Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
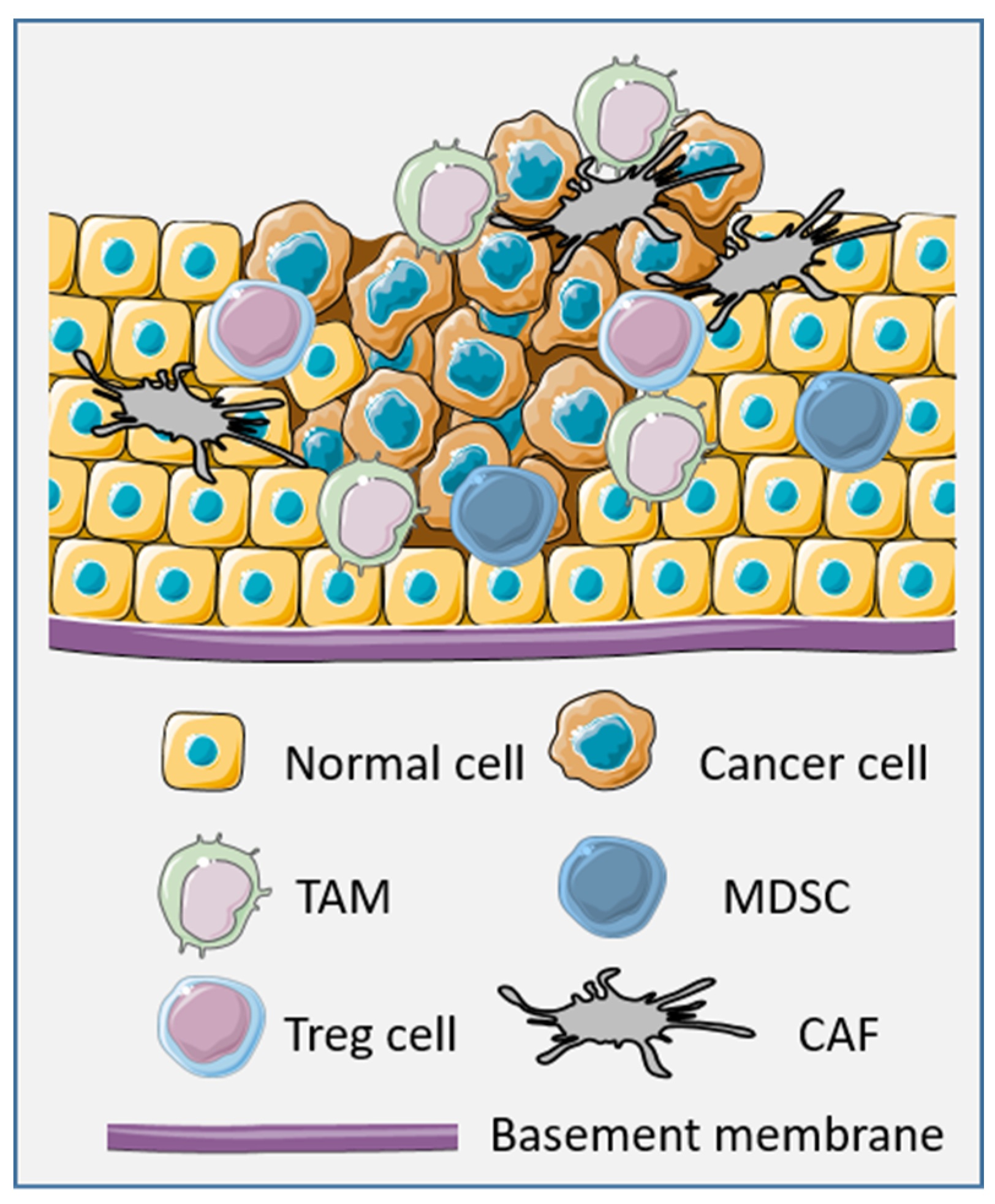
:1. Introduction
2. TAMs
2.1. Overview on Normal Macrophages and Their Physiological Functions
2.2. Tumor-Associated Macrophages
2.3. TAM Targeting
2.3.1. Differentiation and Depletion
2.3.2. Reprogramming
2.3.3. Inhibition of Recruitment
3. T-Regs
3.1. Overview on T-Regs and Their Physiological Functions
3.2. Tumor-Infiltrating T-Regs
3.3. T-Reg Targeting
3.3.1. Antibodies Depleting Tumor-Infiltrating T-Regs
3.3.2. Tumor-Infiltrating T-Reg Vaccine Approaches
3.3.3. Small Molecules for T-Reg Depletion or Functional Modulation
4. MSCs
4.1. Overview on Normal MSCs and Their Physiological Functions
4.2. MSCs in Cancer
4.3. MSC Targeting
4.3.1. Inhibition of Recruitment
4.3.2. Reprogramming
4.3.3. Blocking Functions
4.3.4. Blocking the Cell-to-Cell Contact
4.3.5. Inhibition of the MSC Immune Suppressor Capacities
5. CAFs
5.1. Overview on Normal Fibroblasts and Their Physiological Functions
5.2. Cancer-Associated Fibroblasts
5.3. CAFs Targeting
5.3.1. Depletion
5.3.2. Reprogramming
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Kim, K.; Marquez-Palencia, M.; Malladi, S. Metastatic Latency, a Veiled Threat. Front. Immunol. 2019, 10, 1836. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Frydrychowicz, M.; Boruczkowski, M.; Kolecka-Bednarczyk, A.; Dworacki, G. The Dual Role of Treg in Cancer. Scand. J. Immunol. 2017, 86, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeffel, G.; Ginhoux, F. Ontogeny of Tissue-Resident Macrophages. Front. Immunol. 2015, 6, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabe, Y.; Medzhitov, R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 2014, 157, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Takeda, K.; Tanaka, T.; Shi, W.; Matsumoto, M.; Minami, M.; Kashiwamura, S.; Nakanishi, K.; Yoshida, N.; Kishimoto, T.; Akira, S. Essential role of Stat6 in IL-4 signalling. Nature 1996, 380, 627–630. [Google Scholar] [CrossRef]
- Pilling, D.; Fan, T.; Huang, D.; Kaul, B.; Gomer, R.H. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS ONE 2009, 4, e7475. [Google Scholar] [CrossRef]
- Andersen, M.N.; Etzerodt, A.; Graversen, J.H.; Holthof, L.C.; Moestrup, S.K.; Hokland, M.; Møller, H.J. STAT3 inhibition specifically in human monocytes and macrophages by CD163-targeted corosolic acid-containing liposomes. Cancer Immunol. Immunother. 2019, 68, 489–502. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Yamamoto, Y.; Akasaki, Y.; Sasaki, H. Dual role of macrophage in tumor immunity. Immunotherapy 2018, 10, 899–909. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 597. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.; Alexander, P. Cooperation of immune lymphoid cells with macrophages in tumour immunity. Nature 1970, 228, 620–622. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Goswami, K.K.; Ghosh, T.; Ghosh, S.; Sarkar, M.; Bose, A.; Baral, R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell. Immunol. 2017, 316, 1–10. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.-C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729. [Google Scholar] [CrossRef]
- Kato, Y.; Tabata, K.; Kimura, T.; Yachie-Kinoshita, A.; Ozawa, Y.; Yamada, K.; Ito, J.; Tachino, S.; Hori, Y.; Matsuki, M.; et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS ONE 2019, 14, e0212513. [Google Scholar] [CrossRef]
- Jung, M.; Mertens, C.; Tomat, E.; Brüne, B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, L.; Gong, C.; Shi, H.; Zeng, Y.; Wang, X.; Zhao, Y.; Wei, Y. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef]
- Hume, D.A.; MacDonald, K.P.A. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 2012, 119, 1810–1820. [Google Scholar] [CrossRef]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef]
- Moughon, D.L.; He, H.; Schokrpur, S.; Jiang, Z.K.; Yaqoob, M.; David, J.; Lin, C.; Iruela-Arispe, M.L.; Dorigo, O.; Wu, L. Macrophage Blockade Using CSF1R Inhibitors Reverses the Vascular Leakage Underlying Malignant Ascites in Late-Stage Epithelial Ovarian Cancer. Cancer Res. 2015, 75, 4742–4752. [Google Scholar] [CrossRef] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase I Study of Emactuzumab Single Agent or in Combination with Paclitaxel in Patients with Advanced/Metastatic Solid Tumors Reveals Depletion of Immunosuppressive M2-like Macrophages. Ann. Oncol. 2019, 30, 1381–1392. [Google Scholar] [CrossRef]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef]
- Lu, X.; Meng, T. Depletion of tumor-associated macrophages enhances the anti-tumor effect of docetaxel in a murine epithelial ovarian cancer. Immunobiology 2019, 224, 355–361. [Google Scholar] [CrossRef]
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro-Oncology 2016, 18, 797–806. [Google Scholar] [CrossRef]
- Michaelson, M.D.; Smith, M.R. Bisphosphonates for treatment and prevention of bone metastases. J. Clin. Oncol. 2005, 23, 8219–8224. [Google Scholar] [CrossRef] [PubMed]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjällman, A.H.M.; Ballmer-Hofer, K.; Schwendener, R.A. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Schulte, B.C.; Zhou, Y.; Haribhai, D.; Mackinnon, A.C.; Plaza, J.A.; Williams, C.B.; Hwang, S.T. Depletion of M2-like tumor-associated macrophages delays cutaneous T-cell lymphoma development in vivo. J. Invest. Dermatol. 2014, 134, 2814–2822. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, K.; Zenmyo, M.; Watari, K.; Iguchi, H.; Fotovati, A.; Kimura, Y.N.; Hosoi, F.; Shoda, T.; Nagata, K.; Osada, H.; et al. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci. 2008, 99, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [PubMed]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A.; Mordoh, J.; Wainstok, R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Invest. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef]
- Diel, I.J.; Solomayer, E.F.; Costa, S.D.; Gollan, C.; Goerner, R.; Wallwiener, D.; Kaufmann, M.; Bastert, G. Reduction in new metastases in breast cancer with adjuvant clodronate treatment. N. Engl. J. Med. 1998, 339, 357–363. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Anguille, S.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F. Bisphosphonates for cancer treatment: Mechanisms of action and lessons from clinical trials. Pharmacol. Ther. 2016, 158, 24–40. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Liguori, M.; Buracchi, C.; Pasqualini, F.; Bergomas, F.; Pesce, S.; Sironi, M.; Grizzi, F.; Mantovani, A.; Belgiovine, C.; Allavena, P. Functional TRAIL receptors in monocytes and tumor-associated macrophages: A possible targeting pathway in the tumor microenvironment. Oncotarget 2016, 7, 41662–41676. [Google Scholar] [CrossRef] [Green Version]
- Georgoudaki, A.-M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [Green Version]
- Sommariva, M.; Le Noci, V.; Storti, C.; Bianchi, F.; Tagliabue, E.; Balsari, A.; Sfondrini, L. Activation of NK cell cytotoxicity by aerosolized CpG-ODN/poly(I:C) against lung melanoma metastases is mediated by alveolar macrophages. Cell. Immunol. 2017, 313, 52–58. [Google Scholar] [CrossRef]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. IFNγ and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016, 6, 400–413. [Google Scholar] [CrossRef]
- Maeda, A.; Digifico, E.; Andon, F.T.; Mantovani, A.; Allavena, P. Poly(I:C) stimulation is superior than Imiquimod to induce the antitumoral functional profile of tumor-conditioned macrophages. Eur. J. Immunol. 2019, 49, 801–811. [Google Scholar] [CrossRef]
- Yin, T.; He, S.; Wang, Y. Toll-like receptor 7/8 agonist, R848, exhibits antitumoral effects in a breast cancer model. Mol. Med. Rep. 2015, 12, 3515–3520. [Google Scholar] [CrossRef]
- Weihrauch, M.R.; Richly, H.; von Bergwelt-Baildon, M.S.; Becker, H.J.; Schmidt, M.; Hacker, U.T.; Shimabukuro-Vornhagen, A.; Holtick, U.; Nokay, B.; Schroff, M.; et al. Phase I clinical study of the toll-like receptor 9 agonist MGN1703 in patients with metastatic solid tumours. Eur. J. Cancer 2015, 51, 146–156. [Google Scholar] [CrossRef]
- Lizotte, P.H.; Baird, J.R.; Stevens, C.A.; Lauer, P.; Green, W.R.; Brockstedt, D.G.; Fiering, S.N. Attenuated Listeria monocytogenes reprograms M2-polarized tumor-associated macrophages in ovarian cancer leading to iNOS-mediated tumor cell lysis. Oncoimmunology 2014, 3, e28926. [Google Scholar] [CrossRef]
- Tutt, A.L.; O’Brien, L.; Hussain, A.; Crowther, G.R.; French, R.R.; Glennie, M.J. T Cell Immunity to Lymphoma Following Treatment with Anti-CD40 Monoclonal Antibody. J. Immunol. 2002, 168, 2720–2728. [Google Scholar] [CrossRef] [Green Version]
- Rüter, J.; Antonia, S.J.; Burris, H.A.; Huhn, R.D.; Vonderheide, R.H. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol. Ther. 2010, 10, 983–993. [Google Scholar] [CrossRef] [Green Version]
- Dheilly, E.; Moine, V.; Broyer, L.; Salgado-Pires, S.; Johnson, Z.; Papaioannou, A.; Cons, L.; Calloud, S.; Majocchi, S.; Nelson, R.; et al. Selective Blockade of the Ubiquitous Checkpoint Receptor CD47 Is Enabled by Dual-Targeting Bispecific Antibodies. Mol. Ther. 2017, 25, 523–533. [Google Scholar] [CrossRef]
- Mullins, D.W.; Burger, C.J.; Elgert, K.D. Paclitaxel enhances macrophage IL-12 production in tumor-bearing hosts through nitric oxide. J. Immunol. 1999, 162, 6811–6818. [Google Scholar]
- Kortylewski, M.; Swiderski, P.; Herrmann, A.; Wang, L.; Kowolik, C.; Kujawski, M.; Lee, H.; Scuto, A.; Liu, Y.; Yang, C.; et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 2009, 27, 925–932. [Google Scholar] [CrossRef] [Green Version]
- Binnemars-Postma, K.; Bansal, R.; Storm, G.; Prakash, J. Targeting the Stat6 pathway in tumor-associated macrophages reduces tumor growth and metastatic niche formation in breast cancer. FASEB J. 2018, 32, 969–978. [Google Scholar] [CrossRef]
- Squadrito, M.L.; Etzrodt, M.; De Palma, M.; Pittet, M.J. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol. 2013, 34, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Sonda, N.; Simonato, F.; Peranzoni, E.; Calì, B.; Bortoluzzi, S.; Bisognin, A.; Wang, E.; Marincola, F.M.; Naldini, L.; Gentner, B.; et al. miR-142-3p prevents macrophage differentiation during cancer-induced myelopoiesis. Immunity 2013, 38, 1236–1249. [Google Scholar] [CrossRef]
- Baer, C.; Squadrito, M.L.; Laoui, D.; Thompson, D.; Hansen, S.K.; Kiialainen, A.; Hoves, S.; Ries, C.H.; Ooi, C.-H.; De Palma, M. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 2016, 18, 790–802. [Google Scholar] [CrossRef]
- Chen, W.; Ma, T.; Shen, X.; Xia, X.; Xu, G.; Bai, X.; Liang, T. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012, 72, 1363–1372. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef]
- Chakraborty, P.; Chatterjee, S.; Ganguly, A.; Saha, P.; Adhikary, A.; Das, T.; Chatterjee, M.; Choudhuri, S.K. Reprogramming of TAM toward proimmunogenic type through regulation of MAP kinases using a redox-active copper chelate. J. Leukoc. Biol. 2012, 91, 609–619. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Chen, H.; Huang, K.; Liu, X.; Qiu, M.; Liu, Y.; Yang, Y.; Yang, J. CYP4 × 1 Inhibition by Flavonoid CH625 Normalizes Glioma Vasculature through Reprogramming TAMs via CB2 and EGFR-STAT3 Axis. J. Pharmacol. Exp. Ther. 2018, 365, 72–83. [Google Scholar] [CrossRef]
- Tan, H.-Y.; Wang, N.; Man, K.; Tsao, S.-W.; Che, C.-M.; Feng, Y. Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis. 2015, 6, e1942. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef]
- Perry, C.J.; Muñoz-Rojas, A.R.; Meeth, K.M.; Kellman, L.N.; Amezquita, R.A.; Thakral, D.; Du, V.Y.; Wang, J.X.; Damsky, W.; Kuhlmann, A.L.; et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J. Exp. Med. 2018, 215, 877–893. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Q.; Zeng, S.; Vitiello, G.A.; Seifert, A.M.; Medina, B.D.; Beckman, M.J.; Loo, J.K.; Santamaria-Barria, J.; Maltbaek, J.H.; Param, N.J.; et al. Macrophages and CD8+ T Cells Mediate the Antitumor Efficacy of Combined CD40 Ligation and Imatinib Therapy in Gastrointestinal Stromal Tumors. Cancer Immunol. Res. 2018, 6, 434–447. [Google Scholar] [CrossRef]
- Smith, D.A.; Conkling, P.; Richards, D.A.; Nemunaitis, J.J.; Boyd, T.E.; Mita, A.C.; de La Bourdonnaye, G.; Wages, D.; Bexon, A.S. Antitumor activity and safety of combination therapy with the Toll-like receptor 9 agonist IMO-2055, erlotinib, and bevacizumab in advanced or metastatic non-small cell lung cancer patients who have progressed following chemotherapy. Cancer Immunol. Immunother. 2014, 63, 787–796. [Google Scholar] [CrossRef]
- Xu, A.; Zhang, L.; Yuan, J.; Babikr, F.; Freywald, A.; Chibbar, R.; Moser, M.; Zhang, W.; Zhang, B.; Fu, Z.; et al. TLR9 agonist enhances radiofrequency ablation-induced CTL responses, leading to the potent inhibition of primary tumor growth and lung metastasis. Cell. Mol. Immunol. 2018. [Google Scholar] [CrossRef]
- Ma, B.; Cheng, H.; Mu, C.; Geng, G.; Zhao, T.; Luo, Q.; Ma, K.; Chang, R.; Liu, Q.; Gao, R.; et al. The SIAH2-NRF1 axis spatially regulates tumor microenvironment remodeling for tumor progression. Nat. Commun. 2019, 10, 1034. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef]
- Verzella, D.; Bennett, J.; Fischietti, M.; Thotakura, A.K.; Recordati, C.; Pasqualini, F.; Capece, D.; Vecchiotti, D.; D’Andrea, D.; Di Francesco, B.; et al. GADD45β Loss Ablates Innate Immunosuppression in Cancer. Cancer Res. 2018, 78, 1275–1292. [Google Scholar] [CrossRef]
- Wei, X.; Nie, S.; Liu, H.; Sun, J.; Liu, J.; Li, J.; Li, S.; Wang, S.; Han, S.; Wang, J.; et al. Angiopoietin-like protein 2 facilitates non-small cell lung cancer progression by promoting the polarization of M2 tumor-associated macrophages. Am. J. Cancer Res. 2017, 7, 2220–2233. [Google Scholar]
- Racioppi, L.; Nelson, E.R.; Huang, W.; Mukherjee, D.; Lawrence, S.A.; Lento, W.; Masci, A.M.; Jiao, Y.; Park, S.; York, B.; et al. CaMKK2 in myeloid cells is a key regulator of the immune-suppressive microenvironment in breast cancer. Nat. Commun. 2019, 10, 2450. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.; Mao, C.; Sun, M.; Dominah, G.; Chen, L.; Zhuang, Z. Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol. Immunol. 2018, 94, 27–35. [Google Scholar] [CrossRef]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389. [Google Scholar] [CrossRef]
- Bartneck, M.; Schrammen, P.L.; Möckel, D.; Govaere, O.; Liepelt, A.; Krenkel, O.; Ergen, C.; McCain, M.V.; Eulberg, D.; Luedde, T.; et al. The CCR2+ Macrophage Subset Promotes Pathogenic Angiogenesis for Tumor Vascularization in Fibrotic Livers. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 371–390. [Google Scholar] [CrossRef]
- Gui, P.; Ben-Neji, M.; Belozertseva, E.; Dalenc, F.; Franchet, C.; Gilhodes, J.; Labrousse, A.; Bellard, E.; Golzio, M.; Poincloux, R.; et al. The Protease-Dependent Mesenchymal Migration of Tumor-Associated Macrophages as a Target in Cancer Immunotherapy. Cancer Immunol. Res. 2018, 6, 1337–1351. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Kitajima, S.; Kohno, S.; Yoshida, A.; Tange, S.; Sasaki, S.; Okada, N.; Nishimoto, Y.; Muranaka, H.; Nagatani, N.; et al. Retinoblastoma inactivation induces a protumoral microenvironment via enhanced CCL2 secretion. Cancer Res. 2019, 79, 3903–3915. [Google Scholar] [CrossRef]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef]
- Yang, Z.; Li, H.; Wang, W.; Zhang, J.; Jia, S.; Wang, J.; Wei, J.; Lei, D.; Hu, K.; Yang, X. CCL2/CCR2 Axis Promotes the Progression of Salivary Adenoid Cystic Carcinoma via Recruiting and Reprogramming the Tumor-Associated Macrophages. Front. Oncol. 2019, 9, 231. [Google Scholar] [CrossRef] [Green Version]
- Bonapace, L.; Coissieux, M.-M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Vela, M.; Aris, M.; Llorente, M.; Garcia-Sanz, J.A.; Kremer, L. Chemokine receptor-specific antibodies in cancer immunotherapy: Achievements and challenges. Front. Immunol. 2015, 6, 12. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Papadopoulos, K.; Fong, P.C.; Patnaik, A.; Messiou, C.; Olmos, D.; Wang, G.; Tromp, B.J.; Puchalski, T.A.; Balkwill, F.; et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 1041–1050. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.-P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Leftin, A.; Ben-Chetrit, N.; Joyce, J.A.; Koutcher, J.A. Imaging endogenous macrophage iron deposits reveals a metabolic biomarker of polarized tumor macrophage infiltration and response to CSF1R breast cancer immunotherapy. Sci. Rep. 2019, 9, 857. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Gluck, L.; Martin, L.P.; Olszanski, A.J.; Tolcher, A.W.; Ngarmchamnanrith, G.; Rasmussen, E.; Amore, B.M.; Nagorsen, D.; Hill, J.S.; et al. First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5703–5710. [Google Scholar] [CrossRef]
- Cassier, P.A.; Italiano, A.; Gomez-Roca, C.A.; Le Tourneau, C.; Toulmonde, M.; Cannarile, M.A.; Ries, C.; Brillouet, A.; Müller, C.; Jegg, A.M.; et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: A dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 2015, 16, 949–956. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, L.; Hao, X.; Liu, Y.; Zhang, J.; Ning, Z.; Shi, Y. Phase I dose-escalation study of chiauranib, a novel angiogenic, mitotic, and chronic inflammation inhibitor, in patients with advanced solid tumors. J. Hematol. Oncol. 2019, 12, 9. [Google Scholar] [CrossRef]
- Rosenbaum, E.; Kelly, C.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Movva, S.; Condy, M.; Adamson, T.; Mcfadyen, C.R.; et al. A Phase I Study of Binimetinib (MEK162) Combined with Pexidartinib (PLX3397) in Patients with Advanced Gastrointestinal Stromal Tumor. Oncologist 2019, 24, 1–8. [Google Scholar] [CrossRef]
- Hegab, A.E.; Ozaki, M.; Kameyama, N.; Gao, J.; Kagawa, S.; Yasuda, H.; Soejima, K.; Yin, Y.; Guzy, R.D.; Nakamura, Y.; et al. Effect of FGF/FGFR pathway blocking on lung adenocarcinoma and its cancer-associated fibroblasts. J. Pathol. 2019. [Google Scholar] [CrossRef]
- Xia, Y.; Wei, Y.; Li, Z.-Y.; Cai, X.-Y.; Zhang, L.-L.; Dong, X.-R.; Zhang, S.; Zhang, R.-G.; Meng, R.; Zhu, F.; et al. Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages. Brain Behav. Immun. 2019, 81, 111–121. [Google Scholar] [CrossRef]
- Piao, C.; Zhang, W.-M.; Li, T.-T.; Zhang, C.-C.; Qiu, S.; Liu, Y.; Liu, S.; Jin, M.; Jia, L.-X.; Song, W.-C.; et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp. Cell Res. 2018, 366, 127–138. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef]
- Tang, Q.; Henriksen, K.J.; Boden, E.K.; Tooley, A.J.; Ye, J.; Subudhi, S.K.; Zheng, X.X.; Strom, T.B.; Bluestone, J.A. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J. Immunol. 2003, 171, 3348–3352. [Google Scholar] [CrossRef]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef]
- Read, S.; Malmström, V.; Powrie, F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 2000, 192, 295–302. [Google Scholar] [CrossRef]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef]
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-γ Drives Treg Fragility to Promote Anti-tumor Immunity. Cell 2017, 169, 1130–1141. [Google Scholar] [CrossRef]
- Asano, T.; Meguri, Y.; Yoshioka, T.; Kishi, Y.; Iwamoto, M.; Nakamura, M.; Sando, Y.; Yagita, H.; Koreth, J.; Kim, H.T.; et al. PD-1 modulates regulatory T-cell homeostasis during low-dose interleukin-2 therapy. Blood 2017, 129, 2186–2197. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; McMichael, E.L.; Shayan, G.; Li, J.; Chen, K.; Srivastava, R.; Kane, L.P.; Lu, B.; Ferris, R.L. Novel Effector Phenotype of Tim-3+ Regulatory T Cells Leads to Enhanced Suppressive Function in Head and Neck Cancer Patients. Clin. Cancer Res. 2018, 24, 4529–4538. [Google Scholar] [CrossRef]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.-F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef]
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635–646. [Google Scholar] [CrossRef]
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M.J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 2007, 8, 1353–1362. [Google Scholar] [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef]
- Viguier, M.; Lemaître, F.; Verola, O.; Cho, M.-S.; Gorochov, G.; Dubertret, L.; Bachelez, H.; Kourilsky, P.; Ferradini, L. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J. Immunol. 2004, 173, 1444–1453. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Novak, A.J.; Stenson, M.J.; Witzig, T.E.; Ansell, S.M. Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood 2006, 107, 3639–3646. [Google Scholar] [CrossRef]
- Liyanage, U.K.; Moore, T.T.; Joo, H.-G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef]
- Woo, E.Y.; Chu, C.S.; Goletz, T.J.; Schlienger, K.; Yeh, H.; Coukos, G.; Rubin, S.C.; Kaiser, L.R.; June, C.H. Regulatory CD4+CD25+ T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001, 61, 4766–4772. [Google Scholar]
- Bluestone, J.A.; Liu, W.; Yabu, J.M.; Laszik, Z.G.; Putnam, A.; Belingheri, M.; Gross, D.M.; Townsend, R.M.; Vincenti, F. The effect of costimulatory and interleukin 2 receptor blockade on regulatory T cells in renal transplantation. Am. J. Transplant. 2008, 8, 2086–2096. [Google Scholar] [CrossRef]
- Rech, A.J.; Mick, R.; Martin, S.; Recio, A.; Aqui, N.A.; Powell, D.J.; Colligon, T.A.; Trosko, J.A.; Leinbach, L.I.; Pletcher, C.H.; et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci. Transl. Med. 2012, 4, 134ra62. [Google Scholar] [CrossRef]
- Berlato, C.; Khan, M.N.; Schioppa, T.; Thompson, R.; Maniati, E.; Montfort, A.; Jangani, M.; Canosa, M.; Kulbe, H.; Hagemann, U.B.; et al. A CCR4 antagonist reverses the tumor-promoting microenvironment of renal cancer. J. Clin. Invest. 2017, 127, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Kurose, K.; Ohue, Y.; Wada, H.; Iida, S.; Ishida, T.; Kojima, T.; Doi, T.; Suzuki, S.; Isobe, M.; Funakoshi, T.; et al. Phase Ia Study of FoxP3+ CD4 Treg Depletion by Infusion of a Humanized Anti-CCR4 Antibody, KW-0761, in Cancer Patients. Clin. Cancer Res. 2015, 21, 4327–4336. [Google Scholar] [CrossRef]
- Rapp, M.; Wintergerst, M.W.M.; Kunz, W.G.; Vetter, V.K.; Knott, M.M.L.; Lisowski, D.; Haubner, S.; Moder, S.; Thaler, R.; Eiber, S.; et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J. Exp. Med. 2019, 216, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
- Schoenhals, J.E.; Cushman, T.R.; Barsoumian, H.B.; Li, A.; Cadena, A.P.; Niknam, S.; Younes, A.I.; Caetano, M.D.S.; Cortez, M.A.; Welsh, J.W. Anti-glucocorticoid-induced Tumor Necrosis Factor-Related Protein (GITR) Therapy Overcomes Radiation-Induced Treg Immunosuppression and Drives Abscopal Effects. Front. Immunol. 2018, 9, 2170. [Google Scholar] [CrossRef]
- Zappasodi, R.; Sirard, C.; Li, Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.; et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019, 25, 759–766. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Rosen, D.B.; Grein, J.; Tedesco, D.; Joyce-Shaikh, B.; Ueda, R.; Semana, M.; Bauer, M.; Bang, K.; Stevenson, C.; et al. GITR Agonism Enhances Cellular Metabolism to Support CD8+ T-cell Proliferation and Effector Cytokine Production in a Mouse Tumor Model. Cancer Immunol. Res. 2018, 6, 1199–1211. [Google Scholar] [CrossRef]
- Marangoni, F.; Zhang, R.; Mani, V.; Thelen, M.; Ali Akbar, N.J.; Warner, R.D.; Äijö, T.; Zappulli, V.; Martinez, G.J.; Turka, L.A.; et al. Tumor Tolerance-Promoting Function of Regulatory T Cells Is Optimized by CD28, but Strictly Dependent on Calcineurin. J. Immunol. 2018, 200, 3647–3661. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Fuhrman, C.A.; Yeh, W.-I.; Seay, H.R.; Saikumar Lakshmi, P.; Chopra, G.; Zhang, L.; Perry, D.J.; McClymont, S.A.; Yadav, M.; Lopez, M.-C.; et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J. Immunol. 2015, 195, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Ohno, T.; Nishii, N.; Harada, K.; Yagita, H.; Azuma, M. Differential contribution of three immune checkpoint (VISTA, CTLA-4, PD-1) pathways to antitumor responses against squamous cell carcinoma. Oral Oncol. 2016, 57, 54–60. [Google Scholar] [CrossRef]
- Foss, F.M. DAB(389)IL-2 (denileukin diftitox, ONTAK): A new fusion protein technology. Clin Lymphoma 2000, 1 (Suppl. 1), S27–S31. [Google Scholar] [CrossRef]
- Cheung, L.S.; Fu, J.; Kumar, P.; Kumar, A.; Urbanowski, M.E.; Ihms, E.A.; Parveen, S.; Bullen, C.K.; Patrick, G.J.; Harrison, R.; et al. Second-generation IL-2 receptor-targeted diphtheria fusion toxin exhibits antitumor activity and synergy with anti-PD-1 in melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 3100–3105. [Google Scholar] [CrossRef]
- Franco-Molina, M.A.; Miranda-Hernández, D.F.; Mendoza-Gamboa, E.; Zapata-Benavides, P.; Coronado-Cerda, E.E.; Sierra-Rivera, C.A.; Saavedra-Alonso, S.; Taméz-Guerra, R.S.; Rodríguez-Padilla, C. Silencing of Foxp3 delays the growth of murine melanomas and modifies the tumor immunosuppressive environment. Onco Targets Ther. 2016, 9, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Miguel, A.; Sendra, L.; Noé, V.; Ciudad, C.J.; Dasí, F.; Hervas, D.; Herrero, M.J.; Aliño, S.F. Silencing of Foxp3 enhances the antitumor efficacy of GM-CSF genetically modified tumor cell vaccine against B16 melanoma. Onco Targets Ther. 2017, 10, 503–514. [Google Scholar] [CrossRef]
- Mousavi Niri, N.; Memarnejadian, A.; Pilehvar-Soltanahmadi, Y.; Agha Sadeghi, M.; Mahdavi, M.; Kheshtchin, N.; Arab, S.; Namdar, A.; Jadidi, F.; Zarghami, N.; et al. Improved Anti-Treg Vaccination Targeting Foxp3 Efficiently Decreases Regulatory T Cells in Mice. J. Immunother. 2016, 39, 269–275. [Google Scholar] [CrossRef]
- Namdar, A.; Mirzaei, R.; Memarnejadian, A.; Boghosian, R.; Samadi, M.; Mirzaei, H.R.; Farajifard, H.; Zavar, M.; Azadmanesh, K.; Elahi, S.; et al. Prophylactic DNA vaccine targeting Foxp3+ regulatory T cells depletes myeloid-derived suppressor cells and improves anti-melanoma immune responses in a murine model. Cancer Immunol. Immunother. 2018, 67, 367–379. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef]
- Ge, Y.; Domschke, C.; Stoiber, N.; Schott, S.; Heil, J.; Rom, J.; Blumenstein, M.; Thum, J.; Sohn, C.; Schneeweiss, A.; et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol. Immunother. 2012, 61, 353–362. [Google Scholar] [CrossRef]
- Scurr, M.; Pembroke, T.; Bloom, A.; Roberts, D.; Thomson, A.; Smart, K.; Bridgeman, H.; Adams, R.; Brewster, A.; Jones, R.; et al. Low-Dose Cyclophosphamide Induces Antitumor T-Cell Responses, which Associate with Survival in Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 6771–6780. [Google Scholar] [CrossRef]
- Le, D.T.; Jaffee, E.M. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: A current perspective. Cancer Res. 2012, 72, 3439–3444. [Google Scholar] [CrossRef]
- Ohkura, N.; Hamaguchi, M.; Morikawa, H.; Sugimura, K.; Tanaka, A.; Ito, Y.; Osaki, M.; Tanaka, Y.; Yamashita, R.; Nakano, N.; et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012, 37, 785–799. [Google Scholar] [CrossRef]
- Ali, K.; Soond, D.R.; Pineiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [CrossRef]
- Ahmad, S.; Abu-Eid, R.; Shrimali, R.; Webb, M.; Verma, V.; Doroodchi, A.; Berrong, Z.; Samara, R.; Rodriguez, P.C.; Mkrtichyan, M.; et al. Differential PI3Kδ Signaling in CD4+ T-cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res. 2017, 77, 1892–1904. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. TGF-beta: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, J.; Gu, Q.; Huang, M.; Zhang, W.; Guo, J.; Zhou, X. Reciprocal Expression of IL-35 and IL-10 Defines Two Distinct Effector Treg Subsets that Are Required for Maintenance of Immune Tolerance. Cell Rep. 2017, 21, 1853–1869. [Google Scholar] [CrossRef]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef]
- Arce Vargas, F.; Furness, A.J.S.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Miranda Rota, E.; Dahan, R.; Georgiou, A.; Sledzinska, A.; et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017, 46, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Cominal, J.G.; da Costa Cacemiro, M.; Pinto-Simões, B.; Kolb, H.-J.; Malmegrim, K.C.R.; de Castro, F.A. Emerging Role of Mesenchymal Stromal Cell-Derived Extracellular Vesicles in Pathogenesis of Haematological Malignancies. Stem Cells Int. 2019, 2019, 6854080. [Google Scholar] [CrossRef]
- He, S.; Gleason, J.; Fik-Rymarkiewicz, E.; DiFiglia, A.; Bharathan, M.; Morschauser, A.; Djuretic, I.; Xu, Y.; Krakovsky, M.; Jankovic, V.; et al. Human Placenta-Derived Mesenchymal Stromal-Like Cells Enhance Angiogenesis via T Cell-Dependent Reprogramming of Macrophage Differentiation. Stem Cells 2017, 35, 1603–1613. [Google Scholar] [CrossRef] [Green Version]
- Najar, M.; Raicevic, G.; Fayyad-Kazan, H.; Bron, D.; Toungouz, M.; Lagneaux, L. Mesenchymal stromal cells and immunomodulation: A gathering of regulatory immune cells. Cytotherapy 2016, 18, 160–171. [Google Scholar] [CrossRef]
- Khakoo, A.Y.; Pati, S.; Anderson, S.A.; Reid, W.; Elshal, M.F.; Rovira, I.I.; Nguyen, A.T.; Malide, D.; Combs, C.A.; Hall, G.; et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J. Exp. Med. 2006, 203, 1235–1247. [Google Scholar] [CrossRef]
- Qiao, L.; Xu, Z.-L.; Zhao, T.-J.; Ye, L.-H.; Zhang, X.-D. Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett. 2008, 269, 67–77. [Google Scholar] [CrossRef]
- Castells, M.; Thibault, B.; Mery, E.; Golzio, M.; Pasquet, M.; Hennebelle, I.; Bourin, P.; Mirshahi, M.; Delord, J.P.; Querleu, D.; et al. Ovarian ascites-derived Hospicells promote angiogenesis via activation of macrophages. Cancer Lett. 2012, 326, 59–68. [Google Scholar] [CrossRef]
- Corre, J.; Hébraud, B.; Bourin, P. Concise review: Growth differentiation factor 15 in pathology: A clinical role? Stem Cells Transl. Med. 2013, 2, 946–952. [Google Scholar] [CrossRef]
- McLean, K.; Tan, L.; Bolland, D.E.; Coffman, L.G.; Peterson, L.F.; Talpaz, M.; Neamati, N.; Buckanovich, R.J. Leukemia inhibitory factor functions in parallel with interleukin-6 to promote ovarian cancer growth. Oncogene 2019, 38, 1576–1584. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.-T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Invest. 2013, 123, 1542–1555. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, L.; Li, Y.; Zhang, X.; Gu, J.; Yan, Y.; Xu, X.; Wang, M.; Qian, H.; Xu, W. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett. 2012, 315, 28–37. [Google Scholar] [CrossRef]
- Li, N.; Hua, J. Interactions between mesenchymal stem cells and the immune system. Cell. Mol. Life Sci. 2017, 74, 2345–2360. [Google Scholar] [CrossRef]
- Castells, M.; Milhas, D.; Gandy, C.; Thibault, B.; Rafii, A.; Delord, J.-P.; Couderc, B. Microenvironment mesenchymal cells protect ovarian cancer cell lines from apoptosis by inhibiting XIAP inactivation. Cell Death Dis. 2013, 4, e887. [Google Scholar] [CrossRef]
- Le Naour, A.; Couderc, B. Role of MSCs in antitumor drug resistance. In Mesenchymal Stromal Cells as Tumor Stromal Modulators; Bolontrade, M., García, M., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 295–333. [Google Scholar]
- Rafii, A.; Mirshahi, P.; Poupot, M.; Faussat, A.-M.; Simon, A.; Ducros, E.; Mery, E.; Couderc, B.; Lis, R.; Capdet, J.; et al. Oncologic trogocytosis of an original stromal cells induces chemoresistance of ovarian tumours. PLoS ONE 2008, 3, e3894. [Google Scholar] [CrossRef]
- Madar, S.; Goldstein, I.; Rotter, V. ’Cancer associated fibroblasts’—more than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Vangala, G.; Imhoff, F.M.; Squires, C.M.L.; Cridge, A.G.; Baird, S.K. Mesenchymal stem cell homing towards cancer cells is increased by enzyme activity of cathepsin D. Exp. Cell Res. 2019, 383, 111494. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; Martin, E.E.; Anwar, T.; Arellano-Garcia, C.; Medhora, N.; Lama, A.; Chen, Y.-C.; Tanager, K.S.; Yoon, E.; Kidwell, K.M.; et al. Mesenchymal Stem Cell-Induced DDR2 Mediates Stromal-Breast Cancer Interactions and Metastasis Growth. Cell Rep. 2017, 18, 1215–1228. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Elango, R.; Al-Toub, M.; Manikandan, M.; Al-Rikabi, A.; Harkness, L.; Ditzel, N.; Atteya, M.; Hamam, R.; Alfayez, M.; et al. Neoplastic Transformation of Human Mesenchymal Stromal Cells Mediated via LIN28B. Sci. Rep. 2019, 9, 8101. [Google Scholar] [CrossRef] [Green Version]
- Favreau, M.; Menu, E.; Gaublomme, D.; Vanderkerken, K.; Faict, S.; Maes, K.; De Bruyne, E.; Govindarajan, S.; Drennan, M.; Van Calenbergh, S.; et al. Leptin receptor antagonism of iNKT cell function: A novel strategy to combat multiple myeloma. Leukemia 2017, 31, 2678–2685. [Google Scholar] [CrossRef]
- Mishra, P.J.; Banerjee, D. Activation and Differentiation of Mesenchymal Stem Cells. Methods Mol. Biol. 2017, 1554, 201–209. [Google Scholar]
- Pasquier, J.; Gosset, M.; Geyl, C.; Hoarau-Véchot, J.; Chevrot, A.; Pocard, M.; Mirshahi, M.; Lis, R.; Rafii, A.; Touboul, C. CCL2/CCL5 secreted by the stroma induce IL-6/PYK2 dependent chemoresistance in ovarian cancer. Mol. Cancer 2018, 17, 47–51. [Google Scholar] [CrossRef]
- Naour, A.L.; Prat, M.; Thibault, B.; Mével, R.; Lemaitre, L.; Leray, H.; Joubert, M.-V.; Coulson, K.; Golzio, M.; Lefevre, L.; et al. Tumor cells educate mesenchymal stromal cells to release chemoprotective and immunomodulatory factors. J. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef]
- Thibault, B.; Genre, L.; Le Naour, A.; Broca, C.; Mery, E.; Vuagniaux, G.; Delord, J.P.; Wiedemann, N.; Couderc, B. DEBIO 1143, an IAP inhibitor, reverses carboplatin resistance in ovarian cancer cells and triggers apoptotic or necroptotic cell death. Sci. Rep. 2018, 8, 17862. [Google Scholar] [CrossRef]
- DiPersio, J.F.; Erba, H.P.; Larson, R.A.; Luger, S.M.; Tallman, M.S.; Brill, J.M.; Vuagniaux, G.; Rouits, E.; Sorensen, J.M.; Zanna, C. Oral Debio1143 (AT406), an antagonist of inhibitor of apoptosis proteins, combined with daunorubicin and cytarabine in patients with poor-risk acute myeloid leukemia--results of a phase I dose-escalation study. Clin. Lymphoma Myeloma Leuk 2015, 15, 443–449. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Hass, R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun. Signal 2018, 16, 2. [Google Scholar] [CrossRef]
- Alfaro, D.; Zapata, A.G. Eph/Ephrin-mediated stimulation of human bone marrow mesenchymal stromal cells correlates with changes in cell adherence and increased cell death. Stem Cell Res. Ther. 2018, 9, 172. [Google Scholar] [CrossRef]
- Ullah, M.; Akbar, A.; Ng, N.N.; Concepcion, W.; Thakor, A.S. Mesenchymal stem cells confer chemoresistance in breast cancer via a CD9 dependent mechanism. Oncotarget 2019, 10, 3435–3450. [Google Scholar] [CrossRef] [Green Version]
- Studeny, M.; Marini, F.C.; Dembinski, J.L.; Zompetta, C.; Cabreira-Hansen, M.; Bekele, B.N.; Champlin, R.E.; Andreeff, M. Mesenchymal stem cells: Potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J. Natl. Cancer Inst. 2004, 96, 1593–1603. [Google Scholar] [CrossRef]
- Yoon, A.-R.; Hong, J.; Li, Y.; Shin, H.C.; Lee, H.; Kim, H.S.; Yun, C.-O. Mesenchymal stem cell-mediated delivery of an oncolytic adenovirus enhances antitumor efficacy in hepatocellular carcinoma. Cancer Res. 2019, 79, 4503–4514. [Google Scholar] [CrossRef]
- Yin, Z.; Jiang, K.; Li, R.; Dong, C.; Wang, L. Multipotent mesenchymal stromal cells play critical roles in hepatocellular carcinoma initiation, progression and therapy. Mol. Cancer 2018, 17, 178. [Google Scholar] [CrossRef] [Green Version]
- de Araujo Farias, V.; O’Valle, F.; Serrano-Saenz, S.; Anderson, P.; Andrés, E.; López-Peñalver, J.; Tovar, I.; Nieto, A.; Santos, A.; Martín, F.; et al. Exosomes derived from mesenchymal stem cells enhance radiotherapy-induced cell death in tumor and metastatic tumor foci. Mol. Cancer 2018, 17, 122. [Google Scholar] [CrossRef]
- Xie, C.; Du, L.-Y.; Guo, F.; Li, X.; Cheng, B. Exosomes derived from microRNA-101-3p-overexpressing human bone marrow mesenchymal stem cells suppress oral cancer cell proliferation, invasion, and migration. Mol. Cell. Biochem. 2019, 458, 11–26. [Google Scholar] [CrossRef]
- Melzer, C.; Rehn, V.; Yang, Y.; Bähre, H.; von der Ohe, J.; Hass, R. Taxol-Loaded MSC-Derived Exosomes Provide a Therapeutic Vehicle to Target Metastatic Breast Cancer and Other Carcinoma Cells. Cancers (Basel) 2019, 11, 798. [Google Scholar] [CrossRef]
- Parsonage, G.; Filer, A.D.; Haworth, O.; Nash, G.B.; Rainger, G.E.; Salmon, M.; Buckley, C.D. A stromal address code defined by fibroblasts. Trends Immunol. 2005, 26, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, N.; Horiuchi, A.; Osada, R.; Imai, T.; Wang, C.; Chen, X.; Konishi, I. Nuclear expression of S100A4 is associated with aggressive behavior of epithelial ovarian carcinoma: An important autocrine/paracrine factor in tumor progression. Cancer Sci. 2006, 97, 1061–1069. [Google Scholar] [CrossRef]
- Rinn, J.L.; Bondre, C.; Gladstone, H.B.; Brown, P.O.; Chang, H.Y. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006, 2, e119. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Marsh, T.; Pietras, K.; McAllister, S.S. Fibroblasts as architects of cancer pathogenesis. Biochim. Biophys. Acta 2013, 1832, 1070–1078. [Google Scholar] [CrossRef]
- Bainbridge, P. Wound healing and the role of fibroblasts. J. Wound Care 2013, 22, 407–408. [Google Scholar]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Muller, G.A.; Kalbacher, H.; Salant, D.J.; Muller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef] [Green Version]
- Procopio, M.G.; Laszlo, C.; Al Labban, D.; Kim, D.E.; Bordignon, P.; Jo, S.H.; Goruppi, S.; Menietti, E.; Ostano, P.; Ala, U.; et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat. Cell Biol. 2015, 17, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model Mech. 2018, 11. [Google Scholar] [CrossRef]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef]
- Walter, K.; Omura, N.; Hong, S.M.; Griffith, M.; Goggins, M. Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol. Ther. 2008, 7, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Klein, G. Toward a genetics of cancer resistance. Proc. Natl. Acad. Sci. USA 2009, 106, 859–863. [Google Scholar] [CrossRef] [Green Version]
- Ferdek, P.E.; Jakubowska, M.A. Biology of pancreatic stellate cells-more than just pancreatic cancer. Pflug. Arch. 2017, 469, 1039–1050. [Google Scholar] [CrossRef]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Invest. 2018, 128, 26–35. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; Andre, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar]
- Biffi, G.; Tuveson, D.A. A FATal Combination: Fibroblast-Derived Lipids and Cancer-Derived Autotaxin Promote Pancreatic Cancer Growth. Cancer Discov. 2019, 9, 578–580. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lovrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- De Wever, O.; Nguyen, Q.D.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Glanz, S.; Raz, Y.; Avivi, C.; Barshack, I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem. Biophys. Res. Commun. 2013, 437, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.J.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, K.S.; Omar, I.S.; Kwong, S.C.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/STAT-3/c-Myc pathway. Am. J. Cancer Res. 2016, 6, 200–213. [Google Scholar] [PubMed]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef]
- Moatassim-Billah, S.; Duluc, C.; Samain, R.; Jean, C.; Perraud, A.; Decaup, E.; Cassant-Sourdy, S.; Bakri, Y.; Selves, J.; Schmid, H.; et al. Anti-metastatic potential of somatostatin analog SOM230: Indirect pharmacological targeting of pancreatic cancer-associated fibroblasts. Oncotarget 2016, 7, 41584–41598. [Google Scholar] [CrossRef] [Green Version]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Laklai, H.; Weaver, V.M. Force Matters: Biomechanical Regulation of Cell Invasion and Migration in Disease. Trends Cell Biol. 2016, 26, 486–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labernadie, A.; Kato, T.; Brugues, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; Gonzalez-Tarrago, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Maris, P.; Blomme, A.; Palacios, A.P.; Costanza, B.; Bellahcene, A.; Bianchi, E.; Gofflot, S.; Drion, P.; Trombino, G.E.; Di Valentin, E.; et al. Asporin Is a Fibroblast-Derived TGF-beta1 Inhibitor and a Tumor Suppressor Associated with Good Prognosis in Breast Cancer. PLoS Med. 2015, 12, e1001871. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.H.; Hwang-Verslues, W.W.; Chang, Y.C.; Chen, C.C.; Hsiao, M.; Jeng, Y.M.; Chang, K.J.; Lee, E.Y.; Shew, J.Y.; Lee, W.H. Activation of Robo1 signaling of breast cancer cells by Slit2 from stromal fibroblast restrains tumorigenesis via blocking PI3K/Akt/beta-catenin pathway. Cancer Res. 2012, 72, 4652–4661. [Google Scholar] [CrossRef]
- Hill, R.; Song, Y.; Cardiff, R.D.; Van Dyke, T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 2005, 123, 1001–1011. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Lo, A.; Li, C.P.; Buza, E.L.; Blomberg, R.; Govindaraju, P.; Avery, D.; Monslow, J.; Hsiao, M.; Pure, E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight 2017, 2, 92232. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Hofheinz, R.D.; Weisser, A.; Willer, A.; Hehlmann, R.; Hochhaus, A. Treatment of a patient with advanced esophageal cancer with a combination of mitomycin C and capecitabine: Activation of the thymidine phosphorylase as active principle? Onkologie 2003, 26, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Narra, K.; Mullins, S.R.; Lee, H.O.; Strzemkowski-Brun, B.; Magalong, K.; Christiansen, V.J.; McKee, P.A.; Egleston, B.; Cohen, S.J.; Weiner, L.M.; et al. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 2007, 6, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Saleh, M.; Bendell, J.; Infante, J.R.; Jones, S.; Kurkjian, C.D.; Moore, K.M.; Kazakin, J.; Abbadessa, G.; Wang, Y.; et al. A phase I dose escalation study of oral c-MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann. Oncol. 2014, 25, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Parikh, P.K.; Ghate, M.D. Recent advances in the discovery of small molecule c-Met Kinase inhibitors. Eur. J. Med. Chem. 2018, 143, 1103–1138. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Schnittert, J.; Heinrich, M.A.; Kuninty, P.R.; Storm, G.; Prakash, J. Reprogramming tumor stroma using an endogenous lipid lipoxin A4 to treat pancreatic cancer. Cancer Lett. 2018, 420, 247–258. [Google Scholar] [CrossRef]
- Brennen, W.N.; Rosen, D.M.; Wang, H.; Isaacs, J.T.; Denmeade, S.R. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J. Natl. Cancer Inst. 2012, 104, 1320–1334. [Google Scholar] [CrossRef]
- Gao, W.; Xiang, B.; Meng, T.T.; Liu, F.; Qi, X.R. Chemotherapeutic drug delivery to cancer cells using a combination of folate targeting and tumor microenvironment-sensitive polypeptides. Biomaterials 2013, 34, 4137–4149. [Google Scholar] [CrossRef]
- De Vlieghere, E.; Gremonprez, F.; Verset, L.; Marien, L.; Jones, C.J.; De Craene, B.; Berx, G.; Descamps, B.; Vanhove, C.; Remon, J.P.; et al. Tumor-environment biomimetics delay peritoneal metastasis formation by deceiving and redirecting disseminated cancer cells. Biomaterials 2015, 54, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Ulland, T.K.; Cervantes-Barragan, L.; Bando, J.K.; Robinette, M.L.; Wang, Q.; White, A.J.; Gilfillan, S.; Cella, M.; Colonna, M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat. Immunol. 2017, 18, 995–1003. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laplagne, C.; Domagala, M.; Le Naour, A.; Quemerais, C.; Hamel, D.; Fournié, J.-J.; Couderc, B.; Bousquet, C.; Ferrand, A.; Poupot, M. Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression. Int. J. Mol. Sci. 2019, 20, 4719. https://doi.org/10.3390/ijms20194719
Laplagne C, Domagala M, Le Naour A, Quemerais C, Hamel D, Fournié J-J, Couderc B, Bousquet C, Ferrand A, Poupot M. Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression. International Journal of Molecular Sciences. 2019; 20(19):4719. https://doi.org/10.3390/ijms20194719
Chicago/Turabian StyleLaplagne, Chloé, Marcin Domagala, Augustin Le Naour, Christophe Quemerais, Dimitri Hamel, Jean-Jacques Fournié, Bettina Couderc, Corinne Bousquet, Audrey Ferrand, and Mary Poupot. 2019. "Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression" International Journal of Molecular Sciences 20, no. 19: 4719. https://doi.org/10.3390/ijms20194719
APA StyleLaplagne, C., Domagala, M., Le Naour, A., Quemerais, C., Hamel, D., Fournié, J. -J., Couderc, B., Bousquet, C., Ferrand, A., & Poupot, M. (2019). Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression. International Journal of Molecular Sciences, 20(19), 4719. https://doi.org/10.3390/ijms20194719