń, M.A.; Szentpetery, Z.; Caley, M.P.; Papadakis, E.S.; Mackenzie, I.C.; Brennan, C.H.; O’Toole, E.A. The receptor tyrosine kinase Axl regulates cell–cell adhesion and stemness in cutaneous squamous cell carcinoma. Oncogene 2013, 33, 4185. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, C.; Serrels, B.; Proby, C.; Cunningham, D.L.; Findlay, J.E.; Baillie, G.S.; Heath, J.K.; Frame, M.C. Eps8 controls Src- and FAK-dependent phenotypes in squamous carcinoma cells. J. Cell Sci. 2014, 127, 5303–5316. [Google Scholar] [CrossRef] [PubMed]
- Dayal, J.H.S.; Cole, C.L.; Pourreyron, C.; Watt, S.A.; Lim, Y.Z.; Salas-Alanis, J.C.; Murrell, D.F.; McGrath, J.A.; Stieger, B.; Jahoda, C.; et al. Type VII collagen regulates expression of OATP1B3, promotes front-to-rear polarity and increases structural organisation in 3D spheroid cultures of RDEB tumour keratinocytes. J. Cell Sci. 2014, 127, 740. [Google Scholar] [CrossRef] [PubMed]
- Kopecki, Z.; Yang, G.N.; Jackson, J.E.; Melville, E.L.; Calley, M.P.; Murrell, D.F.; Darby, I.A.; O’Toole, E.A.; Samuel, M.S.; Cowin, A.J. Cytoskeletal protein Flightless I inhibits apoptosis, enhances tumor cell invasion and promotes cutaneous squamous cell carcinoma progression. Oncotarget 2015, 6, 36426–36440. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.L.; Caley, M.P.; Moore, K.; Szentpetery, Z.; Marsh, S.T.; Murrell, D.F.; Kim, M.H.; Avari, M.; McGrath, J.A.; Cerio, R.; et al. Suppression of TGFβ and Angiogenesis by Type VII Collagen in Cutaneous SCC. JNCI J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef]
- Rose, A.M.; Spender, L.C.; Stephen, C.; Mitchell, A.; Rickaby, W.; Bray, S.; Evans, A.T.; Dayal, J.; Purdie, K.J.; Harwood, C.A.; et al. Reduced SMAD2/3 activation independently predicts increased depth of human cutaneous squamous cell carcinoma. Oncotarget 2018, 9, 14552–14566. [Google Scholar] [CrossRef] [PubMed]
- Abikhair Burgo, M.; Roudiani, N.; Chen, J.; Santana, A.L.; Doudican, N.; Proby, C.; Felsen, D.; Carucci, J.A. Ruxolitinib inhibits cyclosporine-induced proliferation of cutaneous squamous cell carcinoma. JCI Insight 2018, 3, e120750. [Google Scholar] [CrossRef] [PubMed]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 2018, 9, 53. [Google Scholar] [CrossRef]
- Kalin, J.H.; Eroglu, A.; Liu, H.; Holtzclaw, W.D.; Leigh, I.; Proby, C.M.; Fahey, J.W.; Cole, P.A.; Dinkova-Kostova, A.T. Investigation into the use of histone deacetylase inhibitor MS-275 as a topical agent for the prevention and treatment of cutaneous squamous cell carcinoma in an SKH-1 hairless mouse model. PLoS ONE 2019, 14, e0213095. [Google Scholar] [CrossRef]






| Cell line | Tumour | Immune Status | Immune Therapy 1 | Site | SCC Histology | Age | Sex | Germline DNA 2 | Organotypic Invasion | Transwell Migration | Tumorigenicity 3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PM1 # | pre-malignant lesion | RT | A,P | scalp | dysplastic | 45 | M | dermal fibroblasts | ND | ND | 0/10 4, 6/6 |
| MET1 # | primary | RT | A,P | dorsum left hand | MD | 45 | M | dermal fibroblasts | ND | 40–100% | 5/10 4, 6/6 |
| MET2 # | recurrence of MET1 | RT | A,P | dorsum left hand | MD | 45 | M | dermal fibroblasts | ND | 40–50% | 7/10 4 |
| MET4 # | metastasis of MET 1/2 | RT | A,P | left axillary lymph node | metastasis | 46 | M | dermal fibroblasts | ND | 40–60% | 5/10 4; 6/6 |
| SCC T9 # | unrelated primary of MET patient # | RT | A,P | right hand | WD | 45 | M | dermal fibroblasts | ND | ND | ND |
| SCC IC1 | primary | IC | N/A | right temple | MD | 77 | M | lymphocytes | 30–50% | 14/14 5; 6/6 | |
| SCC IC1MET | metastasis of IC1 | IC | N/A | right preauricular lymph node | metastasis | 77 | M | lymphocytes | ND | 10–50% | 6/6 |
| SCC IC8 | primary | IC (PUVA) | N/A | buttock | PD | 51 | F | lymphocytes | 20–25% | ND | |
| SCC IC12 | primary | IC | N/A | left calf | MD-PD | 87 | F | lymphocytes | ND | ND | ND |
| SCC IC18 | primary | IC | N/A | right ear | MD | 81 | M | lymphocytes | ND | ND | ND |
| SCC IC19 | primary | IC | N/A | scalp | MD | 81 | M | lymphocytes | ND | 20–50% | ND |
| SCC T1 | primary | RT | C, P | forearm | WD | 61 | M | N/A | ND | 30–50% | 0/4 5 |
| SCC T2 | primary | CT | A, C, P | hand | WD | 66 | M | lymphocytes | ND | ND | 2/3 5 |
| SCC T8 | primary | RT | C, P | ear | PD | 67 | M | lymphocytes | ND | ND | 3/4 5 |
| SCC T10 | primary | RT | A *, C, P | left shin | MD | 60 | M | lymphocytes | ND | ND | ND |
| SCC T11 | primary | RT | A *, C, P | back | PD | 48 | M | lymphocytes | ND | ND | ND |
| Cell Line | AMEL | CSF1PO | D13S317 | D16S539 | D18S51 | D19S433 | D21S11 | D2S1338 | D3S1358 | D5S818 | D7S820 | D8S1179 | FGA | TH01 | TPOX | vWA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SCC IC1 | X, Y | 12, 14 | 9, 13 | 9, 13 | 15, 17 | 15, 16.2 | 29, 30 | 19, 25 | 14, 17 | 11, 13 | 8, 11 | 13, 13 | 21, 24 | 9.3, 9.3 | 8, 8 | 16, 16 |
| SCC IC1MET | X, Y | 12, 14 | 9, 13 | 9, 13 | 15, 17 | 15, 16.2 | 29, 30 | 19, 25 | 14, 17 | 11, 13 | 8, 11 | 13, 13 | 21, 24 | 9.3, 9.3 | 8, 8 | 16, 16 |
| PM1 | X, Y | 9, 13 | 8, 12 | 11, 13 | 18, 19 | 14, 14 | 30.2, 33.2 | 17, 21 | 14, 14 | 11, 12 | 7, 8 | 12, 15 | 24, 24 | 8, 9.3 | 9, 11 | 18, 19 |
| MET1 | X, Y | 13, 13 | 8, 12 | 11, 13 | 18, 19 | 14, 14 | 30.2, 33.2 | 17, 21 | 16, 16 | 11, 12 | 7, 8 | 12, 15 | 24, 24 | 9.3, 9.3 | 9, 11 | 18, 19 |
| MET2 | X, Y | 13, 13 | 8, 12 | 11, 13 | 18, 19 | 14, 14 | 30.2, 33.2 | 17, 21 | 16, 16 | 11, 12 | 7, 8 | 12, 15 | 24, 24 | 9.3, 9.3 | 9, 11 | 18, 19 |
| MET4 | X, Y | 13, 13 | 8, 12 | 11, 13 | 18, 19 | 14, 14 | 30.2, 33.2 | 17, 21 | 16, 16 | 11, 12 | 7, 8 | 12, 15 | 24, 24 | 9.3, 9.3 | 9, 11 | 18, 19 |
| SCC T9 | X, Y | 9, 13 | 8, 12 | 11, 13 | 18, 19 | 14, 14 | 30.2, 33.2 | 17, 21 | 14, 16 | 11, 12 | 7, 8 | 12, 15 | 24, 24 | 8, 9.3 | 9, 11 | 18, 19 |
| SCC IC8 | X, X | 12, 13 | 11, 11 | 11, 12 | 13, 18 | 13.1, 14 | 29, 29 | 17, 21 | 16, 16 | 13, 13 | 8, 12 | 16, 16 | 19, 23 | 9, 9 | 8, 8 | 15, 16 |
| SCC IC12 | X, X | 12, 12 | 11, 12 | 12, 13 | 15, 16 | 13.1, 14 | 28, 31.2 | 17, 17 | 15, 15 | 12, 13 | 10, 11 | 11, 12 | 22, 24 | 6, 10 | 10, 11 | 17, 19 |
| SCC IC18 | X, X | 11, 11 | 10, 11 | 9, 13 | 12, 12 | 13.1, 15 | 28, 32.2 | 17, 18 | 16, 17 | 13, 13 | 9, 11 | 8, 12 | 21, 21 | 6, 8 | 9, 9 | 14, 17 |
| SCC IC19 | X, X | 11, 11 | 10, 12 | 9, 12 | 11, 18 | 13.1, 15.2 | 32.2, 32.2 | 19, 21 | 15, 15 | 12, 12 | 9, 12 | 13, 15 | 20, 22 | 6, 7 | 11, 11 | 16, 17 |
| SCC T1 | X, Y | 12, 12 | 9, 12 | 12, 13 | 12, 14 | 14, 15 | 30, 32.2 | 18, 20 | 17, 17 | 12, 13 | 9, 10 | 13, 13 | 21, 24 | 9, 9.3 | 9, 12 | 14, 16 |
| SCC T2 | X, Y | 10, 10 | 11, 14 | 10, 11 | 12, 13 | 14, 16 | 28, 31 | 17, 20 | 15, 15 | 11, 12 | ND | 13, 15 | 22, 22 | 6, 6 | 8, 9 | 15, 18 |
| SCC T8 | X, X | 10, 10 | 13, 13 | 11, 12 | 12, 13 | 14, 15 | 31.2, 32.2 | 25, 25 | 14, 15 | 12, 12 | 11, 12 | 13, 13 | 21, 23 | 8, 8 | 11, 11 | 14, 19 |
| SCC T10 | X, Y | 10, 11 | 8, 9 | 9, 10 | 13, 19 | 15, 15 | 29, 30 | 22, 24 | 16, 17 | 11, 11 | ND | 10, 15 | 24, 25 | 9, 9 | 8, 11 | 14, 17 |
| SCC T11 | X, Y | 10, 12 | 11, 12 | 12, 13 | 11, 13 | 12, 14 | 29, 30 | 17, 19 | 14, 16 | 11, 11 | 7, 12 | 11, 15 | 19, 20 | 6, 9.3 | 8, 8 | 18, 18 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, S.; Purdie, K.J.; Wang, J.; Harwood, C.A.; Proby, C.M.; Pourreyron, C.; Mladkova, N.; Nagano, A.; Dhayade, S.; Athineos, D.; et al. A Unique Panel of Patient-Derived Cutaneous Squamous Cell Carcinoma Cell Lines Provides a Preclinical Pathway for Therapeutic Testing. Int. J. Mol. Sci. 2019, 20, 3428. https://doi.org/10.3390/ijms20143428
Hassan S, Purdie KJ, Wang J, Harwood CA, Proby CM, Pourreyron C, Mladkova N, Nagano A, Dhayade S, Athineos D, et al. A Unique Panel of Patient-Derived Cutaneous Squamous Cell Carcinoma Cell Lines Provides a Preclinical Pathway for Therapeutic Testing. International Journal of Molecular Sciences. 2019; 20(14):3428. https://doi.org/10.3390/ijms20143428
Chicago/Turabian StyleHassan, Sakinah, Karin J. Purdie, Jun Wang, Catherine A. Harwood, Charlotte M. Proby, Celine Pourreyron, Nikol Mladkova, Ai Nagano, Sandeep Dhayade, Dimitris Athineos, and et al. 2019. "A Unique Panel of Patient-Derived Cutaneous Squamous Cell Carcinoma Cell Lines Provides a Preclinical Pathway for Therapeutic Testing" International Journal of Molecular Sciences 20, no. 14: 3428. https://doi.org/10.3390/ijms20143428
APA StyleHassan, S., Purdie, K. J., Wang, J., Harwood, C. A., Proby, C. M., Pourreyron, C., Mladkova, N., Nagano, A., Dhayade, S., Athineos, D., Caley, M., Mannella, V., Blyth, K., Inman, G. J., & Leigh, I. M. (2019). A Unique Panel of Patient-Derived Cutaneous Squamous Cell Carcinoma Cell Lines Provides a Preclinical Pathway for Therapeutic Testing. International Journal of Molecular Sciences, 20(14), 3428. https://doi.org/10.3390/ijms20143428