1. Introduction
Cancer ranks second among the common primary causes of death worldwide, and it is anticipated to have accounted for 9.6 million deaths in 2018 [
1]. The design of potent targeted therapies involving monoclonal antibodies to target specifically cancer cells constitute a promising strategy to treat tumors in cancer patients [
2].
“Evading apoptosis” is an important hallmark of cancer progression and most apoptotic programs divide into the intrinsic or the extrinsic pathway. The extrinsic pathway of apoptosis is triggered by signals that employ extracellular death receptors (DR), and the manipulation of these receptor activation mechanisms has been considered as a tempting therapeutic strategy to induce, exclusively, apoptosis in tumor cells [
3]. Among DRs, death receptor 5 (DR5), also known as TRAIL-R2 or KILLER receptor, is the most promising candidate to develop a targeted therapy against cancer, since its expression level is significantly higher in cancer cells compared to that of healthy cells. Therefore, its triggering may potentially mediate selective activation of apoptosis in cancer cells and thus their killing [
4].
DR5 is a type I transmembrane protein consisting of three regions: an extracellular, a transmembrane and an intracellular part. The last part comprises a homologous cytoplasmic sequence of the death domain for interaction with Fas associated death domain (FADD) and other members of the apoptosis signaling pathway. The extracellular region has 2–4 concatenated cysteine-rich domains (CRDs) that are responsible for ligand binding [
5]. The N-terminal CRD1 of DR5 forms a pre-ligand association domain (PLAD), while a GXXXG motif in the transmembrane helix (TMH) harbors the interaction sites for the dimeric state of inactive DR5 that inhibits apoptosis signaling in absence of TNF-related apoptosis-inducing ligand (TRAIL) [
6].
TRAIL is an important actor within the apoptosis scenario. It is a 281–amino acid type II transmembrane protein that belongs to the Tumor Necrosis Factor (TNF) superfamily, targets death receptors (DR5 and DR4), membrane attached decoy receptors (DcR1 and DcR2), and also a soluble decoy receptor (Osteoprotegrin). These three decoy receptors compete with the death receptors for TRAIL binding and are thus blocking the apoptotic signals [
7]. The extracellular part of membrane-attached TRAIL can be shed after limited proteolysis by cysteine proteases. The self-assembling TRAIL protein forms a noncovalent–bonded homo-trimer with a pyramid-like architecture. Each monomer contains one single cysteine (Cys-230), which is involved in the chelation of a zinc ion in the corner of the pyramid, that is requisite for the native structure, stability and, thus, the biological activity of TRAIL [
8]. TRAIL also contains a significant number of aromatic amino acid residues, of which eight provide a hydrophobic surface interacting with neighboring subunits and stabilizing the inner core of the trimer [
5,
8].
To date, various strategies have been developed to target TRAIL receptors. An ideal therapeutic agent to activate TRAIL-dependent apoptosis is supposed to have an affinity that is comparable to that of the natural ligand, (i.e., single digit nM affinity or lower), and a prolonged circulation time in the bloodstream. Several drawbacks are encountered with the use of recombinant human TRAIL, such as a specificity for its different types of receptors and therefore leading to various and unpredictable functions in the body, short elimination half-life, and in some cases hepatotoxic effects. Consequently, better alternatives are required, such as DR5 agonistic antibodies capable of interacting only with death receptors. Such antibodies are easy to produce, relatively safe to use, exhibiting improved pharmacokinetic properties compared to recombinant TRAIL, and are highly specific for only one single type of receptor [
4].
Although such agonists demonstrate significant anti-tumor activity in preclinical models, the clinical efficacy in human cancer patients has been disappointingly low. One possible explanation might be that the current classes of therapeutic molecules are insufficiently robust to elicit a significant response in patients. In particular, the dimeric antibody agonists require a secondary cross-linking via Fcγ receptors expressed on immune cells to achieve an optimal clustering of DR5 [
9]. Thus, the need to develop better receptor aggregating and clustering molecules is large, and this stimulated us to design multivalent Nanobodies (Nbs) aiming to produce a significantly stronger DR5 agonist [
10]. A Nb is a recombinant single domain antigen-binding fragment derived from heavy chain–only antibodies circulating in blood of Camelidae [
11]. Nbs are easy to generate, and they bind their cognate antigen with high specificity and affinity [
12]. They have a good biodistribution and a fast tissue penetration. Nbs are robust and easy to engineer into multimeric constructs, and the first Nb-based construct has recently been FDA approved [
13].
The objectives of the present study are manifold: (i) identify high affinity DR5-specific Nbs (ii) that recognize a TRAIL-overlapping-epitope on DR5, (iii) unveil the possible correlation of an increasing valency of Nbs in the cell killing activity, and, finally, (iv) demonstrate that DR5-specific Nbs activate apoptosis in target cells. Overall, we demonstrated that our trivalent DR5-targeting Nbs mimic the activity of the natural TRAIL ligand. Moreover, increasing the valency of the nanobody domains to a tetrameric or pentameric constructs (assisted by pentameric Shiga-like toxin 2a B domain (STx2aB
5) [
14] markedly increased the cell-killing potency on various tumor cell lines. The increased potency was attributed to the faster kinetics of assembling the death-inducing signaling complex and caspase 3/7 activation.
3. Discussion
In view of the importance of exploring new anticancer agents we aimed to identify agonistic Nbs against DR5, a crucial target to transduce apoptotic signaling in cancer cells [
25,
26,
27].
In a previous effort, we generated Nbs against this target [
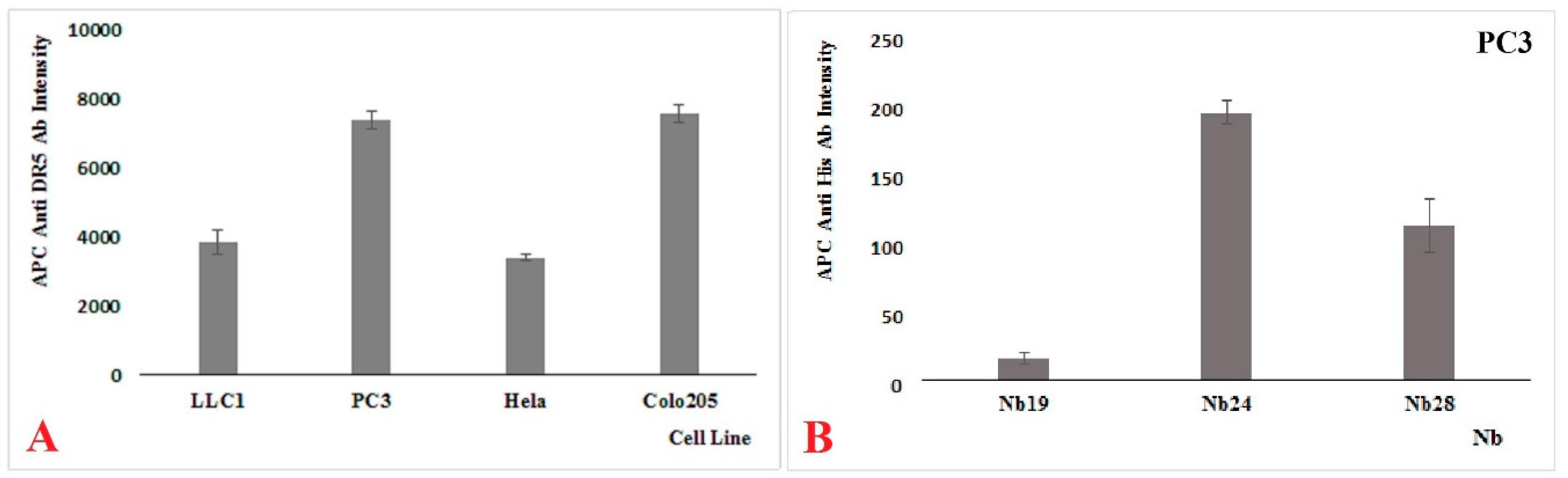
19], and in this report, we focus on two of these Nbs. Both Nbs recognize recombinant DR5 as shown in western blot, and they also recognize Colo205, PC3, and HeLa cells to various degrees in FACS, suggesting that they might recognize the native protein on human cancer cell lines as well. Also, mouse cancer cell line LLC1 was shown to react with our Nbs, indicating a cross reactivity between the mouse and human target. However, our Nbs differed from each other in many ways. Nb24 has a “VH-like” imprint in its FR2 and associates with nM affinity, most probably to a linear epitope on DR5 that is outside the binding site of TRAIL. In contrast, Nb28 has a clear VHH imprint in its FR2 and binds with sub-nM affinity to a conformational epitope that overlaps with that of TRAIL. According to our SPR measurements, the recombinant trimeric TRAIL protein also recognizes the DR5 (monomer and dimer mixture) with an apparent affinity of around 0.5 nM (
Table 1). Interestingly, the monomeric, monovalent Nb28 had nearly identical k
on and k
off rates than the trimeric TRAIL on our DR5 preparations.
Of note, the interaction of Nbs with DR5 and competition with TRAIL was performed with recombinant proteins. Nbs normally are easy to produce recombinantly in E. coli. Although oligomeric Nbs could still be expressed in shake flasks and purified by IMAC and SEC in mg levels per L of culture, we noticed that the expression lowered with each extra Nb within the repeats. Especially oligomeric constructs with Nb24 were sometimes very hard to produce. Therefore, for Nb24-containing constructs we preferred to express only a few hours instead of overnight, and during purification, Tris buffers with elevated salt concentrations (150 mM) seem to give higher yields instead of standard PBS. The production and purification of recombinant DR5 and TRAIL was even more tricky. During SEC of DR5 we noticed the presence of oligomer structures, possibly due to free sulfhydryl groups of the CRD in the bacterial cytoplasm of Shuffle cells that start to oxidize and form intermolecular disulfide bonds. We therefore, used solutions of lower pH (acetate pH 5.0, to reduce the deprotonation of the sulphydryl group) in our buffers. These minor adaptations in protocol resulted in higher production yields, and single symmetrical elution peaks from SEC. The preparation of TRAIL faced serious aggregation problems. It is well established that TRAIL degrades fast and thus has a fast turn-over. So, it is considered to be a very fragile protein. To stabilize our recombinant TRAIL, we used additives including 10% glycerol, 0.01% mannitol, 8% trehalose, 20 µM zinc chloride and 1 mM dithiotreitol.
According to current models, it seems that the trimeric TRAIL protein interacts with up to three DR5 molecules to form a network on the membrane that triggers apoptosis [
6]. To mimic this oligovalency of TRAIL, we generated biparatopic, bivalent, trivalent and tetravalent Nbs with Nb28. The (Gly
4Ser)
3 linkers were chosen to separate the individual Nbs within one molecule, since such an artificial spacer is supposedly non-structured, very flexible and it could separate the individual Nbs by up to 50 Å [
23]. The bivalent Nb and biparatopic Nb construct is capable of binding two molecules within the dimeric DR5, immobilized on the SPR sensor chip, as evidenced from the Rmax values (
Table 1). At this stage, it is not known whether the dimeric DR5 structures in our preparations have the same structure as PLADs, but for sure the DR5 dimer in our preparations comprises two epitopes for Nb24 and also for Nb28. Furthermore, an increased apparent affinity was noticed in SPR for bivalent and biparatopic DR5-specific Nb constructs, which indicates that both entities within the Nb construct were active in antigen binding. The apparent affinity did not increase much for the trimeric Nbs (
Table 1). A bispecific Nb construct whereby the DR5-specific Nb is joined with a serum albumin binding Nb (Nb24-A or Nb24-A) exhibited exactly the same binding parameters to DR5 as the monomeric Nb. Thus, we conclude that the addition of an anti-serum albumin Nb to our constructs will be neutral for DR5 binding, but it might represent a good strategy to increase the blood retention of the Nb construct in future therapeutics.
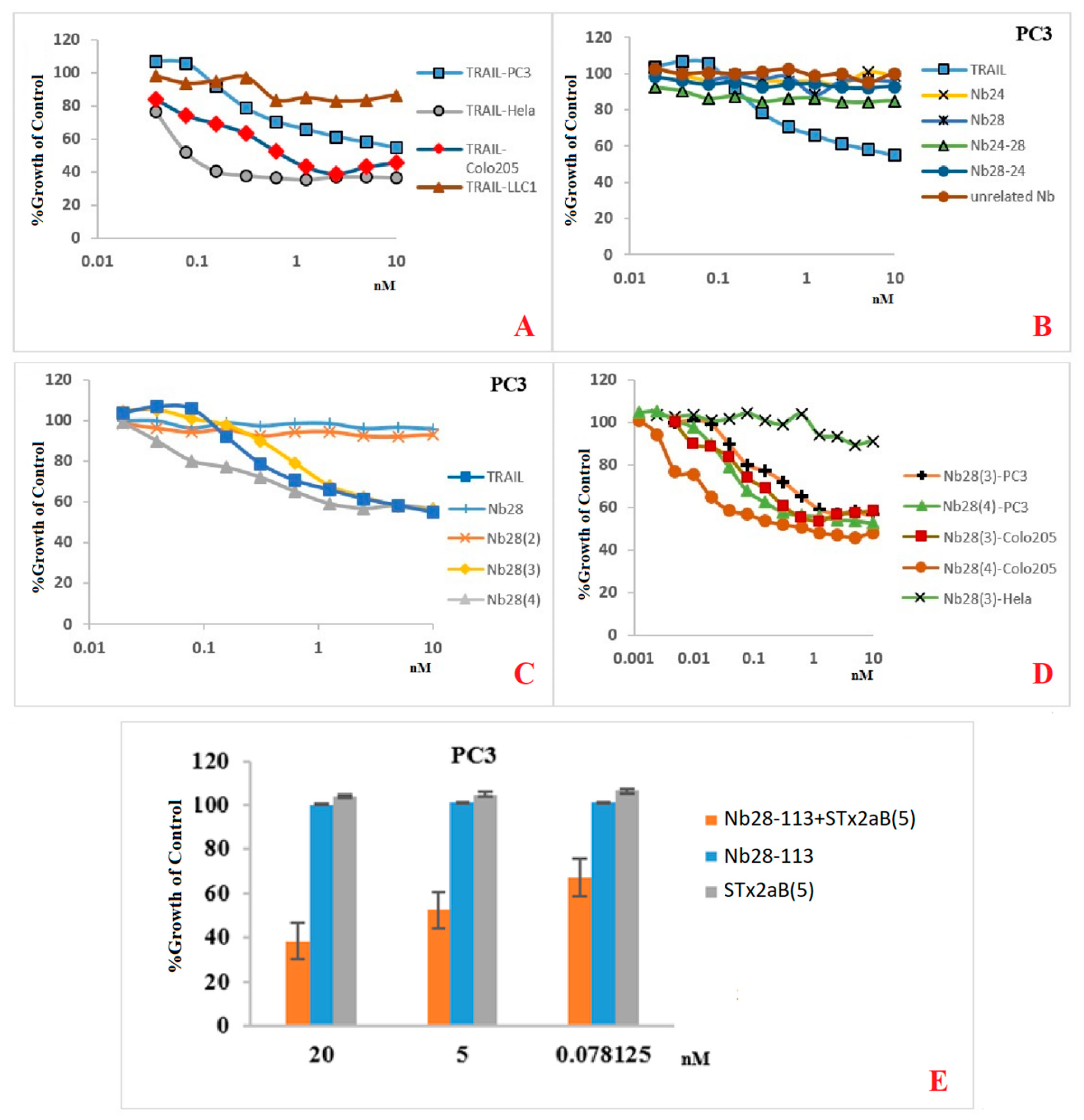
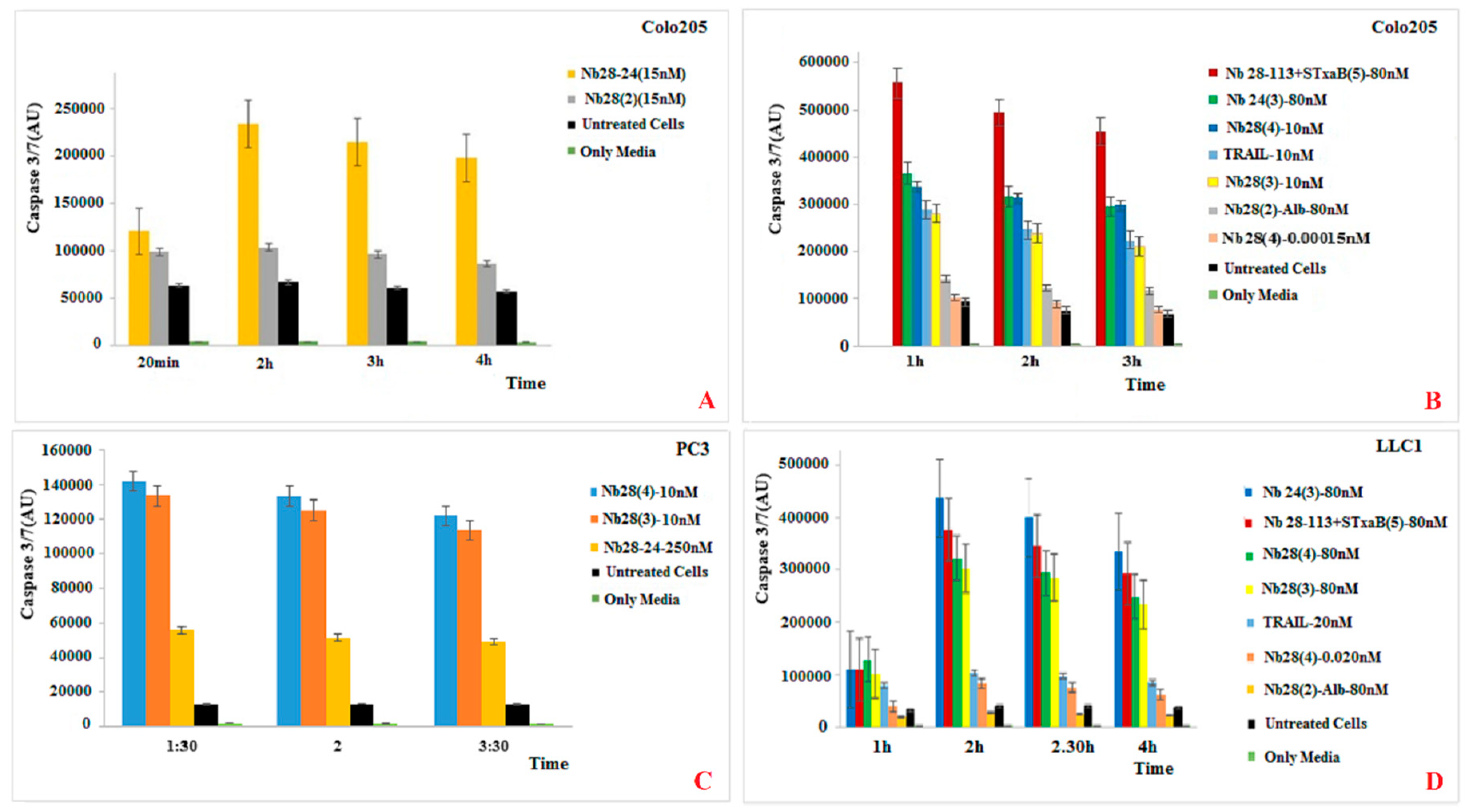
Our recombinant trimeric TRAIL protein was potent in tumor cell killing, confirming its “native-like” folding. Not all cancer cell lines react in the same way to the recombinant TRAIL, which probably reflects the variable abundancy of DR5 on the different cancer cells. Remarkably, the monomeric Nb28 and even the dimeric Nb28 or biparatopic Nbs failed to induce tumor cell–killing activity. We therefore hypothesize that both entities of the bivalent Nb282 will bind to both DR5 molecules within the dimeric PLAD, without destruction of the PLAD conformation and thus without provoking cytoplasmic signaling and killing. In sharp contrast, the Nb283, Nb284 exhibited strong tumor killing capacity in a dose dependent manner and often as good as TRAIL. This suggests that the association of the trivalent or tetravalent Nb28 construct with the cell surface exposed DR5 manages to trigger the clustering of its antigen and to overcome the auto-inactivated state of the PLAD. The cell toxicity of Nb283 and Nb284 was noticed for Colo205 and PC3 cells but not for HeLa, pointing toward the importance of an elevated DR5 exposure on the cell membrane.
Unfortunately, as mentioned earlier, the expression of Nb284 was seriously impeded, and only small amounts could be purified. We therefore explored the generation of a bispecific Nb construct, Nb28-113, where the C-terminal Nb would recognize each single B domain of a pentameric Stx2aB5 compound. Hence, mixing the Nb28-113 and Stx2aB5 will spontaneously generate pentameric Nb28 constructs. Indeed, this mixture was also highly potent in tumor cell killing.
Our data show that the efficacy of TRAIL is highly dependent on the cancer cell line. It even seems that the number of DR5 molecules on the cell surface is not the only determinant for apoptosis. The affinity, the mechanism of interaction and signal transduction might also affect the apoptosis, autophagy or even survival pathways. In addition, TRAIL is capable of binding to five different types of receptors, where DR4 and DR5 are triggering apoptosis and the decoy receptors (DcRs) are surviving receptors. Therefore, the final outcome will depend on the balance between these engaged, different receptors. Our Nbs are supposedly more specific for DR5 alone and less disturbed by presence of DR4 or DcRs than TRAIL (although it is not yet formally demonstrated).
Finally, TRAIL binding to DR5 is expected to induce the apoptotic pathway, which should be witnessed by the activation of caspase 3/7. Indeed, the appearance of caspase 3/7 activity correlates well with the killing activity of TRAIL and of the trimeric, tetrameric or pentameric Nb constructs. We therefore, conclude that the presence of these oligomeric Nb28 constructs binds to the cell surface exposed DR5 molecules and induces cytoplasmic signaling that activates the caspase 3/7 and initiates the apoptotic pathway.
4. Materials & Methods
4.1. Expression and Purification of DR5, TRAIL, and Stx-2aB5
Freshly transformed E. coli SHuffle® T7 Express harboring the recombinant plasmids pET-28a/dr5 (encoding amino acids 56–211) or pET-28a/trail (encoding amino acids 114–281) were inoculated in 5 mL LB broth containing 50 μg/mL kanamycin and cultivated overnight at 30 °C in an orbital shaker at 200 rpm. Then, 1 mL of this pre-culture was used to inoculate 100 mL freshly made TB and grown at 30 °C. The expression was induced with 1 mM IPTG when the culture reached an OD600nm between 0.7–1.0, and incubation was continued for 6 h at 30 °C while shaking.
The cultures were cooled on ice for 5 min and centrifuged at 8000 g for 15 min at 4 °C. After removal of the supernatant, cell pellets were either stored at −20 °C or subjected to further purification. Cell pellets were resuspended in lysis buffer (300 mM NaCl, 10 mM imidazole, 50 mM Tris-HCl, pH 8.0) and sonicated for 10 min (in intervals of 10 s). Samples were then centrifuged at 8000 g for 35 min to remove cell debris. The soluble proteins were applied on HisPur Ni-NTA resin (Thermo Scientific, Waltham, MA, USA), washed extensively with lysis buffer including 30 mM extra imidazole. and recombinant protein was eluted in buffer containing 150 mM imidazole in six separate fractions of 1–2 mL. The fractions containing recombinant protein were pooled and applied on Sephadex-75 column equilibrated in PBS for TRAIL preparation and in 10 mM acetate pH 5.0 for DR5 preparation.
The B subunit of Shiga-like toxin 2a was produced recombinantly in
E. coli BL21. This protein B domain forms spontaneously a homopentamer when expressed and purified as described earlier [
14].
4.2. Expression and Purification of DR5-Binding Nbs
DR5-binding Nbs were identified after immunizing a five-year-old dromedary with soluble DR5 protein, construction of an immune Nb library from blood and lymph nodes in pComb3X vector, and enrichment by phage display and panning following published methods [
20]. After choosing the best DR5-responsive clones in ELISA the chimeric pComb3X phagemid was extracted from TG1
E. coli cells. Its Nb sequence was ligated into pMECS or pHEN6c vectors between PstI and BstEII (pMECS) or EcoR I (pHEN6c) restriction enzyme sites and transformed in
E. coli WK6 cells. The transformed cells were plated on LB agar plates with 2% glucose and 100 µg/mL ampicillin and incubated overnight at 37 °C. After verifying the constructs by nucleotide sequence analysis, a single colony from the plates was cultured in TB medium supplemented with 0.1% (w/v) glucose, 100 μg/mL ampicillin, and 2 mM MgCl
2. The Nb expression was induced with 1 mM IPTG when the culture reached an O.D.
600nm between 0.6–0.9, and incubation was continued overnight at 28 °C while shaking. With the pMECS vector the recombinant protein with a C-terminal hemagglutinin tag and His
6 tag, whereas the recombinant protein expressed from pHEN6 vectors will only carry the His
6 tag. The periplasmic proteins were extracted by osmotic shock and purification of His-tagged Nbs was performed as described before [
14]. The concentration of purified Nbs was measured via UV spectrophotometry on NanoDrop 2000 (Thermo Scientific) using the extinction coefficient predicted with the ExPASy ProtParam tool.
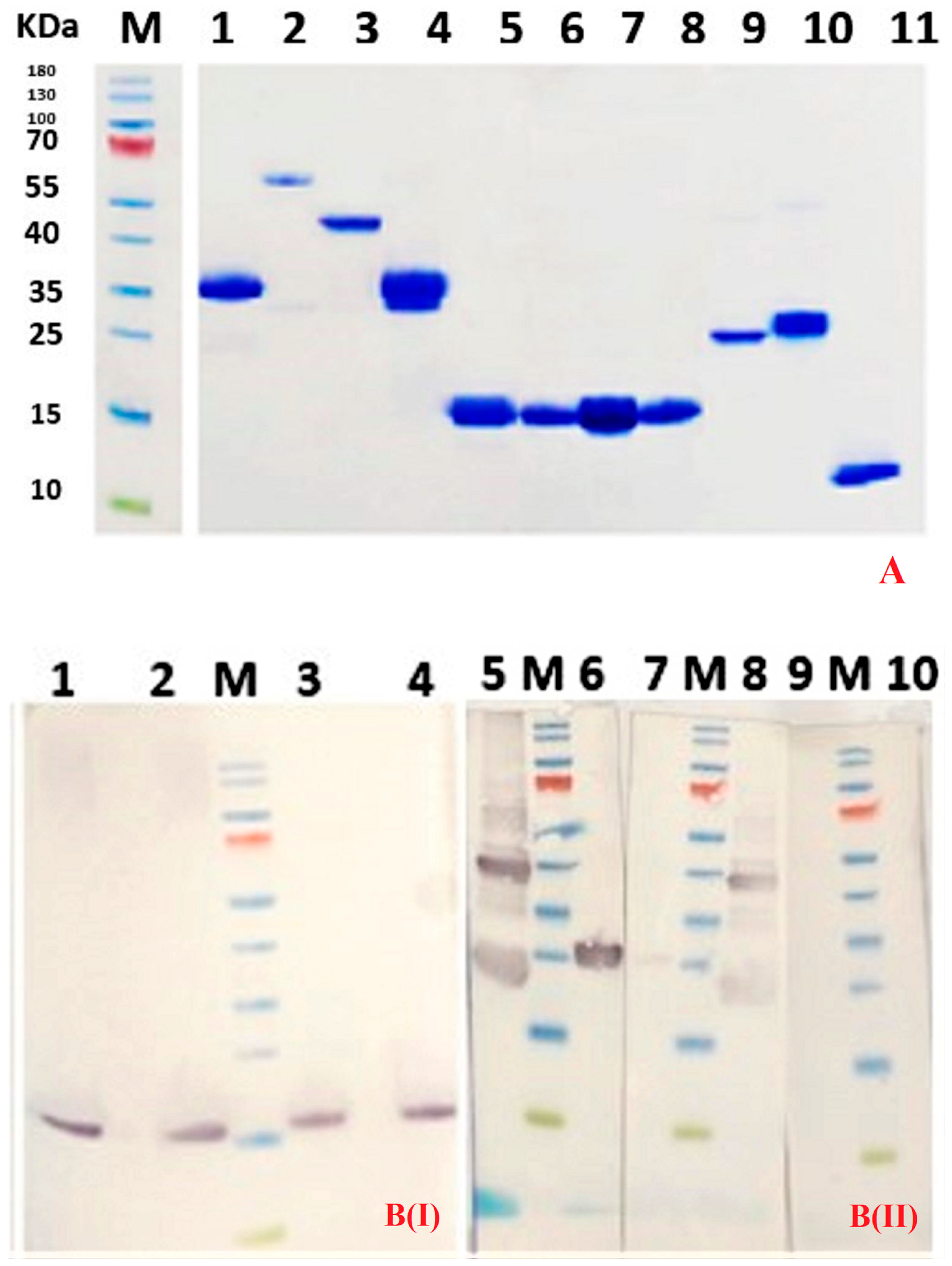
Purity of Nbs was assessed by SDS-PAGE staining with Coomassie blue. A western blot using anti-His Tag monoclonal antibody (Biolegend, San Diego, CA, USA) and goat anti-mouse IgG conjugated to horse radish peroxidase (Sigma Aldrich, St Louis, MO, USA) was used to confirm the identity of the Nb.
The expression of bi, tri, and tetravalent monospecific Nbs and the bispecific Nbs was performed exactly as explained for the monomeric Nb.
4.3. Cloning of Bivalent, Trivalent, or Tetravalent Monospecific Nb Constructs
The plasmid encoding, what will be, the second Nb was digested with PstI and NcoI restriction enzymes, while the gene fragment containing the first Nb sequence was amplified by PCR with primers encoding a (Gly4Ser)3 linker. The amplicon was also digested with the same nucleases. During PCR, a (Gly4Ser)3 encoding linker containing a unique PstI site was added downstream of the Nb template, whereas the primer annealing at the 5′ end of the amplified Nb gene enforced a NcoI restriction enzyme site and removed the PstI site within the framework-1 region of the Nb. The digested products were cleaned and finally ligated using T4 DNA Ligase (Thermo Fisher Scientific). The ligation product was transformed in E. coli WK6 competent cells, plated on LB agar containing glucose (2%) and ampicillin (100 µg/mL), and incubated overnight at 37 °C. A few colonies were picked individually and PCR was performed to confirm the presence of an insert with a size corresponding to the tandem Nb genes separated by the (Gly4Ser)3 linker. The PCR-positive colonies were cultured in LB medium, and their plasmids extracted and sent for sequencing to confirm the presence of the correct insert.
Likewise, for the trivalent and tetravalent constructs, PCR amplified bivalent Nb or trivalent Nb fragments were digested with NcoI and Pst1 and applied on agarose gel. The band corresponding to the bivalent or trivalent Nb was purified from agarose gel and ligated in the pMECS vector containing a Nb cut with the same nucleases to arrive at the trivalent and tetravalent, monospecific Nb constructs, respectively.
4.4. Cloning of Bispecific Nb Constructs
We also replaced one of the DR5-specific Nbs with the Nb SA1 directed against serum albumin of mice and humans or with the Nb113 against the B subunit of Shiga toxin 2a (Stx-2aB5). In this case, a monovalent or bivalent DR5-Nb construct was amplified by PCR, cut by NcoI and PstI, and purified on agarose gel, before ligation in the pHEN6c vector containing the NbSA1 or Nb113, respectively. A (Gly4Ser)3 linker was spacing always the different Nbs. All constructs were confirmed by nucleotide sequencing.
4.5. Western Blot Using Nbs as Probe
DR5 protein (5 µg per lane) was separated by SDS-PAGE under reducing and non-reducing conditions. The proteins were transferred onto Protran 0.45 µm NC nitrocellulose western blotting membrane (Amersham, GE Healthcare, Buckinghamshire, UK) and residual protein binding sites on the membrane were blocked by overnight incubation at 4 °C in 2% skim milk in PBS. The membrane was soaked for 1 h at room temperature (RT) in purified Nb (25 µg/mL). The membrane was washed three times with PBS, and mouse anti-HA tag monoclonal antibody (Biolegend)(1:2000 in blocking buffer) was added and incubated for 1 hr at room temperature to bind to the hemagglutinin-containing Nb. Subsequently, the membrane was washed three times with PBS, and goat anti-mouse IgG HRP-conjugated (Sigma Aldrich) was added (1:2000 in blocking buffer) and incubated for 1 hr at room temperature. Finally, the membrane was washed three times with PBS before adding 18 mg of 4-chloro 1-naphtol in 6 mL ethanol, 30 mL buffer (500 mM NaCl, 25 mM Tris pH 7.5) and 18 µL H2O2 for colorimetric visualization of the DR5 bands.
4.6. Affinity Measurement and Epitope Binning
To determine the binding properties of Nbs to DR5, an SPR experiment was performed on Biacore T200 (GE Healthcare) according to the manufacturer’s instructions. The CM5 sensor chip (GE Healthcare), was used for capturing DR5 at a concentration of about 5 μg/mL and a flow rate of 10 μL/min for 1 min. This resulted in coupling of ~150 RU DR5 to the sensor chip surface. Meanwhile, one flow cell of the sensor chip was left without captured DR5 to provide a reference surface. Nanobodies were prepared at different concentrations starting from 125 nM or 10 nM using a 2-fold serial dilution in running buffer (150 mM NaCl, 0.005% Tween-20, 3.4 mM EDTA, 20 mM HEPES, pH 7.4).
For each Nb construct, sensorgrams were recorded for different analyte concentrations at a flow rate of 30 µL/min and a data collection rate of 1 Hz. Analyte injections were performed with association and dissociation phases of 180 s and 300 s, respectively. Prior to data analysis, reference and zero concentration data were subtracted from the sensorgrams. The collected data were fitted according to a 1:1 Langmuir binding model (one Nb to one monomer of DR5) from the experimental flow cells of a single biosensor chip. All experimental data were treated with Biacore T200 Evaluation Software to calculate the kinetic kon and koff rates and the equilibrium dissociation constant (KD).
For epitope binning, we first injected an excess of Nb-A (a concentration equivalent to 100 times its KD value) for 300 s to saturate all epitopes on the DR5. This was followed by applying a mixture of excess Nb-A and Nb-B (both at a concentration of 100 times their KD value) and monitoring the resonance units (RU) over a period of 300 s. The chip was then injected with buffer without any Nb for 500 s. All possible Nb combinations were tested including all orders of injection.
4.7. Flow Cytometry
The Hela cell line was obtained from dr C. Goyvaerts (VUB, Jette, Belgium). The Colo205 cell line was provided by dr C. Vangestel from UZA Edegem, Belgium. The HeLa, PC3, and LLC1 cell lines were originally obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA).
HeLa (ATCC CCL-2, Colo205 (ATCC CCL-222), PC3 (ATCC CRL-1435), and LLC1 ATC CRL-1642) cells were cultured according to the specifications of the ATCC. The (semi-)adherent PC3, HeLa, and Colo205 cells were collected after trypsinization (ThermoFisher Scientific, Gibco, #25200072) and washed twice with HBSS Buffer (Gibco #14025050). DR5 specific Nbs (10 µ/mL) were mixed with 100 µL cell suspension (106 cells/mL). After incubation for 1 hr, cells were washed with ice-cold HBSS, resuspended with HBSS Buffer, and then APC labelled anti-human CD262 (DR5, TRIAL-R2) antibody (Biolegend) or one of our Nb constructs plus APC labelled anti-His monoclonal antibody (Biolegend) were added. Incubations where performed on ice and washings were at 4 °C, and cells were analyzed on FACS Canto II (BD Biosciences, Erembodegem, Belgium).
4.8. Cell Survival Assay
Cells were plated in a clear-bottom 96-well plate (Sigma Aldrich, Costar, #3903) at 5000 to 10,000 cells/well in dedicated media. The next day, serially diluted Nbs or Apo2L/TRAIL was added to the wells. After 48 h, cell viability was measured using AlamarBlue® (Bio-Rad, # BUF012A, Hercules, CA, USA), and absorbance of the solution in the wells was determined with a standard microplate ELISA reader.
4.9. Caspase Activity
Cells were plated at 5000 to 10,000 cells/well and left to attach overnight at 37 °C. The next day, the 50 µL culture in the wells was supplemented with 50 µL media containing the indicated concentrations of DR5 agonists. Caspase activity was measured using CaspaseGlo-3/7 kit (Promega, #G8202, Madison, WI, USA) according to the suppliers’ instructions.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}