Cachexia Anorexia Syndrome and Associated Metabolic Dysfunction in Peritoneal Metastasis

 , , ,
, , ,
Abstract
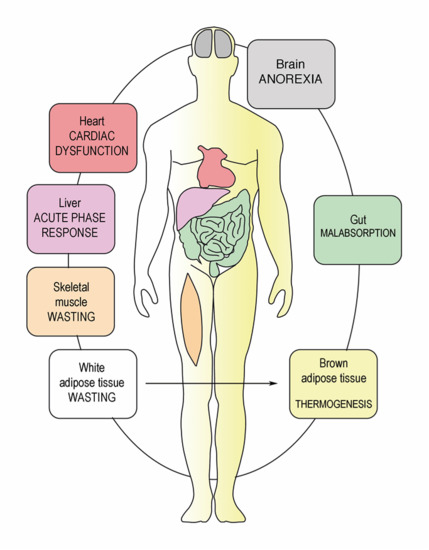
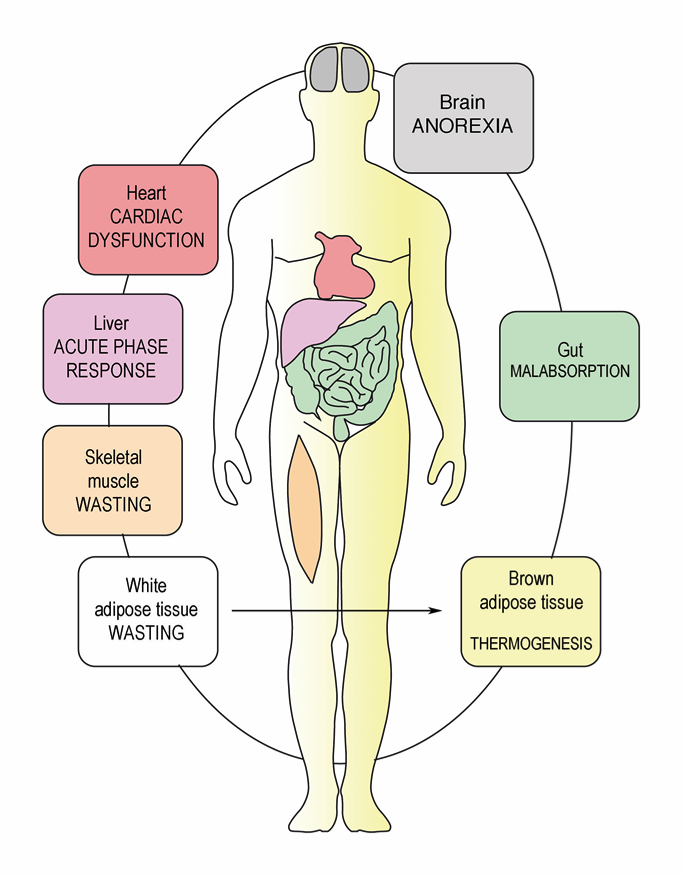
1. Introduction
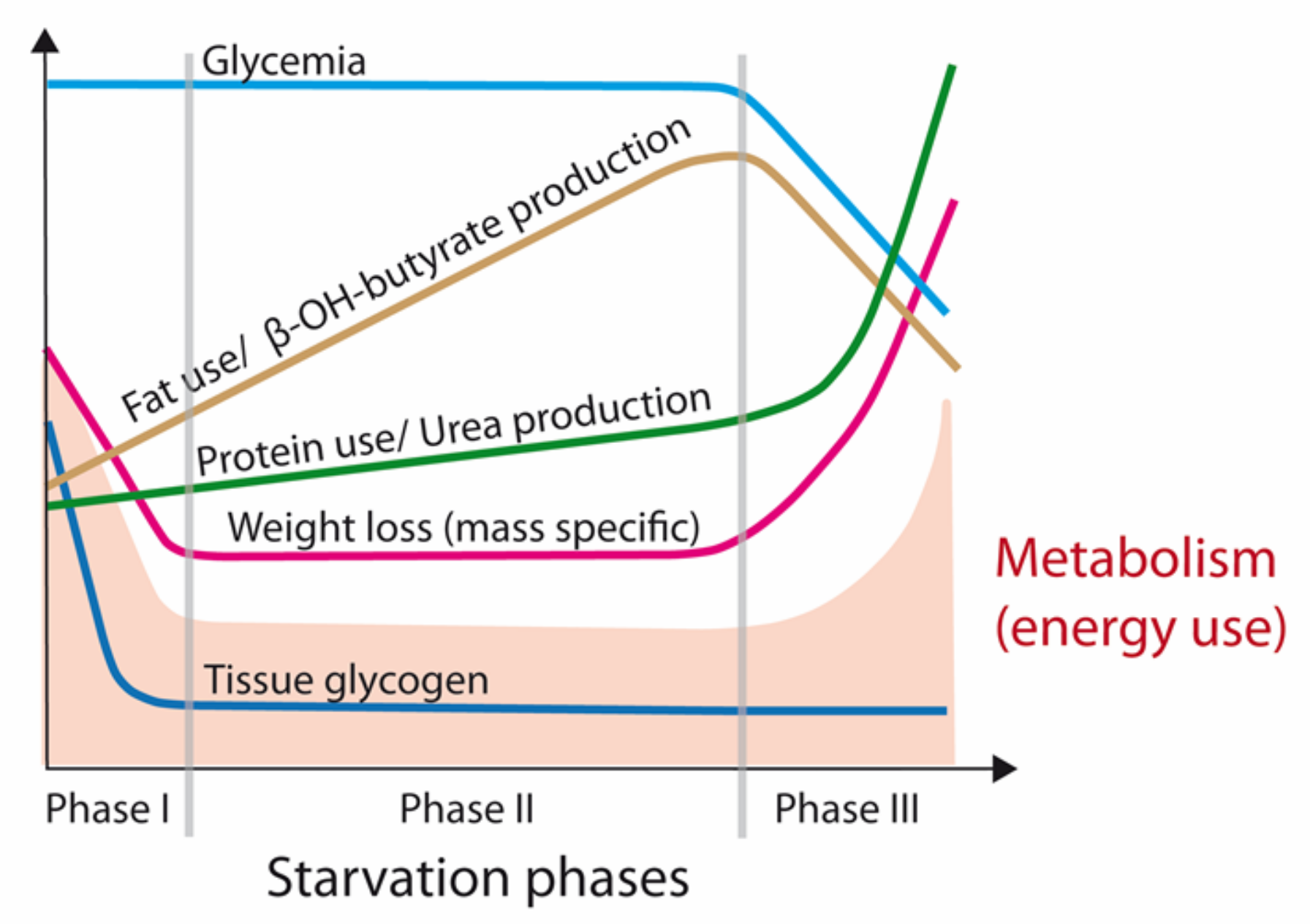
2. Physiology of Starvation
3. Cancer Cachexia Anorexia Syndrome (CAS)
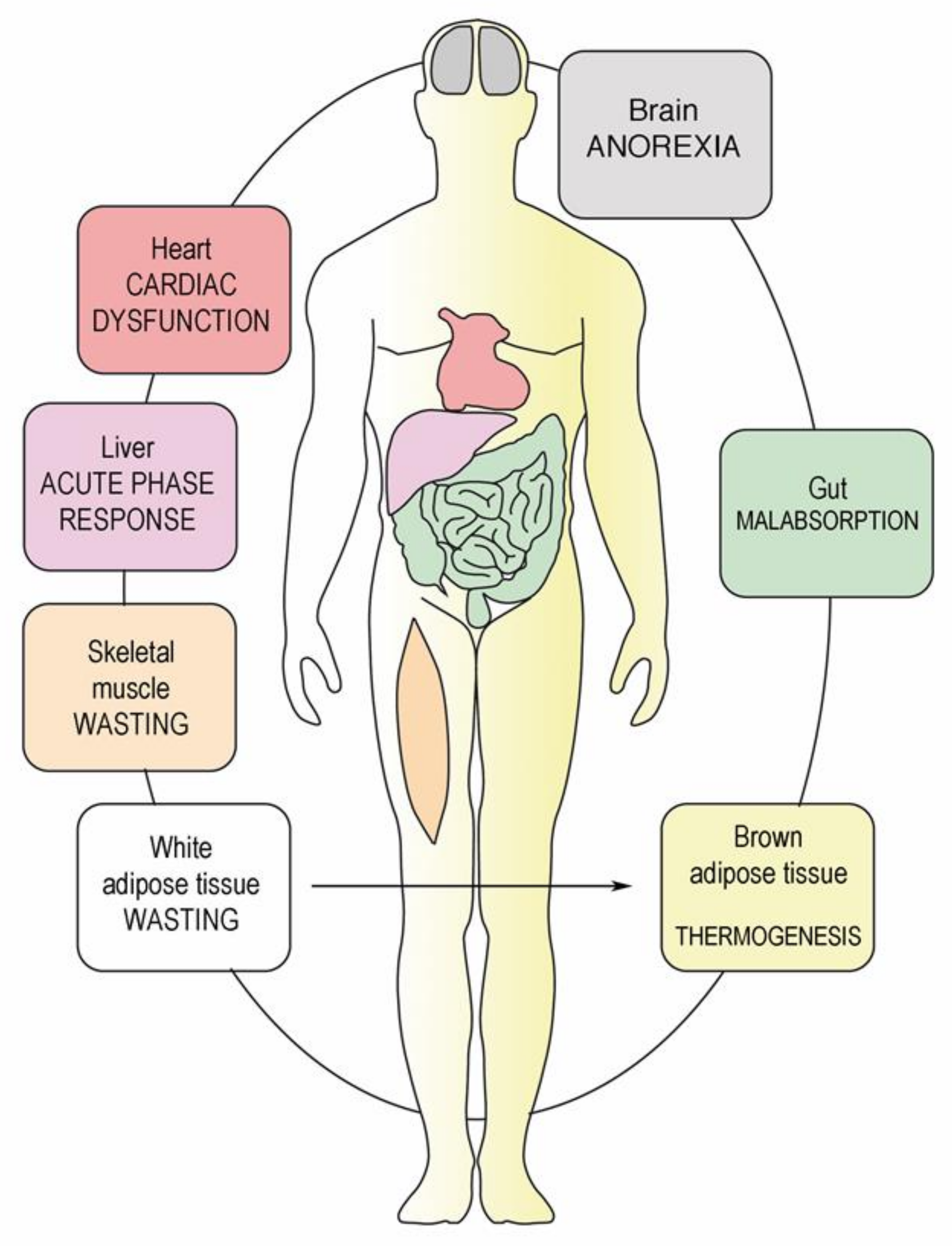
4. Metabolic Dysfunction in Peritoneal Metastasis
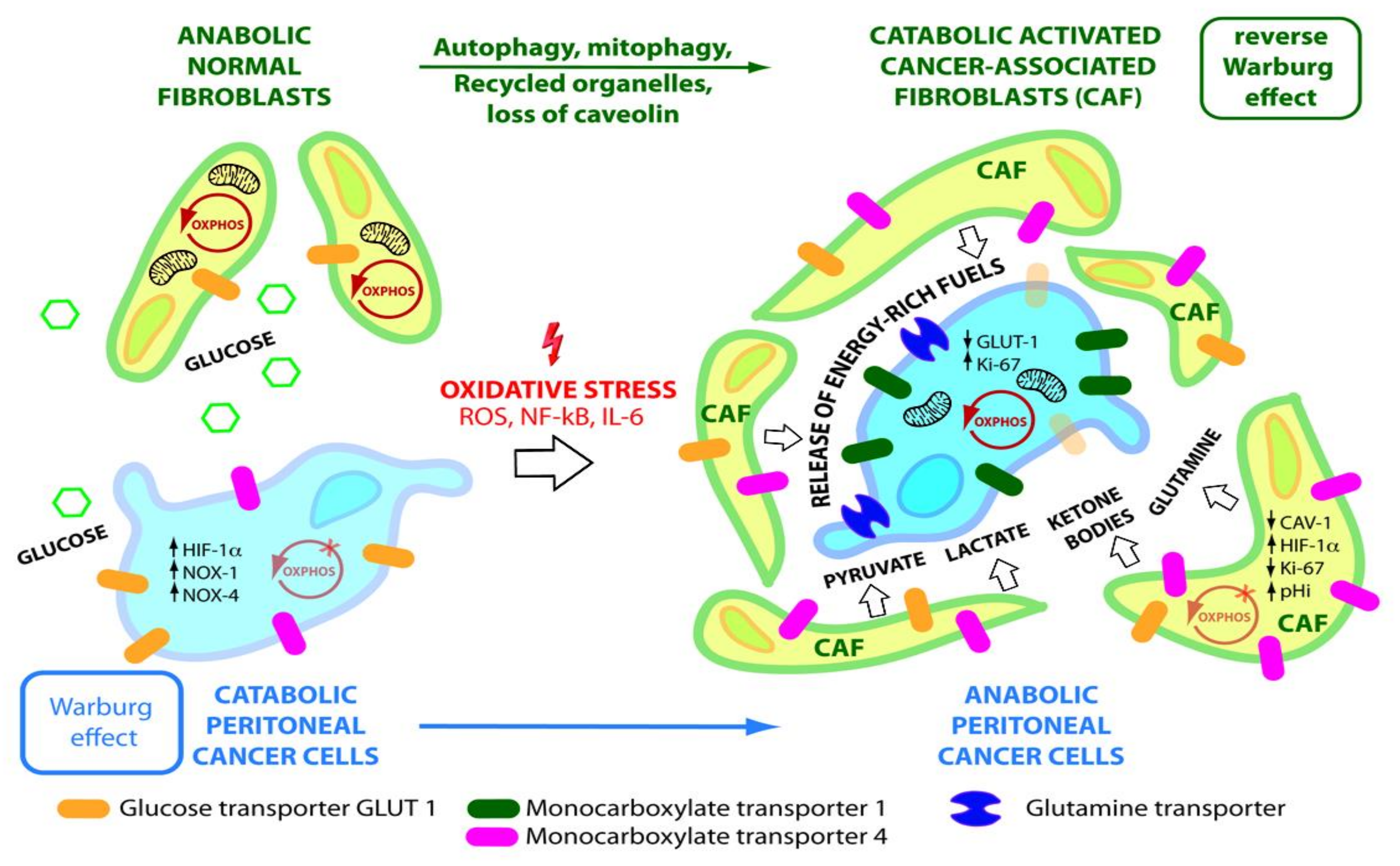
5. Reverse Warburg Effect
6. Host Inflammatory Response and CAS
7. Catabolism of Fat Tissue
8. Breakdown of Muscle Fibers
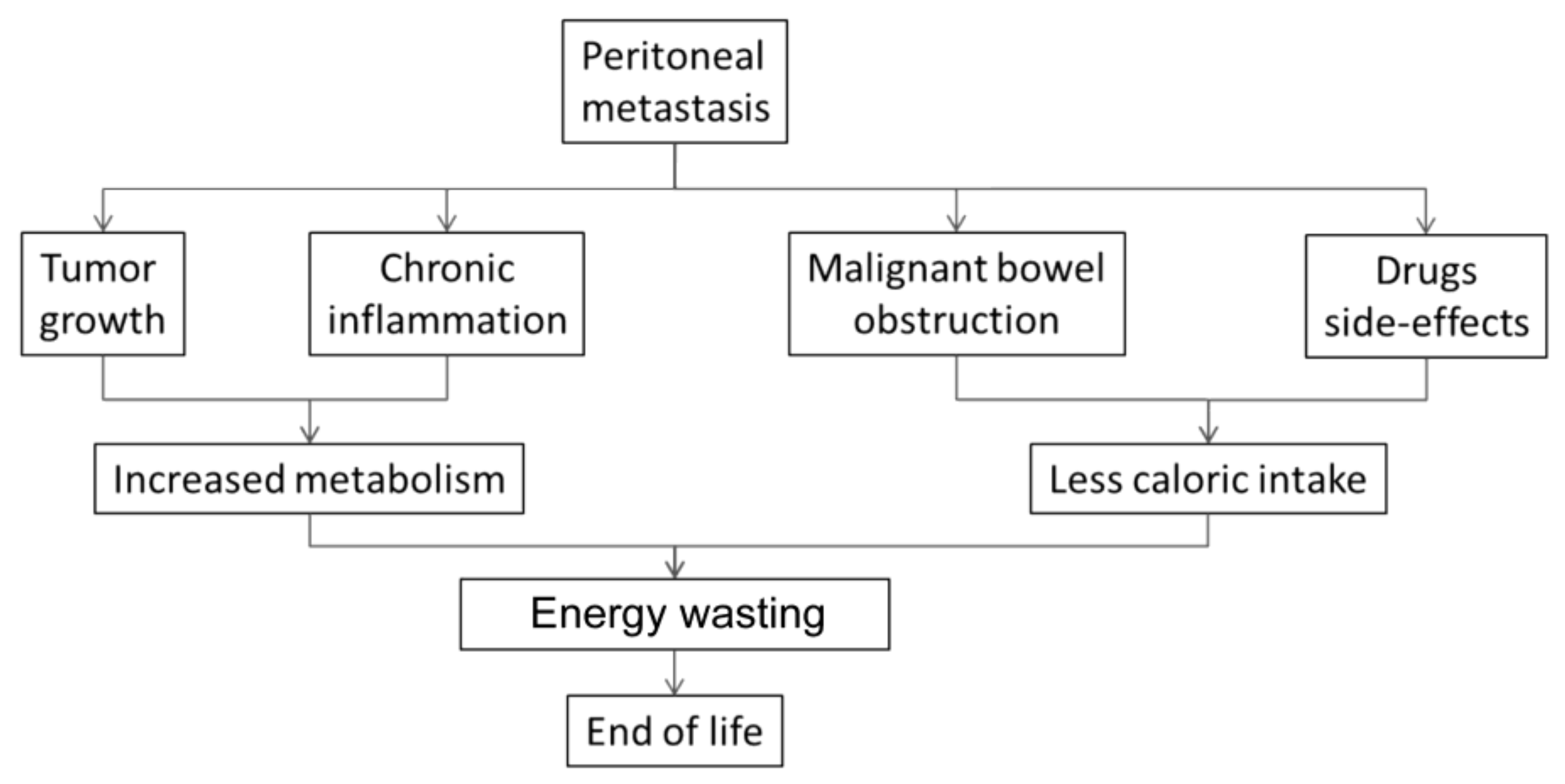
9. Multifactorial Etiology of Cachexia-Anorexia Syndrome in Peritoneal Metastasis
9.1. Bowel Invasion
9.2. Drug-Related Side Effects
9.3. Omental Metastasis
9.4. Systemic Palliative Chemotherapy
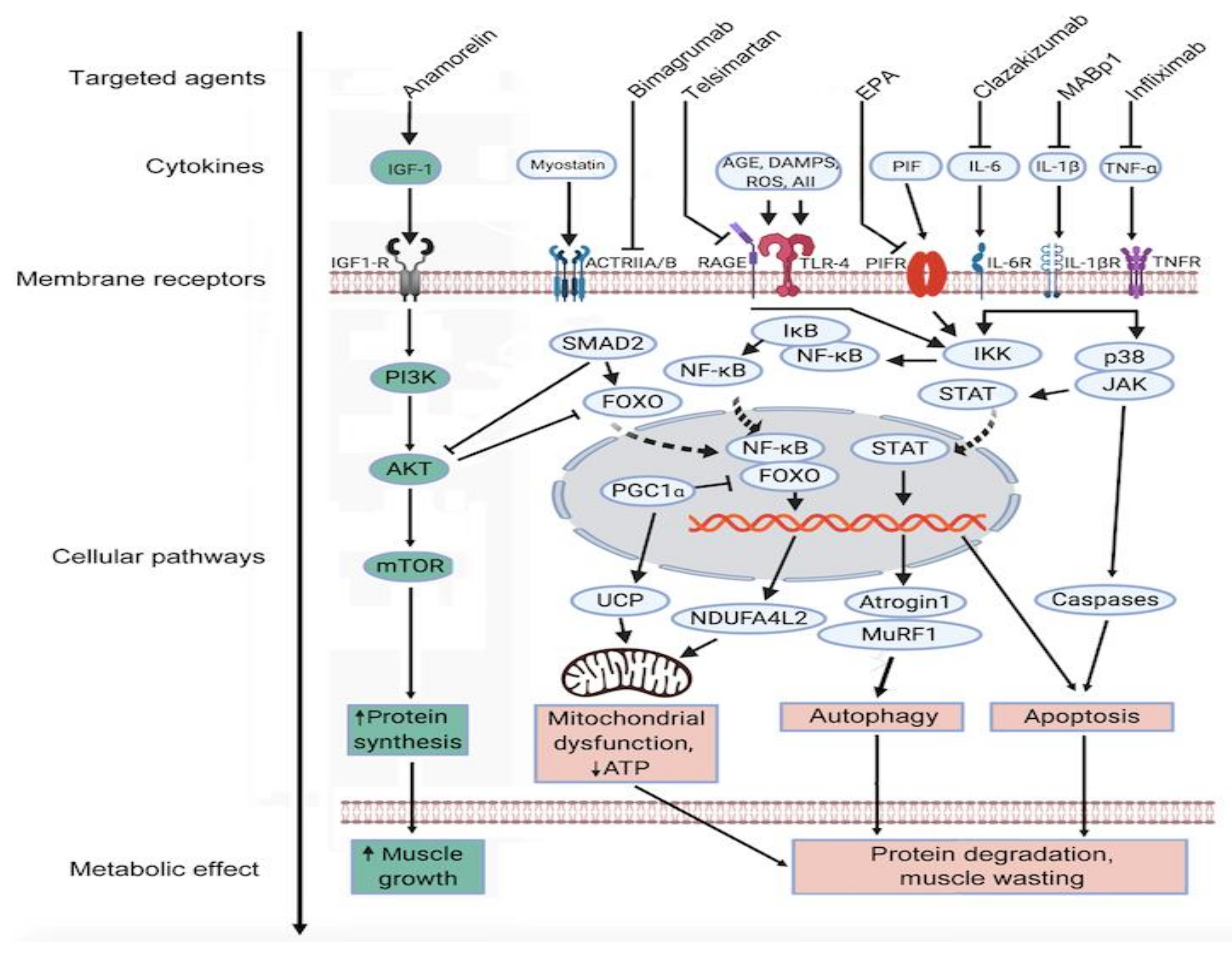
10. Therapy of CAS in PM Patients
11. Conclusions and Outlook
Author Contributions
Funding
Disclosure
Conflicts of Interest
References
- Dev, R. Measuring cachexia-diagnostic criteria. Ann. Palliat. Med. 2019, 8, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Nordhausen, K.; Solass, W.; Demtroeder, C.; Tempfer, C.B.; Reymond, M. Cachexia-anorexia syndrome in patients with peritoneal metastasis: An observational study. Pleura Peritoneum 2016, 1, 57–63. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suzuki, H.; Asakawa, A.; Amitani, H.; Nakamura, N.; Inui, A. Cancer cachexia--pathophysiology and management. J. Gastroenterol. 2013, 48, 574–594. [Google Scholar] [CrossRef]
- Walsh, D.; Donnelly, S.; Rybicki, L. The symptoms of advanced cancer: Relationship to age, gender, and performance status in 1000 patients. Support. Care Cancer 2000, 8, 175–179. [Google Scholar] [CrossRef]
- Childs, D.S.; Jatoi, A. A hunger for hunger: A review of palliative therapies for cancer-associated anorexia. Ann. Palliat. Med. 2019, 8, 50–58. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Lambert, L.A.; Wiseman, J. Palliative Management of Peritoneal Metastases. Ann. Surg. Oncol 2018, 25, 2165–2171. [Google Scholar] [CrossRef]
- Klein, C.; Stiel, S.; Bukki, J.; Ostgathe, C. Pharmacological treatment of malignant bowel obstruction in severely ill and dying patients: A systematic literature review. Schmerz 2012, 26, 587–599. [Google Scholar] [CrossRef]
- Norman, K.; Pichard, C.; Lochs, H.; Pirlich, M. Prognostic impact of disease-related malnutrition. Clin. Nutr. 2008, 27, 5–15. [Google Scholar] [CrossRef]
- Maeda, O.; Ando, T.; Ishiguro, K.; Watanabe, O.; Miyahara, R.; Nakamura, M.; Funasaka, K.; Kazuhiro, F.; Ando, Y.; Goto, H. Safety of repeated cell-free and concentrated ascites reinfusion therapy for malignant ascites from gastrointestinal cancer. Mol. Clin. Oncol. 2014, 2, 1103–1106. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.K.; Makuch, R.; Clamon, G.H.; Feld, R.; Weiner, R.S.; Moran, E.; Blum, R.; Shepherd, F.A.; Jeejeebhoy, K.N.; DeWys, W.D. Limited impact of total parenteral nutrition on nutritional status during treatment for small cell lung cancer. Cancer Res. 1985, 45, 3347–3353. [Google Scholar] [PubMed]
- Rigaud, D.; Hassid, J.; Meulemans, A.; Poupard, A.T.; Boulier, A. A paradoxical increase in resting energy expenditure in malnourished patients near death: The king penguin syndrome. Am. J. Clin. Nutr. 2000, 72, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Secor, S.M.; Carey, H.V. Integrative Physiology of Fasting. Compr. Physiol. 2016, 6, 773–825. [Google Scholar] [CrossRef] [PubMed]
- Soeters, M.R.; Soeters, P.B.; Schooneman, M.G.; Houten, S.M.; Romijn, J.A. Adaptive reciprocity of lipid and glucose metabolism in human short-term starvation. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1397–E1407. [Google Scholar] [CrossRef] [PubMed]
- Belkhou, R.; Bechet, D.; Cherel, Y.; Galluser, M.; Ferrara, M.; le Maho, Y. Effect of fasting and thyroidectomy on cysteine proteinase activities in liver and muscle. Biochim. Biophys. Acta 1994, 1199, 195–201. [Google Scholar] [CrossRef]
- Goodman, M.N.; Larsen, P.R.; Kaplan, M.M.; Aoki, T.T.; Young, V.R.; Ruderman, N.B. Starvation in the rat. II. Effect of age and obesity on protein sparing and fuel metabolism. Am. J. Physiol 1980, 239, E277–E286. [Google Scholar] [CrossRef]
- McCue, M.D.; Terblanche, J.S.; Benoit, J.B. Learning to starve: Impacts of food limitation beyond the stress period. J. Exp. Biol. 2017, 220, 4330–4338. [Google Scholar] [CrossRef]
- Friesen, D.E.; Baracos, V.E.; Tuszynski, J.A. Modeling the energetic cost of cancer as a result of altered energy metabolism: Implications for cachexia. Biol. Med. Model. 2015, 12, 17. [Google Scholar] [CrossRef]
- Cao, D.X.; Wu, G.H.; Zhang, B.; Quan, Y.J.; Wei, J.; Jin, H.; Jiang, Y.; Yang, Z.A. Resting energy expenditure and body composition in patients with newly detected cancer. Clin. Nutr. 2010, 29, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Hilal, Z.; Rezniczek, G.A.; Klenke, R.; Dogan, A.; Tempfer, C.B. Nutritional status, cachexia, and anorexia in women with peritoneal metastasis and intraperitoneal chemotherapy: A longitudinal analysis. J. Gynecol. Oncol. 2017, 28, e80. [Google Scholar] [CrossRef] [PubMed]
- Steinert, R.; Hantschick, M.; Vieth, M.; Gastinger, I.; Kuhnel, F.; Lippert, H.; Reymond, M.A. Influence of subclinical tumor spreading on survival after curative surgery for colorectal cancer. Arch. Surg. 2008, 143, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.B.; Solass, W.; Archid, R.; Weinreich, F.J.; Konigsrainer, A.; Reymond, M.A. Resistance to anoikis in transcoelomic shedding: The role of glycolytic enzymes. Pleura Peritoneum 2019, 4, 20190003. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Kim, J.M.; Sung, M.K. The Efficacy of Oral Nutritional Intervention in Malnourished Cancer Patients: A Systemic Review. Clin. Nutr. Res. 2016, 5, 219–236. [Google Scholar] [CrossRef]
- Porporato, P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Wilson, R.B. Hypoxia, cytokines and stromal recruitment: Parallels between pathophysiology of encapsulating peritoneal sclerosis, endometriosis and peritoneal metastasis. Pleura Peritoneum 2018, 3, 20180103. [Google Scholar] [CrossRef]
- Rempel, A.; Mathupala, S.P.; Griffin, C.A.; Hawkins, A.L.; Pedersen, P.L. Glucose catabolism in cancer cells: Amplification of the gene encoding type II hexokinase. Cancer Res. 1996, 56, 2468–2471. [Google Scholar]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Kluza, J.; Corazao-Rozas, P.; Touil, Y.; Jendoubi, M.; Maire, C.; Guerreschi, P.; Jonneaux, A.; Ballot, C.; Balayssac, S.; Valable, S.; et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012, 72, 5035–5047. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed. Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal-epithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cachexia and sarcopenia: Mechanisms and potential targets for intervention. Curr. Opin. Pharmacol. 2015, 22, 100–106. [Google Scholar] [CrossRef]
- Capparelli, C.; Whitaker-Menezes, D.; Guido, C.; Balliet, R.; Pestell, T.G.; Howell, A.; Sneddon, S.; Pestell, R.G.; Martinez-Outschoorn, U.; Lisanti, M.P.; et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 2012, 11, 2272–2284. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Roy, A.; Bera, S. CAF cellular glycolysis: Linking cancer cells with the microenvironment. Tumour. Biol. 2016, 37, 8503–8514. [Google Scholar] [CrossRef]
- Shangguan, C.; Gan, G.; Zhang, J.; Wu, J.; Miao, Y.; Zhang, M.; Li, B.; Mi, J. Cancer-associated fibroblasts enhance tumor (18)F-FDG uptake and contribute to the intratumor heterogeneity of PET-CT. Theranostics 2018, 8, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Rath, S.; Adhami, V.M.; Mukhtar, H. Hypoxia driven glycation: Mechanisms and therapeutic opportunities. Semin. Cancer Biol. 2018, 49, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; del Castillo, M.D.; de la Maza, M.P.; Filip, R.; Gugliucci, A.; Luevano-Contreras, C.; Macias-Cervantes, M.H.; Markowicz Bastos, D.H.; Medrano, A.; Menini, T.; et al. Dietary advanced glycation end products and their role in health and disease. Adv. Nutr. 2015, 6, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Livesey, K.M.; Schapiro, N.E.; Lotze, M.T.; Zeh, H.J. The Receptor for Advanced Glycation End-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid. Redox Signal. 2011, 15, 2175–2184. [Google Scholar] [CrossRef]
- Riuzzi, F.; Sorci, G.; Sagheddu, R.; Chiappalupi, S.; Salvadori, L.; Donato, R. RAGE in the pathophysiology of skeletal muscle. J. Cachexia Sarcopenia Muscle 2018, 9, 1213–1234. [Google Scholar] [CrossRef]
- Sessa, L.; Gatti, E.; Zeni, F.; Antonelli, A.; Catucci, A.; Koch, M.; Pompilio, G.; Fritz, G.; Raucci, A.; Bianchi, M.E. The receptor for advanced glycation end-products (RAGE) is only present in mammals, and belongs to a family of cell adhesion molecules (CAMs). PLoS ONE 2014, 9, e86903. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Y. Nonmetabolic functions of metabolic enzymes in cancer development. Cancer Commun. 2018, 38, 63. [Google Scholar] [CrossRef]
- Tazzari, M.; Brich, S.; Tuccitto, A.; Bozzi, F.; Beretta, V.; Spagnuolo, R.D.; Negri, T.; Stacchiotti, S.; Deraco, M.; Baratti, D.; et al. Complex Immune Contextures Characterise Malignant Peritoneal Mesothelioma: Loss of Adaptive Immunological Signature in the More Aggressive Histological Types. J. Immunol. Res. 2018, 2018, 5804230. [Google Scholar] [CrossRef]
- Shrotriya, S.; Walsh, D.; Nowacki, A.S.; Lorton, C.; Aktas, A.; Hullihen, B.; Benanni-Baiti, N.; Hauser, K.; Ayvaz, S.; Estfan, B. Serum C-reactive protein is an important and powerful prognostic biomarker in most adult solid tumors. PLoS ONE 2018, 13, e0202555. [Google Scholar] [CrossRef]
- Banks, W.A. Anorectic effects of circulating cytokines: Role of the vascular blood-brain barrier. Nutrition 2001, 17, 434–437. [Google Scholar] [CrossRef]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol. 2015, 7, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Schcolnik-Cabrera, A.; Chavez-Blanco, A.; Dominguez-Gomez, G.; Duenas-Gonzalez, A. Understanding tumor anabolism and patient catabolism in cancer-associated cachexia. Am. J. Cancer Res. 2017, 7, 1107–1135. [Google Scholar] [PubMed]
- Coss, C.C.; Clinton, S.K.; Phelps, M.A. Cachectic Cancer Patients: Immune to Checkpoint Inhibitor Therapy? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5787–5789. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Mazurak, V.C. Evidence and mechanisms of fat depletion in cancer. Nutrients 2014, 6, 5280–5297. [Google Scholar] [CrossRef] [PubMed]
- Tsoli, M.; Swarbrick, M.M.; Robertson, G.R. Lipolytic and thermogenic depletion of adipose tissue in cancer cachexia. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2016; Volume 54, pp. 68–81. [Google Scholar] [CrossRef]
- Dalal, S. Lipid metabolism in cancer cachexia. Ann. Palliat. Med. 2019, 8, 13–23. [Google Scholar] [CrossRef]
- Das, S.K.; Eder, S.; Schauer, S.; Diwoky, C.; Temmel, H.; Guertl, B.; Gorkiewicz, G.; Tamilarasan, K.P.; Kumari, P.; Trauner, M.; et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011, 333, 233–238. [Google Scholar] [CrossRef]
- Argiles, J.M.; Lopez-Soriano, F.J.; Busquets, S. Mediators of cachexia in cancer patients. Nutrition 2019, 66, 11–15. [Google Scholar] [CrossRef]
- Moley, J.F.; Aamodt, R.; Rumble, W.; Kaye, W.; Norton, J.A. Body cell mass in cancer-bearing and anorexic patients. J. Parenter Enter. Nutr. 1987, 11, 219–222. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers. 2018, 4, 17105. [Google Scholar] [CrossRef]
- Carr, R.M.; Enriquez-Hesles, E.; Olson, R.L.; Jatoi, A.; Doles, J.; Fernandez-Zapico, M.E. Epigenetics of cancer-associated muscle catabolism. Epigenomics 2017, 9, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Schneyer, A.L. The biology of activin: Recent advances in structure, regulation and function. J. Endocrinol. 2009, 202, 1–12. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C. Metabolic Functions of Myostatin and Gdf11. Immunol. Endocr. Metab. Agents Med. Chem. 2010, 10, 217–231. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Loumaye, A.; de Barsy, M.; Nachit, M.; Lause, P.; Frateur, L.; van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J.P. Role of Activin A and myostatin in human cancer cachexia. J. Clin. Endocrinol. Metab. 2015, 100, 2030–2038. [Google Scholar] [CrossRef]
- Busquets, S.; Toledo, M.; Orpi, M.; Massa, D.; Porta, M.; Capdevila, E.; Padilla, N.; Frailis, V.; Lopez-Soriano, F.J.; Han, H.Q.; et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J. Cachexia Sarcopenia Muscle 2012, 3, 37–43. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Stephens, N.A.; Skipworth, R.J.; Macdonald, A.J.; Greig, C.A.; Ross, J.A.; Fearon, K.C. Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. J. Cachexia Sarcopenia Muscle 2011, 2, 111–117. [Google Scholar] [CrossRef]
- Antoun, S.; Lanoy, E.; Iacovelli, R.; Albiges-Sauvin, L.; Loriot, Y.; Merad-Taoufik, M.; Fizazi, K.; di Palma, M.; Baracos, V.E.; Escudier, B. Skeletal muscle density predicts prognosis in patients with metastatic renal cell carcinoma treated with targeted therapies. Cancer 2013, 119, 3377–3384. [Google Scholar] [CrossRef]
- Aubrey, J.; Esfandiari, N.; Baracos, V.E.; Buteau, F.A.; Frenette, J.; Putman, C.T.; Mazurak, V.C. Measurement of skeletal muscle radiation attenuation and basis of its biological variation. Acta Physiol. 2014, 210, 489–497. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Kelley, D.E.; Thaete, F.L.; He, J.; Ross, R. Skeletal muscle attenuation determined by computed tomography is associated with skeletal muscle lipid content. J. Appl. Physiol. 2000, 89, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Prado, C.M.; Baracos, V.E.; McCargar, L.J.; Reiman, T.; Mourtzakis, M.; Tonkin, K.; Mackey, J.R.; Koski, S.; Pituskin, E.; Sawyer, M.B. Sarcopenia as a determinant of chemotherapy toxicity and time to tumor progression in metastatic breast cancer patients receiving capecitabine treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 2920–2926. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, Y.S.; Kim, E.Y.; Jin, W. Prognostic significance of CT-determined sarcopenia in patients with advanced gastric cancer. PLoS ONE 2018, 13, e0202700. [Google Scholar] [CrossRef] [PubMed]
- Lieffers, J.R.; Mourtzakis, M.; Hall, K.D.; McCargar, L.J.; Prado, C.M.; Baracos, V.E. A viscerally driven cachexia syndrome in patients with advanced colorectal cancer: Contributions of organ and tumor mass to whole-body energy demands. Am. J. Clin. Nutr. 2009, 89, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Laghi, A.; Bellini, D.; Rengo, M.; Accarpio, F.; Caruso, D.; Biacchi, D.; Di Giorgio, A.; Sammartino, P. Diagnostic performance of computed tomography and magnetic resonance imaging for detecting peritoneal metastases: Systematic review and meta-analysis. Radiol. Med. 2017, 122, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Goswami, G.; Kammar, P.; Mangal, R.; Shaikh, S.; Patel, M.D.; Bhatt, A. Accuracy of CT Scan in Predicting the Surgical PCI in Patients Undergoing Cytoreductive Surgery with/without HIPEC-a Prospective Single Institution Study. Indian J. Surg. Oncol. 2019, 10, 296–302. [Google Scholar] [CrossRef]
- Drewes, A.M.; Munkholm, P.; Simren, M.; Breivik, H.; Kongsgaard, U.E.; Hatlebakk, J.G.; Agreus, L.; Friedrichsen, M.; Christrup, L.L. Definition, diagnosis and treatment strategies for opioid-induced bowel dysfunction-Recommendations of the Nordic Working Group. Scand. J. Pain 2016, 11, 111–122. [Google Scholar] [CrossRef]
- Muller-Lissner, S.; Bassotti, G.; Coffin, B.; Drewes, A.M.; Breivik, H.; Eisenberg, E.; Emmanuel, A.; Laroche, F.; Meissner, W.; Morlion, B. Opioid-Induced Constipation and Bowel Dysfunction: A Clinical Guideline. Pain Med. 2017, 18, 1837–1863. [Google Scholar] [CrossRef]
- Singh, K.; Kober, K.M.; Paul, S.M.; Hammer, M.; Wright, F.; Conley, Y.P.; Levine, J.D.; Miaskowski, C. Gastrointestinal symptoms are associated with trajectories of chemotherapy-induced nausea. Support. Care Cancer 2019. [Google Scholar] [CrossRef]
- Cherwin, C.H. Gastrointestinal symptom representation in cancer symptom clusters: A synthesis of the literature. Oncol. Nurs. Forum 2012, 39, 157–165. [Google Scholar] [CrossRef]
- Argiles, J.M.; Lopez-Soriano, F.J.; Stemmler, B.; Busquets, S. Therapeutic strategies against cancer cachexia. Eur J. Transl. Myol. 2019, 29, 7960. [Google Scholar] [CrossRef] [PubMed]
- Dev, R.; Wong, A.; Hui, D.; Bruera, E. The Evolving Approach to Management of Cancer Cachexia. Oncology 2017, 31, 23–32. [Google Scholar] [PubMed]
- Garcia, J.M. What is next after anamorelin? Curr. Opin. Support. Palliat. Care 2017, 11, 266–271. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.; Creedon, B. Pharmacological treatment of bowel obstruction in cancer patients. Expert Opin. Pharmacother. 2011, 12, 2205–2214. [Google Scholar] [CrossRef]
- Sharkey, K.A.; Wiley, J.W. The Role of the Endocannabinoid System in the Brain-Gut Axis. Gastroenterology 2016, 151, 252–266. [Google Scholar] [CrossRef]
- Jatoi, A.; Windschitl, H.E.; Loprinzi, C.L.; Sloan, J.A.; Dakhil, S.R.; Mailliard, J.A.; Pundaleeka, S.; Kardinal, C.G.; Fitch, T.R.; Krook, J.E.; et al. Dronabinol versus megestrol acetate versus combination therapy for cancer-associated anorexia: A North Central Cancer Treatment Group study. J. Clin. Oncol. 2002, 20, 567–573. [Google Scholar] [CrossRef]
- Cannabis In Cachexia Study, G.; Strasser, F.; Luftner, D.; Possinger, K.; Ernst, G.; Ruhstaller, T.; Meissner, W.; Ko, Y.D.; Schnelle, M.; Reif, M.; et al. Comparison of orally administered cannabis extract and delta-9-tetrahydrocannabinol in treating patients with cancer-related anorexia-cachexia syndrome: A multicenter, phase III, randomized, double-blind, placebo-controlled clinical trial from the Cannabis-In-Cachexia-Study-Group. J. Clin. Oncol. 2006, 24, 3394–3400. [Google Scholar] [CrossRef]
- Malik, Z.; Baik, D.; Schey, R. The role of cannabinoids in regulation of nausea and vomiting, and visceral pain. Curr. Gastroenterol. Rep. 2015, 17, 429. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Starvation Response | Cancer Cachexia Anorexia Syndrome (CAS) |
|---|---|
| Physiologic response to low energy intake | Pathologic response (inflammation, cytokines, hormonal changes) |
| Preserved appetite and ghrelin response | Ghrelin resistance and loss of appetite |
| Decreased REE | Increased REE |
| Use of fat > muscle stores | Use of both fat and muscle for energy production |
| Protein catabolism reduced | Increased protein turnover |
| Reversible | Difficult to reverse |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Archid, R.; Solass, W.; Tempfer, C.; Königsrainer, A.; Adolph, M.; Reymond, M.A.; Wilson, R.B. Cachexia Anorexia Syndrome and Associated Metabolic Dysfunction in Peritoneal Metastasis. Int. J. Mol. Sci. 2019, 20, 5444. https://doi.org/10.3390/ijms20215444
Archid R, Solass W, Tempfer C, Königsrainer A, Adolph M, Reymond MA, Wilson RB. Cachexia Anorexia Syndrome and Associated Metabolic Dysfunction in Peritoneal Metastasis. International Journal of Molecular Sciences. 2019; 20(21):5444. https://doi.org/10.3390/ijms20215444
Chicago/Turabian StyleArchid, Rami, Wiebke Solass, Clemens Tempfer, Alfred Königsrainer, Michael Adolph, Marc A. Reymond, and Robert B. Wilson. 2019. "Cachexia Anorexia Syndrome and Associated Metabolic Dysfunction in Peritoneal Metastasis" International Journal of Molecular Sciences 20, no. 21: 5444. https://doi.org/10.3390/ijms20215444
APA StyleArchid, R., Solass, W., Tempfer, C., Königsrainer, A., Adolph, M., Reymond, M. A., & Wilson, R. B. (2019). Cachexia Anorexia Syndrome and Associated Metabolic Dysfunction in Peritoneal Metastasis. International Journal of Molecular Sciences, 20(21), 5444. https://doi.org/10.3390/ijms20215444