ILC2 Activation by Protozoan Commensal Microbes


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The ILC Lineage
1.1. The Family of Innate Lymphoid Cells
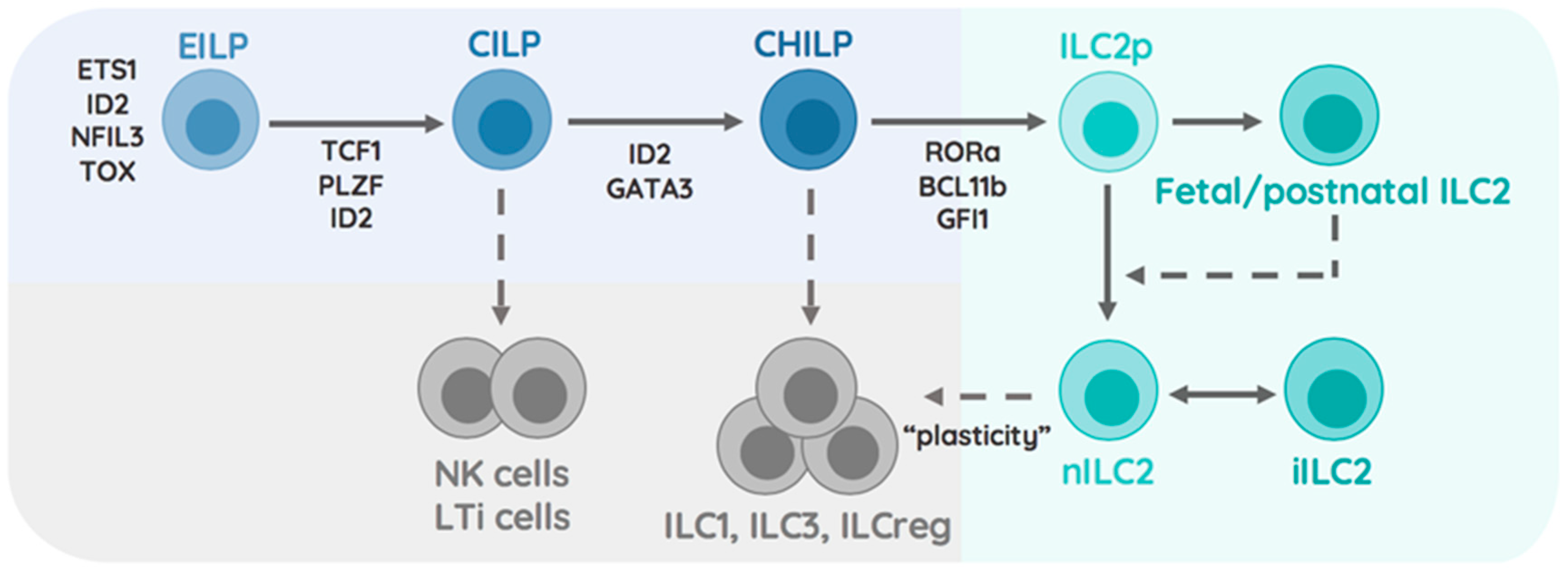
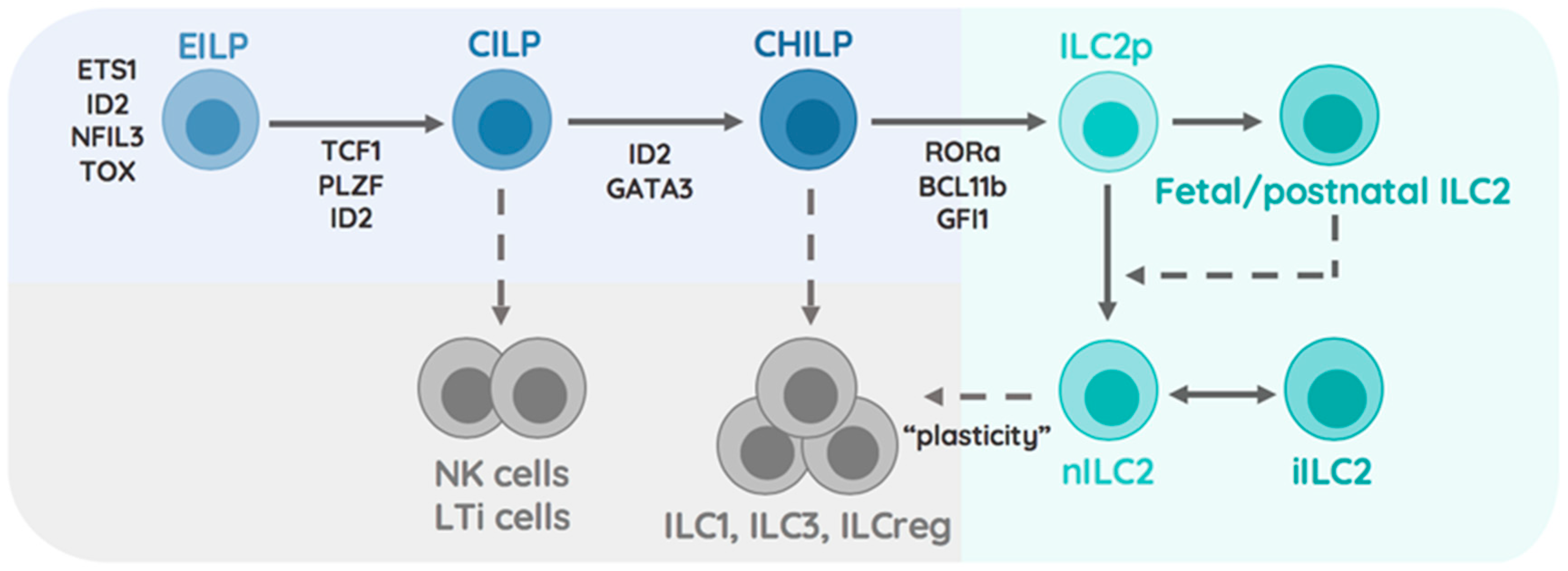
1.2. Transcriptional Specification of the ILC2 Lineage
1.3. ILC2 Heterogeneity Across Tissues
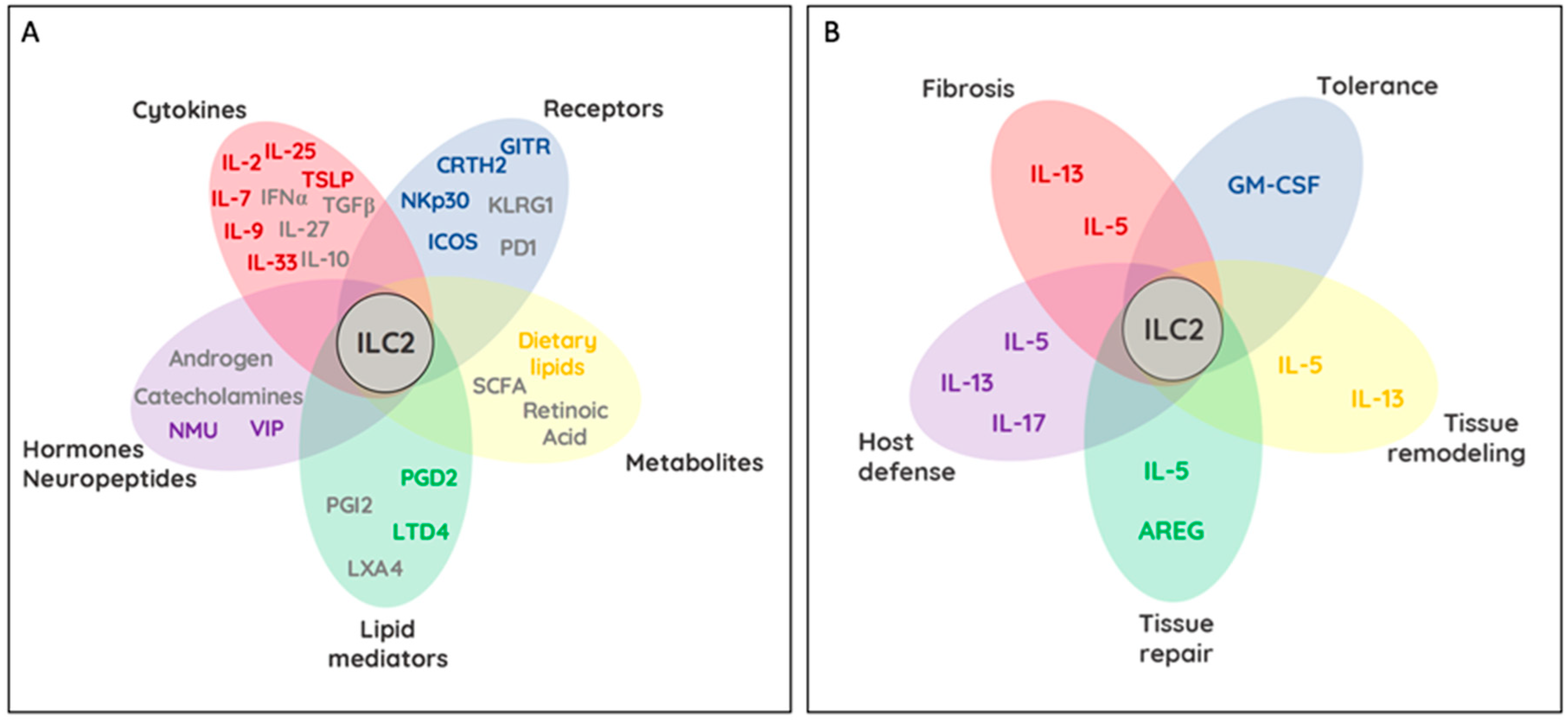
2. Activation and Inhibition of ILC2
2.1. Cytokine Receptor Profiles
2.2. Activating and Inhibiting Surface Receptors on ILC2
2.2.1. GITR (TNFRSF18)
2.2.2. CRTH2
2.2.3. ICOS/ICOSL (CD278)
2.2.4. KLRG1
2.2.5. PD-1 (CD279)
2.2.6. NKp30 (NCR3/CD337)
2.3. Lipid-Driven Modulation of ILC2
2.3.1. Leukotrienes
2.3.2. Prostaglandins
2.3.3. Lipoxins
2.4. Regulation of ILC2 by Metabolites
2.5. Neuronal and Hormonal Modulation of ILC2
2.6. Hormone-Mediated Sex Differences in ILC2 Function
2.7. Control of ILC2 function by MicroRNA
3. ILC2-Tissue Crosstalk
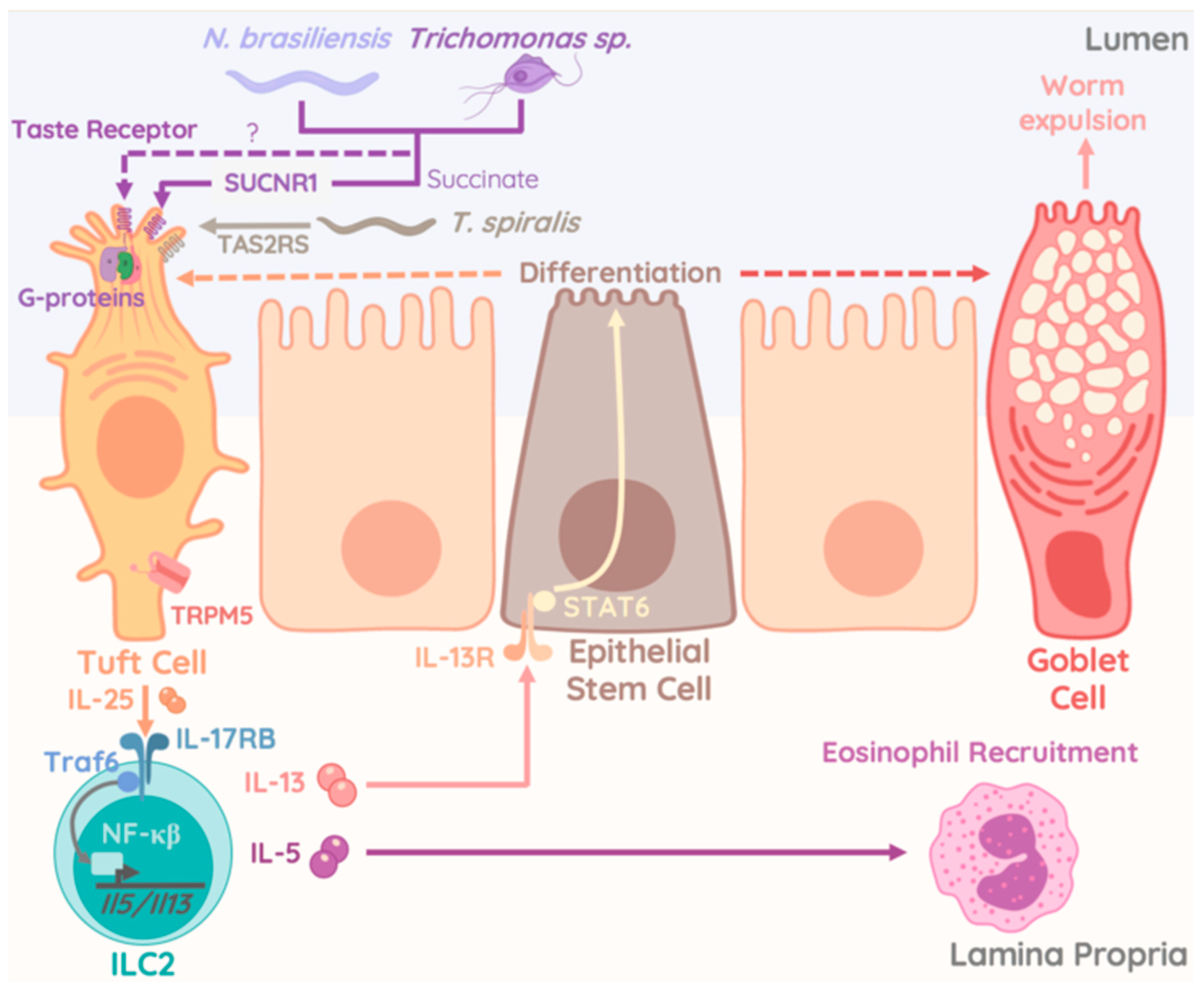
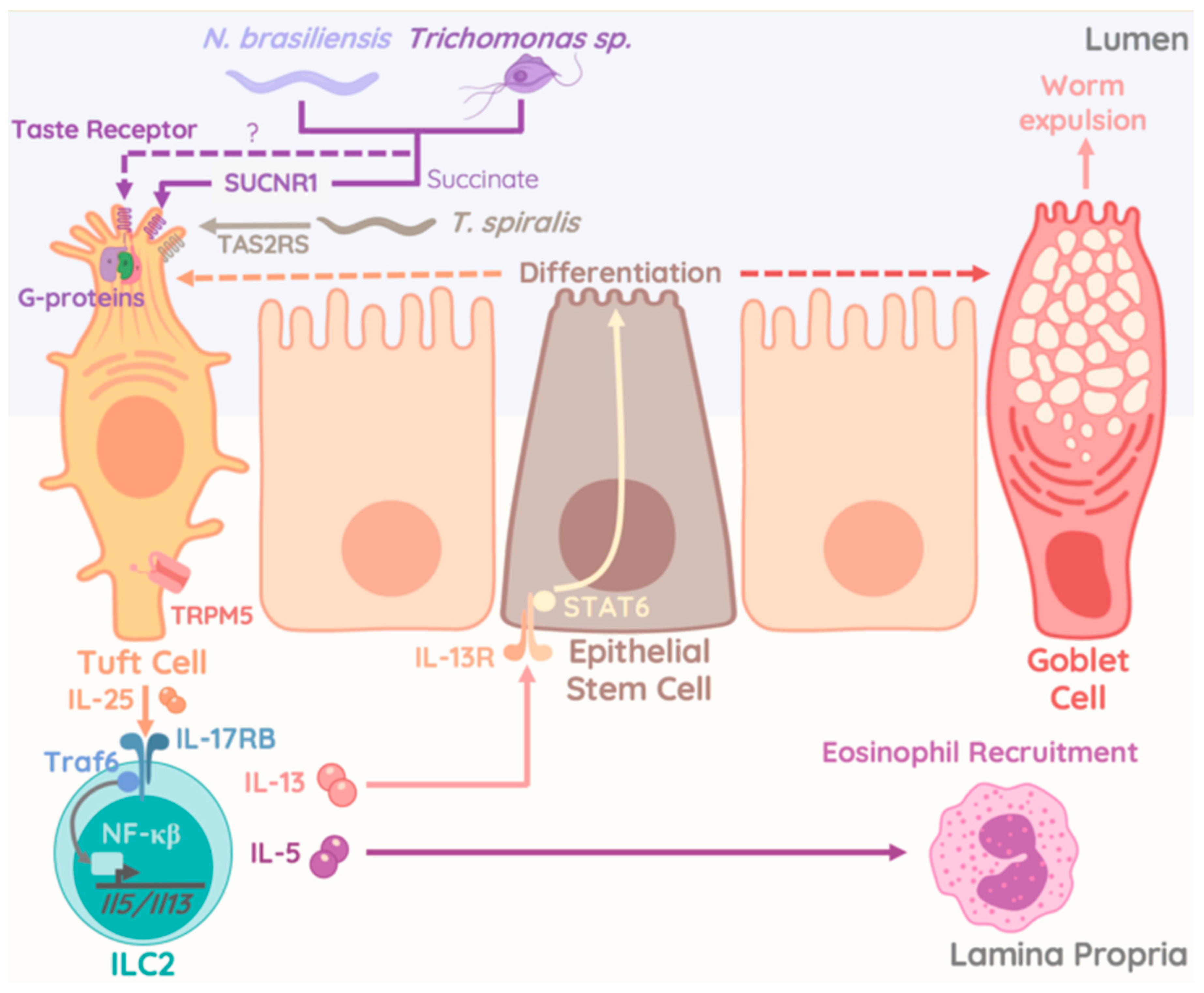
4. Protozoan Commensals as Regulator of ILC2 Activity
- Are there several pools of ILC2 precursors?
- What is the contribution of postnatal and adult ILC2 to tissue homeostasis and host defense?
- Which are ILC2 specific tissue niches?
- What is the role of Tuft cell subsets across tissues in regulating ILC2 activity?
- Exploring the potential of ILC2s in immunotherapy during host immunity.
- Identifying lineage determining transcription factors in human ILC2 for the use in iPSC technology.
- Developing ILC2-specific Cre/reporter lines for faithful tracking of ILC2 subset or ILC2 at distinct developmental stages.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.X.; Leonard, W.J. The Common Cytokine Receptor gamma Chain Family of Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028449. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Takatori, H.; Kanno, Y.; Watford, W.T.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 2009, 206, 35–41. [Google Scholar] [CrossRef]
- Kim, C.H.; Hashimoto-Hill, S.; Kim, M. Migration and Tissue Tropism of Innate Lymphoid Cells. Trends Immunol. 2016, 37, 68–79. [Google Scholar] [CrossRef]
- Mortha, A.; Burrows, K. Cytokine Networks between Innate Lymphoid Cells and Myeloid Cells. Front. Immunol. 2018, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, M.G.; McDonald, B.D.; Verhoef, P.A.; Bendelac, A. A committed precursor to innate lymphoid cells. Nature 2014, 508, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Klose, C.S.N.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeife, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef]
- Cherrier, D.E.; Serafini, N.; Di Santo, J.P. Innate Lymphoid Cell Development: A T Cell Perspective. Immunity 2018, 48, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Spencer, S.P.; Belkaid, Y. Dietary and commensal derived nutrients: Shaping mucosal and systemic immunity. Curr. Opin. Immunol. 2012, 24, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Hoyler, T.; Klose, C.S.; Souabni, A.; Turqueti-Neves, A.; Pfeifer, D.; Rawlins, E.L.; Voehringer, D.; Busslinger, M.; Diefenbach, A. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012, 37, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.; MacLaren, A.; Romanish, M.T.; Gold, M.J.; McNagny, K.M.; Takei, F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity 2012, 37, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Walker, J.A.; Jolin, H.E.; Drynan, L.F.; Hams, E.; Camelo, A.; Barlow, J.L.; Neill, D.R.; Panova, V.; Koch, U.; et al. Transcription factor RORalpha is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Spooner, C.J.; Lesch, J.; Yan, D.; Khan, A.A.; Abbas, A.; Ramirez-Carrozzi, V.; Zhou, M.; Soriano, R.; Eastham-Anderson, J.; Diehl, L.; et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat. Immunol. 2013, 14, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.C.; Yashiro-Ohtani, Y.; del Bianco, C.; Knoblock, D.M.; Blacklow, S.C.; Pear, W.S. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity 2007, 27, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Monticelli, L.A.; Saenz, S.A.; Chi, A.W.; Sonnenberg, G.F.; Tang, J.; de Obaldia, M.E.; Bailis, W.; Bryson, J.L.; Toscano, K.; et al. T cell factor 1 is required for group 2 innate lymphoid cell generation. Immunity 2013, 38, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Mielke, L.A.; Groom, J.R.; Rankin, L.C.; Seillet, C.; Masson, F.; Putoczki, T.; Belz, G.T. TCF-1 controls ILC2 and NKp46+RORgammat+ innate lymphocyte differentiation and protection in intestinal inflammation. J. Immunol. 2013, 191, 4383–4391. [Google Scholar] [CrossRef]
- Walker, J.A.; Oliphant, C.J.; Englezakis, A.; Yu, Y.; Clare, S.; Rodewald, H.R.; Belz, G.; Liu, P.; Fallon, P.G.; McKenzie, A.N. Bcl11b is essential for group 2 innate lymphoid cell development. J. Exp. Med. 2015, 212, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wang, C.; Clare, S.; Wang, J.; Lee, S.C.; Brandt, C.; Burke, S.; Lu, L.; He, D.; Jenkins, N.A.; et al. The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J. Exp. Med. 2015, 212, 865–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolterink, R.G.K.; Serafini, N.; van Nimwegen, M.; Vosshenrich, C.A.; de Bruijn, M.J.; Pereira, D.F.; Fernandes, H.V.; Hendriks, R.W.; di Santo, J.P. Essential, dose-dependent role for the transcription factor Gata3 in the development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10240–10245. [Google Scholar] [CrossRef] [PubMed]
- Antignano, F.; Braam, M.; Hughes, M.R.; Chenery, A.L.; Burrows, K.; Gold, M.J.; Oudhoff, M.J.; Rattray, D.; Halim, T.Y.; Cait, A.; et al. G9a regulates group 2 innate lymphoid cell development by repressing the group 3 innate lymphoid cell program. J. Exp. Med. 2016, 213, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Gury-BenAri, M.; Thaiss, C.A.; Serafini, N.; Winter, D.R.; Giladi, A.; Lara-Astiaso, D.; Levy, M.; Salame, T.M.; Weiner, A.; David, E.; et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell 2016, 166, 1231–1246.e13. [Google Scholar] [CrossRef] [PubMed]
- Ricardo-Gonzalez, R.R.; van Dyken, S.J.; Schneider, C.; Lee, J.; Nussbaum, J.C.; Liang, H.E.; Vaka, D.; Eckalbar, W.L.; Molofsky, A.B.; Erle, D.J.; et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 2018, 19, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mao, K.; Chen, X.; Sun, M.A.; Kawabe, T.; Li WLUsher, N.; Zhu, J.; Urban, J.F., Jr.; Paul, W.E.; et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 2018, 359, 114–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.; Lee, J.; Koga, S.; Ricardo-Gonzalez, R.R.; Nussbaum, J.C.; Smith, L.K.; Villeda, S.A.; Liang, H.E.; Locksley, R.M. Tissue-Resident Group 2 Innate Lymphoid Cells Differentiate by Layered Ontogeny and In Situ Perinatal Priming. Immunity 2019, 50, 1425–1438.e5. [Google Scholar] [CrossRef]
- Van Dyken, S.J.; Nussbaum, J.C.; Lee, J.; Molofsky, A.B.; Liang, H.E.; Pollack, J.L.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Marson, A.; et al. A tissue checkpoint regulates type 2 immunity. Nat. Immunol. 2016, 17, 1381–1387. [Google Scholar] [CrossRef] [Green Version]
- Duerr, C.U.; McCarthy, C.D.; Mindt, B.C.; Rubio, M.; Meli, A.P.; Pothlichet, J.; Eva, M.M.; Gauchat, J.F.; Qureshi, S.T.; Mazer, B.D.; et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat. Immunol. 2016, 17, 65–75. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, X.; Pasha, M.A.; Siebel, C.W.; Costello, A.; Haczku, A.; MacNamara, K.; Liang, T.; Zhu, J.; Bhandoola, A.; et al. Cutting Edge: Notch Signaling Promotes the Plasticity of Group-2 Innate Lymphoid Cells. J. Immunol. 2017, 198, 1798–1803. [Google Scholar] [CrossRef] [Green Version]
- Camelo, A.; Rosignoli, G.; Ohne, Y.; Stewart, R.A.; Overed-Sayer, C.; Sleeman, M.A.; May, R.D. IL-33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Adv. 2017, 1, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, C.; Hirota, K.; Stieglitz, B.; van Snick, J.; Tolaini, M.; Lahl, K.; Sparwasser, T.; Helmby, H.; Stockinger, B. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat. Immunol. 2011, 12, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.E.; Morrison, P.J.; Wilhelm, C.; Wilson, M.; Ahlfors, H.; Renauld, J.C.; Panzer, U.; Helmby, H.; Stockinger, B. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 2013, 210, 2951–2965. [Google Scholar] [CrossRef] [PubMed]
- Rigas, D.; Lewis, G.; Aron, J.L.; Wang, B.; Banie, H.; Sankaranarayanan, I.; Galle-Treger, L.; Maazi, H.; Lo, R.; Freeman, G.J.; et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J. Allergy Clin. Immunol. 2017, 139, 1468–1477.e2. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xia, P.; Chen, Y.; Qu, Y.; Xiong, Z.; Ye, B.; Du, Y.; Tian, Y.; Yin, Z.; Xu, Z.; et al. Regulatory Innate Lymphoid Cells Control Innate Intestinal Inflammation. Cell 2017, 171, 201–216.e18. [Google Scholar] [CrossRef] [Green Version]
- Moro, K.; Kabata, H.; Tanabe, M.; Koga, S.; Takeno, N.; Mochizuki, M.; Fukunaga, K.; Asano, K.; Betsuyaku, T.; Koyasu, S. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat. Immunol. 2016, 17, 76–86. [Google Scholar] [CrossRef]
- Lim, A.I.; Menegatti, S.; Bustamante, J.; le Bourhis, L.; Allez, M.; Rogge, L.; Casanova, J.L.; Yssel, H.; di Santo, J.P. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J. Exp. Med. 2016, 213, 569–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bal, S.M.; Bernink, J.H.; Nagasawa, M.; Groot, J.; Shikhagaie, M.M.; Golebski, K.; van Drunen, C.M.; Lutter, R.; Jonkers, R.E.; Hombrink, P.; et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat. Immunol. 2016, 17, 636–645. [Google Scholar] [CrossRef]
- Watts, T.H. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005, 23, 23–68. [Google Scholar] [CrossRef]
- Nocentini, G.; Giunchi, L.; Ronchetti, S.; Krausz, L.T.; Bartoli, A.; Moraca, R.; Migliorati, G.; Riccardi, C. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 6216–6221. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Voskens, C.J.; Zhang, X.; Schindler, D.G.; Wood, A.; Burch, E.; Wei, Y.; Chen, L.; Tian, G.; Tamada, K.; et al. Fc-dependent expression of CD137 on human NK cells: Insights into “agonistic” effects of anti-CD137 monoclonal antibodies. Blood 2008, 112, 699–707. [Google Scholar] [CrossRef]
- NNagashima, H.; Okuyama, Y.; Fujita, T.; Takeda, T.; Motomura, Y.; Moro, K.; Hidaka, T.; Omori, K.; Sakurai, T.; Machiyama, T.; et al. GITR cosignal in ILC2s controls allergic lung inflammation. J. Allergy Clin. Immunol. 2018, 141, 1939–1943.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galle-Treger, L.; Sankaranarayanan, I.; Hurrell, B.P.; Howard, E.; Lo, R.; Maazi, H.; Lewis, G.; Banie, H.; Epstein, A.L.; Hu, P.; et al. Costimulation of type-2 innate lymphoid cells by GITR promotes effector function and ameliorates type 2 diabetes. Nat. Commun. 2019, 10, 713. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Nussbaum, J.C.; Liang, H.E.; van Dyken, S.J.; Cheng, L.E.; Mohapatra, A.; Chawla, A.; Locksley, R.M. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med. 2013, 210, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Dalmas, E.; Lehmann, F.M.; Dror, E.; Wueest, S.; Thienel, C.; Borsigova, M.; Stawiski, M.; Traunecker, E.; Lucchini, F.C.; Dapito, D.H.; et al. Interleukin-33-Activated Islet-Resident Innate Lymphoid Cells Promote Insulin Secretion through Myeloid Cell Retinoic Acid Production. Immunity 2017, 47, 928–942.e7. [Google Scholar] [CrossRef] [Green Version]
- Nagata, K.; Hirai, H.; Tanaka, K.; Ogawa, K.; Aso, T.; Sugamura, K.; Nakamura, M.; Takano, S. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s). FEBS Lett. 1999, 459, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.E.; Doherty, T.A.; Baum, R.; Broide, D. Prostaglandin D2 regulates human type 2 innate lymphoid cell chemotaxis. J. Allergy Clin. Immunol. 2014, 133, 899–901.e3. [Google Scholar] [CrossRef] [PubMed]
- Wojno, E.D.; Monticelli, L.A.; Tran, S.V.; Alenghat, T.; Osborne, L.C.; Thome, J.J.; Willis, C.; Budelsky, A.; Farber, D.L.; Artis, D. The prostaglandin D(2) receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal. Immunol. 2015, 8, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Salimi, M.; Panse, I.; Mjösberg, J.M.; McKenzie, A.N.; Spits, H.; Klenerman, P.; Ogg, G. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J. Allergy Clin. Immunol. 2014, 133, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Maric, J.; Ravindran, A.; Mazzurana, L.; van Acker, A.; Rao, A.; Kokkinou, E.; Ekoff, M.; Thomas, D.; Fauland, A.; Nilsson, G.; et al. Cytokine-induced endogenous production of prostaglandin D2 is essential for human group 2 innate lymphoid cell activation. J. Allergy Clin. Immunol. 2019, 143, 2202–2214.e5. [Google Scholar] [CrossRef]
- Pettipher, R.; Hunter, M.G.; Perkins, C.M.; Collins, L.P.; Lewis, T.; Baillet, M.; Steiner, J.; Bell, J.; Payton, M.A. Heightened response of eosinophilic asthmatic patients to the CRTH2 antagonist OC000459. Allergy 2014, 69, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, S.K.; Whoriskey, J.S.; Khare, S.D.; Sarmiento, U.; Guo, J.; Horan, T.; Shih, G.; Zhang, M.; Coccia, M.A.; Kohno, T.; et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature 1999, 402, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Maazi, H.; Patel, N.; Sankaranarayanan, I.; Suzuki, Y.; Rigas, D.; Soroosh, P.; Freeman, G.J.; Sharpe, A.H.; Akbari, O. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity 2015, 42, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Paclik, D.; Stehle, C.; Lahmann, A.; Hutloff, A.; Romagnani, C. ICOS regulates the pool of group 2 innate lymphoid cells under homeostatic and inflammatory conditions in mice. Eur. J. Immunol. 2015, 45, 2766–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamachi, F.; Isshiki, T.; Harada, N.; Akiba, H.; Miyake, S. ICOS promotes group 2 innate lymphoid cell activation in lungs. Biochem. Biophys. Res. Commun. 2015, 463, 739–745. [Google Scholar] [CrossRef]
- Hrusch, C.L.; Manns, S.T.; Bryazka, D.; Casaos, J.; Bonham, C.A.; Jaffery, M.R.; Blaine, K.M.; Mills, K.A.M.; Verhoef, P.A.; Adegunsoye, A.O.; et al. ICOS protects against mortality from acute lung injury through activation of IL-5(+) ILC2s. Mucosal. Immunol. 2018, 11, 61–70. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Van Gool, F.; Liang, H.E.; van Dyken, S.J.; Nussbaum, J.C.; Lee, J.; Bluestone, J.A.; Locksley, R.M. Interleukin-33 and Interferon-gamma Counter-Regulate Group 2 Innate Lymphoid Cell Activation during Immune Perturbation. Immunity 2015, 43, 161–174. [Google Scholar] [CrossRef]
- Grundemann, C.; Bauer, M.; Schweier, O.; von Oppen, N.; Lässing, U.; Saudan, P.; Becker, K.F.; Karp, K.; Hanke, T.; Bachmann, M.F.; et al. Cutting edge: Identification of E-cadherin as a ligand for the murine killer cell lectin-like receptor G1. J. Immunol. 2006, 176, 1311–1315. [Google Scholar] [CrossRef]
- Ito, M.; Maruyama, T.; Saito, N.; Koganei, S.; Yamamoto, K.; Matsumoto, N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J. Exp. Med. 2006, 203, 289–295. [Google Scholar] [CrossRef]
- Riedl, E.; Stockl, J.; Majdic, O.; Scheinecker, C.; Knapp, W.; Strobl, H. Ligation of E-cadherin on in vitro-generated immature Langerhans-type dendritic cells inhibits their maturation. Blood 2000, 96, 4276–4284. [Google Scholar]
- Rosshart, S.; Hofmann, M.; Schweier, O.; Pfaff, A.K.; Yoshimoto, K.; Takeuchi, T.; Molnar, E.; Schamel, W.W.; Pircher, H. Interaction of KLRG1 with E-cadherin: New functional and structural insights. Eur. J. Immunol. 2008, 38, 3354–3364. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.; Huang, Y.; Mallett, G.; Stathopoulou, C.; Felizardo, T.C.; Sun, M.A.; Martin, E.L.; Zhu, N.; Woodward, E.L.; Elias, M.S.; et al. PD-1 regulates KLRG1(+) group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1663–1678. [Google Scholar] [CrossRef]
- Yu, Y.; Tsang, J.C.; Wang, C.; Clare, S.; Wang, J.; Chen, X.; Brandt, C.; Kane, L.; Campos, L.S.; Lu, L.; et al. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature 2016, 539, 102–106. [Google Scholar] [CrossRef]
- Schwartz, C.; Khan, A.R.; Floudas, A.; Saunders, S.P.; Hams, E.; Rodewald, H.R.; McKenzie, A.N.J.; Fallon, P.G. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J. Exp. Med. 2017, 214, 2507–2521. [Google Scholar] [CrossRef]
- Oldenhove, G.; Boucquey, E.; Taquin, A.; Acolty, V.; Bonetti, L.; Ryffel, B.; le Bert, M.; Englebert, K.; Boon, L.; Moser, M. PD-1 Is Involved in the Dysregulation of Type 2 Innate Lymphoid Cells in a Murine Model of Obesity. Cell Rep. 2018, 25, 2053–2060.e4. [Google Scholar] [CrossRef] [Green Version]
- Mariotti, F.R.; Quatrini, L.; Munari, E.; Vacca, P.; Moretta, L. Innate Lymphoid Cells: Expression of PD-1 and Other Checkpoints in Normal and Pathological Conditions. Front. Immunol. 2019, 10, 910. [Google Scholar] [CrossRef]
- Guia, S.; Fenis, A.; Vivier, E.; Narni-Mancinelli, E. Activating and inhibitory receptors expressed on innate lymphoid cells. Semin. Immunopathol. 2018, 40, 331–341. [Google Scholar] [CrossRef]
- Brusilovsky, M.; Rosental, B.; Shemesh, A.; Appel, M.Y.; Porgador, A. Human NK cell recognition of target cells in the prism of natural cytotoxicity receptors and their ligands. J. Immunotoxicol. 2012, 9, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnon, T.I.; Achdout, H.; Levi, O.; Markel, G.; Saleh, N.; Katz, G.; Gazit, R.; Gonen-Gross, T.; Hanna, J.; Nahari, E.; et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat. Immunol. 2015, 6, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Mavoungou, E.; Held, J.; Mewono, L.; Kremsner, P.G. A Duffy binding-like domain is involved in the NKp30-mediated recognition of Plasmodium falciparum-parasitized erythrocytes by natural killer cells. J. Infect. Dis. 2007, 195, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Von Strandmann, E.P.; Simhadri, V.R.; von Tresckow, B.; Sasse, S.; Reiners, K.S.; Hansen, H.P.; Rothe, A.; Böll, B.; Simhadri, V.L; Borchmann, P.; et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 2007, 27, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimi, M.; Xue, L.; Jolin, H.; Hardman, C.; Cousins, D.J.; McKenzie, A.N.; Ogg, G.S. Group 2 innate lymphoid cells express functional NKp30 receptor inducing type 2 cytokine production. J. Immunol. 2016, 196, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Trabanelli, S.; Chevalier, M.F.; Martinez-Usatorre, A.; Gomez-Cadena, A.; Salomé, B.; Lecciso, M.; Salvestrini, V.; Verdeil, G.; Racle, J.; Papayannidis, C.; et al. Tumour-derived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat. Commun. 2017, 8, 593. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Dennis, E.A. Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem. 1994, 269, 13057–13060. [Google Scholar]
- Singh, R.K.; Tandon, R.; Dastidar, S.G.; Ray, A. A review on leukotrienes and their receptors with reference to asthma. J. Asthma 2013, 50, 922–931. [Google Scholar] [CrossRef]
- Peres, C.M.; Aronoff, D.M.; Serezani, C.H.; Flamand, N.; Faccioli, L.H.; Peters-Golden, M. Specific leukotriene receptors couple to distinct G proteins to effect stimulation of alveolar macrophage host defense functions. J. Immunol. 2007, 179, 5454–5461. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, D.J.; Breyer, R.M.; Defoe, S.K.; Kargman, S.; Daugherty, B.L.; Waldburger, K.; Liu, Q.; Clements, M.; Zeng, Z.; O’Neill, G.P.; et al. Expression of the cysteinyl leukotriene 1 receptor in normal human lung and peripheral blood leukocytes. Am. J. Respir. Crit. Care Med. 2001, 163, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, K.; Liang, H.; Fanat, A.; Watson, R.; Snider, D.P.; O’Byrne, P.M. Role for cysteinyl leukotrienes in allergen-induced change in circulating dendritic cell number in asthma. J. Allergy Clin. Immunol. 2004, 114, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Boyce, J.A. Mast cells and eicosanoid mediators: A system of reciprocal paracrine and autocrine regulation. Immunol. Rev. 2007, 217, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, C.N.; Fuerst, E.; McDonald, J.; Bowen, H.; Lee, T.H.; Pease, J.E.; Woszczek, G.; Cousins, D.J. Human T(H)2 cells respond to cysteinyl leukotrienes through selective expression of cysteinyl leukotriene receptor 1. J. Allergy Clin. Immunol. 2012, 129, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.A.; Khorram, N.; Lund, S.; Mehta, A.K.; Croft, M.; Broide, D.H. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J. Allergy Clin. Immunol. 2013, 132, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Moltke, J.; O’Leary, C.E.; Barrett, N.A.; Kanaoka, Y.; Austen, K.F.; Locksley, R.M. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J. Exp. Med. 2017, 214, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jakobsson, P.J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [PubMed]
- Claar, D.; Hartert, T.V.; Peebles, R.S., Jr. The role of prostaglandins in allergic lung inflammation and asthma. Expert Rev. Respir. Med. 2015, 9, 55–72. [Google Scholar] [CrossRef]
- Mjosberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055–1062. [Google Scholar] [CrossRef]
- Zhou, W.; Toki, S.; Zhang, J.; Goleniewksa, K.; Newcomb, D.C.; Cephus, J.Y.; Dulek, D.E.; Bloodworth, M.H.; Stier, M.T.; Polosuhkin, V.; et al. Prostaglandin I2 Signaling and Inhibition of Group 2 Innate Lymphoid Cell Responses. Am. J. Respir. Crit. Care Med. 2016, 193, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Fanning, L.B.; Boyce, J.A. Lipid mediators and allergic diseases. Ann. Allergy Asthma Immunol. 2013, 111, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnig, C.; Cernadas, M.; Dutile, S.; Liu, X.; Perrella, M.A.; Kazani, S.; Wechsler, M.E.; Israel, E.; Levy, B.D. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci. Transl. Med. 2013, 5, 174ra26. [Google Scholar] [CrossRef] [PubMed]
- Larange, A.; Cheroutre, H. Retinoic Acid and Retinoic Acid Receptors as Pleiotropic Modulators of the Immune System. Annu. Rev. Immunol. 2016, 34, 369–394. [Google Scholar] [CrossRef] [PubMed]
- Bernink, J.H.; Krabbendam, L.; Germar, K.; de Jong, E.; Gronke, K.; Kofoed-Nielsen, M.; Munneke, J.M.; Hazenberg, M.D.; Villaudy, J.; Buskens, C.J.; et al. Interleukin-12 and -23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 2015, 43, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Taparowsky, E.J.; Kim, C.H. Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity 2015, 43, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Mortha, A.; Chudnovskiy, A.; Hashimoto, D.; Bogunovic, M.; Spencer, S.P.; Belkaid, Y.; Merad, M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014, 343, 1249288. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.; Harrison, O.J.; Schmitt, V.; Pelletier, M.; Spencer, S.P.; Urban, J.F., Jr.; Ploch, M.; Ramalingam, T.R.; Siegel, R.M.; Belkaid, Y. Critical role of fatty acid metabolism in ILC2-mediated barrier protection during malnutrition and helminth infection. J. Exp. Med. 2016, 213, 1409–1418. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Kim, B.S.; Saenz, S.A.; Stine, R.; Monticelli, L.A.; Sonnenberg, G.F.; Thome, J.J.; Farber, D.L.; Lutfy, K.; Seale, P.; et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 2015, 519, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Newland, S.A.; Mohanta, S.; Clement, M.; Taleb, S.; Walker, J.A.; Nus, M.; Sage, A.P.; Yin, C.; Hu, D.; Kitt, L.L.; et al. Type-2 innate lymphoid cells control the development of atherosclerosis in mice. Nat. Commun. 2017, 8, 15781. [Google Scholar] [CrossRef]
- Theiler, A.; Barnthaler, T.; Platzer, W.; Richtig, G.; Peinhaupt, M.; Rittchen, S.; Kargl, J.; Ulven, T.; Marsh, L.M.; Marsche, G.; et al. Butyrate ameliorates allergic airway inflammation by limiting eosinophil trafficking and survival. J. Allergy Clin. Immunol. 2019, 144, 764–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Pavert, S.A.; Olivier, B.J.; Goverse, G.; Vondenhoff, M.F.; Greuter, M.; Beke, P.; Kusser, K.; Höpken, U.E.; Lipp, M.; Niederreither, K.; et al. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat. Immunol. 2009, 10, 1193–1199. [Google Scholar] [CrossRef] [Green Version]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017, 549, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, V.; Chesne, J.; Ribeiro, H.; García-Cassani, B.; Carvalho, T.; Bouchery, T.; Shah, K.; Barbosa-Morais, N.L.; Harris, N.; Veiga-Fernandes, H. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 2017, 549, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, C.S.N.; Mahlakoiv, T.; Moeller, J.B.; Rankin, L.C.; Flamar, A.L.; Kabata, H.; Monticelli, L.A.; Moriyama, S.; Putzel, G.G.; Rakhilin, N.; et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 2017, 549, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.C.; van Dyken, S.J.; von Moltke, J.; Cheng, L.E.; Mohapatra, A.; Molofsky, A.B.; Thornton, E.E.; Krummel, M.F.; Chawla, A.; Liang, H.E.; et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013, 502, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, S.; Brestoff, J.R.; Flamar, A.L.; Moeller, J.B.; Klose, C.S.N.; Rankin, L.C.; Yudanin, N.A.; Monticelli, L.A.; Putzel, G.G.; Rodewald, H.R.; et al. beta2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 2018, 359, 1056–1061. [Google Scholar] [CrossRef]
- Talbot, S.; Abdulnour, R.E.; Burkett, P.R.; Lee, S.; Cronin, S.J.; Pascal, M.A.; Laedermann, C.; Foster, S.L.; Tran, J.V.; Lai, N.; et al. Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron 2015, 87, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartemes, K.; Chen, C.C.; Iijima, K.; Drake, L.; Kita, H. IL-33-Responsive Group 2 Innate Lymphoid Cells Are Regulated by Female Sex Hormones in the Uterus. J. Immunol. 2018, 200, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Kadel, S.; Ainsua-Enrich, E.; Hatipoglu, I.; Turner, S.; Singh, S.; Khan, S.; Kovats, S. A Major Population of Functional KLRG1(-) ILC2s in Female Lungs Contributes to a Sex Bias in ILC2 Numbers. Immunohorizons 2018, 2, 74–86. [Google Scholar] [CrossRef]
- Laffont, S.; Blanquart, E.; Savignac, M.; Cénac, C.; Laverny, G.; Metzger, D.; Girard, J.P.; Belz, G.T.; Pelletier, L.; Seillet, C.; et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Cephus, J.Y.; Stier, M.T.; Fuseini, H.; Yung, J.A.; Toki, S.; Bloodworth, M.H.; Zhou, W.; Goleniewska, K.; Zhang, J.; Garon, S.L.; et al. Testosterone Attenuates Group 2 Innate Lymphoid Cell-Mediated Airway Inflammation. Cell Rep. 2017, 21, 2487–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Entwistle, L.J.; Wilson, M.S. MicroRNA-mediated regulation of immune responses to intestinal helminth infections. Parisite Immunol. 2017, 39, e12406. [Google Scholar] [CrossRef] [PubMed]
- Pua, H.H.; Ansel, K.M. MicroRNA regulation of allergic inflammation and asthma. Curr Opin Immunol. 2015, 36, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pua, H.H.; Steiner, D.F.; Patel, S.; Gonzalez, J.R.; Ortiz-Carpena, J.F.; Kagevama, R.; Chiou, N.T.; Gallman, A.; de Kouchkovsky, D.; Jeker, L.T.; et al. MircoRNAs 24 and 27 Suppress Allergic Inflammation and Terget a Network of Regulators of T Helper 2 Cell-Associated Cytokine Production. Immunity 2016, 44, 821–832. [Google Scholar] [CrossRef]
- Malmhall, C.; Alawieh, S.; Lu, Y.; Sjostrand, M.; Bossios, A.; Eldh, M.; Radinger, M. MicroRNA-155 is essential for T(H)2-mediated allergen-induced eosinophil inflammation in the lung. J. Allergy Clin. Immunol. 2014, 133, 1429–1438. [Google Scholar] [CrossRef]
- Okoye, I.S.; CZieso, S.; Ktistaki, E.; Roderick, K.; Coomes, S.M.; Pelly, V.S.; Kannan, Y.; Perez-Lloret, J.; Zhao, J.L.; Baltimore, D.; et al. Transcriptomics identified a critical role for the Th2 cell-intrisic miR-155 in mediating allergy and antihelminth immunity. Proc. Natl. Acad. Sci. USA 2014, 111, E3081–E3090. [Google Scholar] [CrossRef]
- Johansson, K.; Malmhall, C.; Ramos-Ramirez, P.; Radinger, M. MicroRNA-155 is a critical regulatory of type 2 innate lymphoid cells and IL-33 signaling in experimental models of allergic airway inflammation. J. Allergy Clin. Immunol. 2017, 139, 1007–1016. [Google Scholar] [CrossRef]
- Knolle, M.D.; Chin, S.B.; Rana, B.M.J.; Englezakis, A.; Nakagawa, R.; Fallon, P.G.; Git, A.; McKenzie, A.N.J. MicroNRA-155 Protects Group 2 Innate Lymphoid Cells From Apoptosis to Promote Type-2 Immunity. Front. Immunol. 2018, 9, 2232. [Google Scholar] [CrossRef]
- Singh, P.B.; Pua, H.H.; Happ, H.C.; Schneider, C.; von Moltke, J.; Locksley, R.M.; Baumjohann, D.; Ansel, K.M. MicroRNA regulation of type 2 innate lymphoid cell homeostasis and function in allerfic inflammation. J. Exp. Med. 2017, 214, 3627–3643. [Google Scholar] [CrossRef]
- Mogilyansky, F.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numberous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D.; Reese, T.A.; Huang, X.; Shinkai, K.; Locksley, R.M. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J. Exp. Med. 2006, 203, 1435–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, A.N.J.; Spits, H.; Eberl, G. Innate lymphoid cells in inflammation and immunity. Immunity 2014, 41, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Lefrancais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haenuki, Y.; Matsushita, K.; Futatsugi-Yumikura, S.; Ishii, K.J.; Kawagoe, T.; Imoto, Y.; Fujieda, S.; Yasuda, M.; Hisa, Y.; Akira, S.; et al. A critical role of IL-33 in experimental allergic rhinitis. J. Allergy Clin. Immunol. 2012, 130, 184–194.e11. [Google Scholar] [CrossRef]
- Cohen, E.S.; Scott, I.C.; Majithiya, J.B.; Rapley, L.; Kemp, B.P.; England, E.; Rees, D.G.; Overed-Sayer, C.L.; Woods, J.; Bond, N.J.; et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat. Commun. 2015, 6, 8327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamijo, S.; Takeda, H.; Tokura, T.; Suzuki, M.; Inui, K.; Hara, M.; Matsuda, H.; Matsuda, A.; Oboki, K.; Ohno, T.; et al. IL-33-mediated innate response and adaptive immune cells contribute to maximum responses of protease allergen-induced allergic airway inflammation. J. Immunol. 2013, 190, 4489–4499. [Google Scholar] [CrossRef]
- Kouzaki, H.; Iijima, K.; Kobayashi, T.; O’Grady, S.M.; Kita, H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J. Immunol. 2011, 186, 4375–4387. [Google Scholar] [CrossRef]
- Savinko, T.; Matikainen, S.; Saarialho-Kere, U.; Lehto, M.; Wang, G.; Lehtimäki, S.; Karisola, P.; Reunala, T.; Wolff, H.; Lauerma, A.; et al. IL-33 and ST2 in atopic dermatitis: Expression profiles and modulation by triggering factors. J. Invest. Dermatol. 2012, 132, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, D.K.; Schwarze, V.; Matta, B.M.; Tkachev, V.; Lieberknecht, E.; Liu, Q.; Koehn, B.H.; Pfeifer, D.; Taylor, P.A.; Prinz, G.; et al. The IL-33/ST2 axis augments effector T-cell responses during acute GVHD. Blood 2015, 125, 3183–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, D.E.; Alexander-Brett, J.; Patel, A.C.; Agapov, E.; Dang-Vu, G.; Jin, X.; Wu, K.; You, Y.; Alevy, Y.; Girard, J.P.; et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J. Clin. Investig. 2013, 123, 3967–3982. [Google Scholar] [CrossRef]
- Hardman, C.S.; Panova, V.; McKenzie, A.N. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur. J. Immunol. 2013, 43, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Muto, T.; Kawagoe, T.; Matsumoto, M.; Sasaki, Y.; Matsushita, K.; Taki, Y.; Futatsugi-Yumikura, S.; Tsutsui, H.; Ishii, K.J.; et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc. Natl. Acad. Sci. USA 2012, 109, 3451–3456. [Google Scholar] [CrossRef] [PubMed]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.H.; Ward, C.K.; Criner, G.J.; et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.L.; Peel, S.; Fox, J.; Panova, V.; Hardman, C.S.; Camelo, A.; Bucks, C.; Wu, X.; Kane, C.M.; Neill, D.R.; et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J. Allergy Clin. Immunol. 2013, 132, 933–941. [Google Scholar] [CrossRef]
- Manetti, M.; Ibba-Manneschi, L.; Liakouli, V.; Guiducci, S.; Milia, A.F.; Benelli, G.; Marrelli, A.; Conforti, M.L.; Romano, E.; Giacomelli, R.; et al. The IL1-like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann. Rheum. Dis. 2010, 69, 598–605. [Google Scholar] [CrossRef]
- Masamune, A.; Watanabe, T.; Kikuta, K.; Satoh, K.; Kanno, A.; Shimosegawa, T. Nuclear expression of interleukin-33 in pancreatic stellate cells. Am. J. Physiol. Gastrointest Liver Physiol. 2010, 299, G821–G832. [Google Scholar] [CrossRef] [Green Version]
- Marvie, P.; Lisbonne, M.; L’Helgoualc’h, A.; Rauch, M.; Turlin, B.; Preisser, L.; Bourd-Boittin, K.; Théret, N.; Gascan, H.; Piquet-Pellorce, C.; et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J. Cell Mol. Med. 2010, 14, 1726–1739. [Google Scholar] [CrossRef]
- Sponheim, J.; Pollheimer, J.; Olsen, T.; Balogh, J.; Hammarström, C.; Loos, T.; Kasprzycka, M.; Sørensen, D.R.; Nilsen, H.R.; Küchler, A.M.; et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am. J. Pathol. 2010, 177, 2804–2815. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, N.E.; Xu, D.; Hepworth, M.R.; Liew, F.Y.; Grencis, R.K. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J. Immunol. 2008, 180, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.Y.; Lewkowich, I.P.; Dawson, L.A.; Downey, J.; Yang, Y.; Smith, D.E.; Herbert, D.R. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc. Natl. Acad. Sci. USA 2013, 110, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Price, A.E.; Liang, H.E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef]
- Salmond, R.J.; Mirchandani, A.S.; Besnard, A.G.; Bain, C.C.; Thomson, N.C.; Liew, F.Y. IL-33 induces innate lymphoid cell-mediated airway inflammation by activating mammalian target of rapamycin. J. Allergy Clin. Immunol. 2012, 130, 1159–1166.e6. [Google Scholar] [CrossRef]
- Wills-Karp, M. Interleukin-13 in asthma pathogenesis. Immunol. Rev. 2004, 202, 175–190. [Google Scholar] [CrossRef]
- Halim, T.Y.; Steer, C.A.; Matha, L.; Gold, M.J.; Martinez-Gonzalez, I.; McNagny, K.M.; McKenzie, A.N.; Takei, F. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014, 40, 425–435. [Google Scholar] [CrossRef]
- Hams, E.; Armstrong, M.E.; Barlow, J.L.; Saunders, S.P.; Schwartz, C.; Cooke, G.; Fahy, R.J.; Crotty, T.B.; Hirani, N.; Flynn, R.J.; et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 367–372. [Google Scholar] [CrossRef]
- Li, D.; Guabiraba, R.; Besnard, A.G.; Komai-Koma, M.; Jabir, M.S.; Zhang, L.; Graham, G.J.; Kurowska-Stolarska, M.; Liew, F.Y.; McSharry, C.; et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J. Allergy Clin. Immunol. 2014, 134, 1422–1432.e11. [Google Scholar] [CrossRef]
- Imai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13921–13926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med. 2013, 5, 170ra16. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Razumilava, N.; Gores, G.J.; Walters, S.; Mizuochi, T.; Mourya, R.; Bessho, K.; Wang, Y.H.; Glaser, S.S.; Shivakumar, P.; et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J. Clin. Investig. 2014, 124, 3241–3251. [Google Scholar] [CrossRef] [PubMed]
- McHedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Monga, S.P. PDGFRalpha in liver pathophysiology: Emerging roles in development, regeneration, fibrosis, and cancer. Gene. Expr. 2015, 16, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, M.W.; Jones, S.W.; Cautivo, K.M.; Dubinin, A.; Ortiz-Carpena, J.F.; Farhat, S.; Yu, K.S.; Lee, K.; Wang, C.; Molofsky, A.V.; et al. Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches. Immunity 2019, 50, 707–722.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlakoiv, T.; Flamar, A.L.; Johnston, L.K.; Moriyama, S.; Putzel, G.G.; Bryce, P.J.; Artis, D. Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci. Immunol. 2019, 4, eaax0416. [Google Scholar] [CrossRef] [PubMed]
- Von Moltke, J.; Ji, M.; Liang, H.E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Gerbe, F.; Sidot, E.; Smyth, D.J.; Ohmoto, M.; Matsumoto, I.; Dardalhon, V.; Cesses, P.; Garnier, L.; Pouzolles, M.; Brulin, B.; et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016, 529, 226–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, K.; Nakajima, H.; Suzuki, K.; Kagami, S.; Hirose, K.; Suto, A.; Saito, Y.; Iwamoto, I. Mast cells produce interleukin-25 upon Fc epsilon RI-mediated activation. Blood 2003, 101, 3594–3596. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.M.; Jang, A.S.; Ahn, M.H.; Shin, J.A.; Kim, J.H.; Choi, Y.S.; Rhim, T.Y.; Park, C.S. Interleukin-25 and interleukin-13 production by alveolar macrophages in response to particles. Am. J. Respir. Cell Mol. Biol. 2005, 33, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Zaph, C.; Du, Y.; Saenz, S.A.; Nair, M.G.; Perrigoue, J.G.; Taylor, B.C.; Troy, A.E.; Kobuley, D.E.; Kastelein, R.A.; Cua, D.J.; et al. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J. Exp. Med. 2008, 205, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- Nadjsombati, M.S.; McGinty, J.W.; Lyons-Cohen, M.R.; Jaffe, J.B.; DiPeso, L.; Schneider, C.; Miller, C.N.; Pollack, J.L.; Gowda, G.A.N.; Fontana, M.F.; et al. Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 2018, 49, 33–41.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.; O’Leary, C.E.; von Moltke, J.; Liang, H.E.; Ang, Q.Y.; Turnbaugh, P.J.; Radhakrishnan, S.; Pellizzon, M.; Ma, A.; Locksley, R.M. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell 2018, 174, 271–284.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Guo, L.; Qiu, J.; Chen, X.; Hu-Li, J.; Siebenlist, U.; Williamson, P.R.; Urban, J.F., Jr.; Paul, W.E. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 2015, 16, 161–169. [Google Scholar] [CrossRef]
- Bailey, J.R.; Bland, P.W.; Tarlton, J.F.; Peters, I.; Moorghen, M.; Sylvester, P.A.; Probert, C.S.; Whiting, C.V. IL-13 promotes collagen accumulation in Crohn’s disease fibrosis by down-regulation of fibroblast MMP synthesis: A role for innate lymphoid cells? PLoS ONE 2012, 7, e52332. [Google Scholar] [CrossRef] [PubMed]
- Camelo, A.; Barlow, J.L.; Drynan, L.F.; Neill, D.R.; Ballantyne, S.J.; Wong, S.H.; Pannell, R.; Gao, W.; Wrigley, K.; Sprenkle, J.; et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J. Gastroenterol. 2012, 47, 1198–1211. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Harrison, O.J. Homeostatic Immunity and the Microbiota. Immunity 2017, 46, 562–576. [Google Scholar] [CrossRef]
- Hornef, M. Pathogens, Commensal Symbionts, and Pathobionts: Discovery and Functional Effects on the Host. ILAR J. 2015, 56, 159–162. [Google Scholar] [CrossRef]
- Langford, T.D.; Housley, M.P.; Boes, M.; Chen, J.; Kagnoff, M.F.; Gillin, F.D.; Eckmann, L. Central importance of immunoglobulin A in host defense against Giardia spp. Infect. Immun. 2002, 70, 11–18. [Google Scholar] [CrossRef]
- Koyama, K.; Tamauchi, H.; Tomita, M.; Kitajima, T.; Ito, Y. B-cell activation in the mesenteric lymph nodes of resistant BALB/c mice infected with the murine nematode parasite Trichuris muris. Parasitol. Res. 1999, 85, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Nunez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Chudnovskiy, A.; Mortha, A.; Kana, V.; Kennard, A.; Ramirez, J.D.; Rahman, A.; Remark, R.; Mogno, I.; Ng, R.; Gnjatic, S.; et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 2016, 167, 444–456.e14. [Google Scholar] [CrossRef] [Green Version]
- Stark, D.; Barratt, J.; Chan, D.; Ellis, J.T. Dientamoeba fragilis, the Neglected Trichomonad of the Human Bowel. Clin. Microbiol. Rev. 2016, 29, 553–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escalante, N.K.; Lemire, P.; Cruz Tleugabulova, M.; Prescott, D.; Mortha, A.; Streutker, C.J.; Girardin, S.E.; Philpott, D.J.; Mallevaey, T. The common mouse protozoa Tritrichomonas muris alters mucosal T cell homeostasis and colitis susceptibility. J. Exp. Med. 2016, 213, 2841–2850. [Google Scholar] [CrossRef]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.C.; Chen, Z.H.; Xue, J.B.; Zhao, D.X.; Lu, C.; Li, Y.H.; Li, S.M.; Du, Y.W.; Liu, Q.; Wang, P.; et al. Infection by the parasitic helminth Trichinella spiralis activates a Tas2r-mediated signaling pathway in intestinal tuft cells. Proc. Natl. Acad. Sci. USA 2019, 116, 5564–5569. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, I.; Matha, L.; Steer, C.A.; Ghaedi, M.; Poon, G.F.; Takei, F. Allergen-Experienced Group 2 Innate Lymphoid Cells Acquire Memory-like Properties and Enhance Allergic Lung Inflammation. Immunity 2016, 45, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Vetter, M.; Neurath, M.F. Emerging oral targeted therapies in inflammatory bowel diseases: Opportunities and challenges. Ther. Adv. Gastroenterol. 2017, 10, 773–790. [Google Scholar] [CrossRef]
- Chaudhry, B.Z.; Cohen, J.A.; Conway, D.S. Sphingosine 1-Phophate Receptor Modulators for the Treatment of Multiple Sclerosis. Neurotherapeutics 2017, 14, 859–873. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burrows, K.; Ngai, L.; Wong, F.; Won, D.; Mortha, A. ILC2 Activation by Protozoan Commensal Microbes. Int. J. Mol. Sci. 2019, 20, 4865. https://doi.org/10.3390/ijms20194865
Burrows K, Ngai L, Wong F, Won D, Mortha A. ILC2 Activation by Protozoan Commensal Microbes. International Journal of Molecular Sciences. 2019; 20(19):4865. https://doi.org/10.3390/ijms20194865
Chicago/Turabian StyleBurrows, Kyle, Louis Ngai, Flora Wong, David Won, and Arthur Mortha. 2019. "ILC2 Activation by Protozoan Commensal Microbes" International Journal of Molecular Sciences 20, no. 19: 4865. https://doi.org/10.3390/ijms20194865
APA StyleBurrows, K., Ngai, L., Wong, F., Won, D., & Mortha, A. (2019). ILC2 Activation by Protozoan Commensal Microbes. International Journal of Molecular Sciences, 20(19), 4865. https://doi.org/10.3390/ijms20194865