Abstract
The last century has witnessed rapid domestication of the narrow-leafed lupin (Lupinus angustifolius L.) as a grain legume crop, exploiting discovered alleles conferring low-alkaloid content (iucundus), vernalization independence (Ku and Julius), and reduced pod shattering (lentus and tardus). In this study, a L. angustifolius mapping population was subjected to massive analysis of cDNA ends (MACE). The MACE yielded 4185 single nucleotide polymorphism (SNP) markers for linkage map improvement and 30,595 transcriptomic profiles for expression quantitative trait loci (eQTL) mapping. The eQTL highlighted a high number of cis- and trans-regulated alkaloid biosynthesis genes with gene expression orchestrated by a regulatory agent localized at iucundus locus, supporting the concept that ETHYLENE RESPONSIVE TRANSCRIPTION FACTOR RAP2-7 may control low-alkaloid phenotype. The analysis of Ku shed light on the vernalization response via FLOWERING LOCUS T and FD regulon in L. angustifolius, providing transcriptomic evidence for the contribution of several genes acting in C-repeat binding factor (CBF) cold responsiveness and in UDP-glycosyltransferases pathways. Research on lentus selected a DUF1218 domain protein as a candidate gene controlling the orientation of the sclerified endocarp and a homolog of DETOXIFICATION14 for purplish hue of young pods. An ABCG transporter was identified as a hypothetical contributor to sclerenchyma fortification underlying tardus phenotype.
1. Introduction
The narrow-leafed lupin, Lupinus angustifolius L., is a grain legume crop, appreciated as an organic fertilizer that improves soil structure and productivity, as well as providing a source of protein for human and animals. This species has witnessed rapid domestication during the last century. Several important agronomic traits have been identified and transferred into improved germplasm [1]. These traits include, among others, vernalization independence (overlapping loci Ku and Julius), low-alkaloid content (iucundus), reduced pod shattering (tardus and lentus), soft seededness (mollis), white flower color (leucospermus) and anthracnose resistance (Lanr1).
Vernalization responsiveness is the natural adaptation to climatic conditions, based on the requirement of a prolonged low temperature period during germination to induce flowering [2,3]. Natural dominant mutation in the so-called Ku or Julius loci diminished the need of vernalization and enabled temperature-independent sowing of L. angustifolius [4].
A high level of quinolizidine alkaloids is a typical feature of primitive populations in many lupin species, as these chemical compounds protect plants from pests and fungi [5], however, alkaloids are major antinutritional factors and provide bitter taste [6,7]. Three unlinked low-alkaloid recessive alleles were identified in L. angustifolius, and one of them, iucundus, was extensively implemented in breeding [8,9]. Some germplasm resources having less than 0.01% of grain alkaloid have been developed [10,11].
Shattering of dry pods is natural process of seed dispersal, however, it is a very undesired trait in modern agriculture because it dramatically decreases harvested yield. Two unlinked recessive alleles contribute to reduced pod shattering in L. angustifolius, namely tardus, affecting sclerenchyma strips of the dorsal and ventral pod seams, and lentus, modifying the orientation of the sclerified endocarp of the pod [12,13].
L. angustifolius is natively adapted to the Mediterranean climate which has hot dry summers, because of one of its survival strategies which is impermeability of seed coat to water. Hard-seeded germplasm has a long dormancy period and irregular germination. Recessive soft-seediness allele mollis confers water permeability and efficient seed germination [14]. It is the most difficult domestication L. angustifolius allele for breeding because the desired phenotype is maternally determined [15].
The agronomic potential of L. angustifolius has been reduced by high susceptibility to anthracnose, caused by the pathogenic fungus, Colletotrichum lupini (Bondar) Nirenberg, Feiler and Hagedorn [16]. The resistance to anthracnose in L. angustifolius was revealed to be controlled by several single dominant genes that were discovered in different germplasm resources, namely, Lanr1 in cultivar Tanjil, AnMan in cv. Mandelup, and LanrBo in the breeding line Bo7212 [17,18,19].
Several genes contribute to L. angustifolius seed and flower color. The most widely exploited is the recessive allele leucospermus, affecting anthocyanin synthesis and resulting in bright seeds and white flowers [1].
To generate numerous molecular markers for agronomic trait selection in narrow-leafed lupin breeding programs, microsatellite-anchored fragment length polymorphisms (MFLP) fingerprinting has been exploited [20,21]. Trait-associated markers have been developed for iucundus (marker iucLi) [22], Ku (KuHM1) [23], mollis (MoLi) [15], lentus (LeM1, LeM2 and LeLi) [13,24], and tardus (TaLi, TaM1 and TaM2) [25,26]. Narrow-leafed lupin genomic studies have been greatly facilitated by the incremental development of linkage map carrying sequence-defined markers [27,28,29,30], construction of nuclear genome bacterial artificial chromosome (BAC) libraries [31,32], and assembly of the draft genome sequence [30,33,34].
Recently, a new method of transcriptome-based genotyping-by-sequencing, called massive analysis of cDNA ends (MACE), has been developed [35]. The MACE provides markers anchored in 3′-ends of transcribed sequences, and therefore is directly matching active RNA content of the genome. First implementations highlighted the relevance of the MACE for sequence polymorphism detection, gene expression quantification, transcript-based marker development, and candidate gene identification [36,37,38,39,40,41]. In this study, the MACE protocol was used for development of polymorphic gene-based markers and for quantification of gene expression in mapping population of L. angustifolius. These new data were exploited for construction of a linkage map and for determination of expression quantitative trait loci (eQTLs) related to selected domestication traits.
2. Results and Discussion
2.1. Development of New Polymorphic Markers
The MACE protocol was applied for 89 RILs and for parental lines of L. angustifolius mapping population (83A:476 × P27255), yielding 11,864 markers. A total of 9304 markers were localized within gene sequences whereas 2560 markers were found in loci lacking any annotation. There were 4185 MACE markers retained after application of total missing data threshold (counting heterozygotes and no data scores), followed by inference of consensus segregation for genes represented by several single nucleotide polymorphism, SNPs. There were 3532 genes represented by single markers, four genes were represented by pairs of markers with heterogeneous segregation patterns, and 645 markers were localized in unannotated loci. The annotation of markers is provided in the Table S1.
The MACE is a method providing sequences anchored in the 3′-ends of mRNA and can be used to develop sequence-defined markers, as well as to quantify gene expression [35,36]. In this study both applications of the MACE protocol were exploited, providing molecular markers and gene expression scores related to the same RNA isolates. The MACE marker set was supplemented with 10 newly developed BAC-end derived PCR markers, namely five dCAPS (019A15_3, 026O16_3, 034M08_5, 043N19_3, and 103O20_3), four CAPS (061O23_3, 085K20_5, 085L14_3, and 128I22_5), and one allele-specific PCR marker (085L14_5). The BAC-end based marker allele sequences were deposited in NCBI Genbank under accession numbers (MN518055-MN518073). Information on primer pair sequences, PCR primer annealing temperature, PCR product lengths, enzyme used for polymorphism detection, and restriction product lengths for both alleles is provided in Table S2. For the past 15 years, the use of BAC-derived PCR markers has been a method of choice in studies involving L. angustifolius genome physical and linkage mapping. Because the L. angustifolius karyotype carries numerous small and very uniform chromosomes, the BAC-derived markers have been frequently used as chromosome-specific landmarks to validate physical linkage of particular genome regions, as well as to facilitate assignment of particular chromosomes to linkage groups [31,42,43,44,45,46,47]. The BAC-derived markers have also been exploited for fine mapping of a region carrying a candidate gene for vernalization independence Ku locus, as well as for comparative mapping of genes from isoflavonoid and fatty acid synthesis pathways [48,49,50,51]. In this study, BAC-derived markers were developed to localize on the linkage map some clones identified during our previous studies and confirmed to carry repetitive elements.
2.2. Construction of a Linkage Map
Markers used for linkage map development included 4,185 MACE and 10 BAC-end PCR markers developed in this study as well as previously published data including seven trait loci (Ku, tardus, lentus, mollis, leucospermus, iucundus and Lanr1) [21], eight trait related markers (TaM1, TaM2, LeM1, LeM2, KuHM1, AntjM2, MoA, MoLi) [13,15,24,25,52,53], and 109 BAC-derived markers anchoring particular linkage groups to chromosomes [43,44,45,46,47,49,50,51,54]. The segregation data for markers used for linkage mapping and the calculated χ2 p-values of distortion from expected 1:1 segregation are provided in Table S3. There were 4309 markers localized on the genetic map, which constituted 20 linkage groups, carrying from 144 to 304 markers (215 on average) and 16 markers remained unmapped (Table 1).

Table 1.
Characteristics of the MACE-based L. angustifolius linkage map.
The segregation pattern of 59.7% of the markers was redundant, and therefore the map contains 1735 loci, namely from 60 to 120 loci per linkage group. The lengths of linkage groups vary from 78.5 to 156.38 cM, reaching 2163.63 cM in total. The results of linkage mapping are provided in Table S4. A high percentage of markers matching particular linkage groups with corresponding pseudochromosomes, reaching from 96.6% (chromosome NLL-16) to 100% (chromosomes NLL-14 and NLL-20), highlighted the collinearity between published L. angustifolius draft genome sequence [34] and this version of linkage map (Figure 1a). Major issues were found for chromosomes NLL-16 (block of eight adjacent markers representing ~300 kbp localized in the linkage group NLL-01), NLL-15 (block of six adjacent markers covering ~200 kbp mapped in the linkage group NLL-17), and NLL-12 (five markers mapped in the linkage group NLL-14). As many as 209 unassembled scaffolds were localized on the genetic map, namely from six to 22 scaffold per linkage group (Figure 1b). A comparison of the number of markers assigned to particular chromosomes, scaffolds, and linkage groups is provided in Table S5.
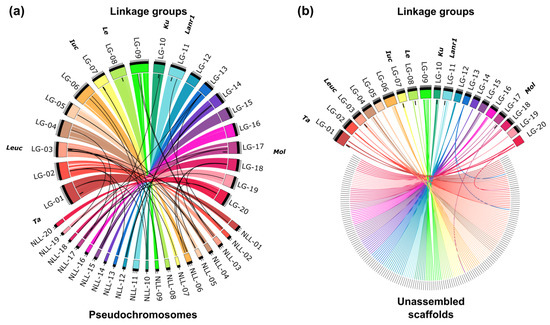
Figure 1.
Collinearity links matching narrow-leafed lupin linkage groups (LG-01–LG-20) and: (a) pseudochromosomes (NLL-01–NLL-20) and (b) unassembled scaffolds. Ribbons symbolize homologous links identified by DNA sequence similarity. Chromosomes and linkage groups are drawn to scale indicated by ticks (10 Mbp and 10 cM). Postions of the following major domestication loci are indicated: Tardus (Ta), leucospermus (Leuc), iucundus (Iuc), lentus (Le), Ku, Lanr1, and mollis (Mol).
Mapping data from the most recent L. angustifolius linkage maps [30,34] were not incorporated to our map due to a limited number of RIL lines common for all three studies (about 70), as well as due to observed inconsistency in segregation patterns between physically linked markers originating from different studies, indicating diverse genetic origin of some RILs having the same numbers assigned, putatively resulting from seed admixture or cross-pollination during seed multiplication. Seeds of the mapping population were shared between the Department of Agriculture and Food Western Australia and the Institute of Plant Genetics, Polish Academy of Sciences, in the year 2003, and maintained independently thereafter. As lupin breeding was recently licensed to the private sector in Australia it may be currently impossible to access original set of RILs developed for this mapping population. Similar issues with possible cross-pollination during mapping population development have also been reported for 43 RILs from the recently published linkage map of yellow lupin, L. luteus [55], as well as for one RIL in white lupin, L. albus [56]. Nevertheless, the total number of RILs used in the most recent L. angustifolius genome mapping study, namely 87 lines with only 78 lines overlapping with previous mapping studies, was too low to provide the high resolution required for significant improvement of genome assembly, and resulted in high marker redundancy, reaching 89.9% [30].
2.3. Gene Expression Profiling, Gene Ontology Enrichment, and Expression Quantitative Trait Loci Mapping
The MACE analysis provided normalized gene expression levels for all RILs analyzed. Namely, 30,595 genes revealed nonzero expression for at least 1 RIL, 25,024 genes for at least 30% of RILs, 23,557 genes for at least 50% of RILs, and 15,686 genes for all RILs. The normalized gene expression values for mapping population and parental lines (83A:476 and P27255) are provided in Table S6. The gene expression patterns in the mapping population were associated with domestication trait segregation (wild alleles used as positive values). Genes with a statistically significant association (FDR p-value threshold of 0.01) were identified for all domestication traits analyzed, namely 98 genes for iucundus, 50 for Ku, 35 for leucospermus, 29 for lentus, 17 for tardus, 11 for Lanr1 and five for mollis. The values of the t-Student test association between domestication trait segregation and gene expression patterns, including FDR correction and statistical significance analysis, are provided in Table S7. The gene ontology (GO) enrichment analysis of genes with expression pattern associated with iucundus trait segregation highlighted lysine biosynthesis and lysine metabolism, as well as cofactor binding and coenzyme binding, as the most overrepresented processes and functions, respectively (Table S8). This was an expected outcome as quinolizidine alkaloids are derived from lysine via a series of chemical reactions [57]. GO analysis for tardus-associated genes revealed iron-sulfur cluster assembly, metallo-sulfur cluster assembly, and cofactor biosynthesis process enrichments. No statistically significant GO enrichments were identified for genes associated with Ku, leucospermus, lentus, mollis and Lanr1 traits.
Composite interval mapping revealed the presence of numerous eQTL peaks close to domestication trait loci. Within a genetic linkage distance of 2 cM from a particular domestication trait locus, from one (mollis) to 61 (iucundus) genes had eQTL peaks localized (Table 2). The LOD values for eQTL permutation test are provided in Table S9, whereas data on eQTL localization on the linkage map are provided in Table S10. Some potential candidate genes were identified for all analyzed loci, except anthracnose resistance locus Lanr1. As plants were not inoculated to allow long-range gene profiling during their development, including the generative phase, genes related to anthracnose were putatively not activated in the experiment. Anthracnose resistance will be addressed in another study. Here, genes identified for iucundus, Ku, lentus, tardus, mollis and leucospermus are discussed.
Table 2.
Expression quantitative trait loci localized near (≤2 cM) major domestication trait loci.
2.4. Genes Identified for Low-Alkaloid Iucundus Locus
The high number of genes revealed for iucundus locus might be related to the complexity of the alkaloid biosynthesis process and the number of genes involved. Taking into consideration the position of gene coding sequences in the genome, only a relatively small fraction of eQTL genes revealed for iucundus (18%) was cis-regulated, whereas the vast majority (74%) was trans-regulated. Furthermore, from 34 genes for iucundus that had association values between their expression patterns and trait segregation above 0.5 or below −0.5, as many as 30 revealed a positive association with wild, high alkaloid phenotype. Moreover, as many as 16 genes highly associated with iucundus revealed to have their major eQTL locus explaining more than 50% of their observed expression variance localized directly at iucundus, or very close to it (Table 3). Many of these genes are hypothesized to be involved in alkaloid biosynthesis process. Such an observation strongly supports a hypothesis that iucundus locus in L. angustifolius encodes a single regulatory agent controlling this complex secondary metabolic pathway and differentiating between high and low quinolizidine alkaloid biosynthesis profiles.
Table 3.
Genes showing the highest gene expression association and eQTL peak co-localization (≤2 cM) with low-alkaloid iucundus locus.
Among the genes with expression positively associated with high alkaloid phenotypes in the RIL population, the highest LOD values of eQTLs were revealed for Lup009726 (OIV96299.1, LOD 37.3), Lup028431 (OIV96574.1, LOD 35.4), Lup015923 (OIW10551.1, LOD 29.4), and Lup007628 (OIV89004.1, LOD 29.2) (Figure 2a). The Lup009726 product revealed 99.5% sequence identity to lysine/ornithine decarboxylase LDC (BAK32797.1) protein which catalyzes the first step of quinolizidine alkaloid biosynthesis [57]. Expression of the LDC gene has been confirmed to be associated with alkaloid content in L. angustifolius by several independent studies [58,59,60]. Lup028431 encodes purine permease transporter 1 (PUP1), PUP proteins which are generally involved in alkaloid biosynthesis and transport. Nicotine uptake permease from Nicotiana tabacum (NtPUP1), for example, affects nicotine metabolism, as well as regulates the ETHYLENE RESPONSE FACTOR 189, a key transcription factor in nicotine biosynthesis pathway [61,62]. Indeed, Lup028431 was selected in another study as potential L. angustifolius quinolizidine alkaloid biosynthetic gene because it revealed similar expression pattern to the LDC gene [63]. Lup015923 has been annotated as MAJOR LATEX PROTEIN 423 (MLP423, AT1G24020), which is hypothesized to be involved in stress responsive activation of biosynthetic pathway of coumestrol, a coumestan isoflavone in soybean [64]. Lup015923 was recently highlighted as one of candidate quinolizidine alkaloid biosynthesis genes in L. angustifolius due to highly elevated expression in bitter P27255 accession [58]. Lup007628 (OIV89004.1) is the ETHYLENE RESPONSIVE TRANSCRIPTION FACTOR RAP2-7, a candidate locus for iucundus, evidenced by a gene expression study involving transcriptome sequencing of four accessions and quantitative RT-PCR profiling of 14 accessions differing in alkaloid content, as well as by molecular marker development and linkage mapping [59,60]. Interestingly, closely located to RAP2.7 at iucundus locus, another cis-regulated component, Fe SUPEROXIDE DISMUTASE 2 (Lup007632, OIV89008.1), revealed similarly high LOD and explained eQTL variance values, but opposite direction of association. Other iucundus-associated genes have included Lup005321 (OIW13431.1) and Lup005322 (OIW13432.1) encoding homologs of DIHYDROFLAVONOL 4-REDUCTASE which is one of the key genes from anthocyanin biosynthesis pathway [65]. The set of genes with highly significant eQTLs localized in the iucundus region also includes a Lup021586 (OIV95196.1) gene encoding HXXXD-type ACYL-TRANSFERASE (LaAT, AB581532.1). The expression profile of LaAT has been highly associated with alkaloid content in L. angustifolius [58,59,60,66]. Moreover, one of the homologs of this gene, LAGI01_35805, has been recently designated as a candidate gene underlying low-alkaloid pauper locus in L. albus, as evidenced by linkage mapping and validation survey in a set of 127 bitter and 23 sweet accessions [56].
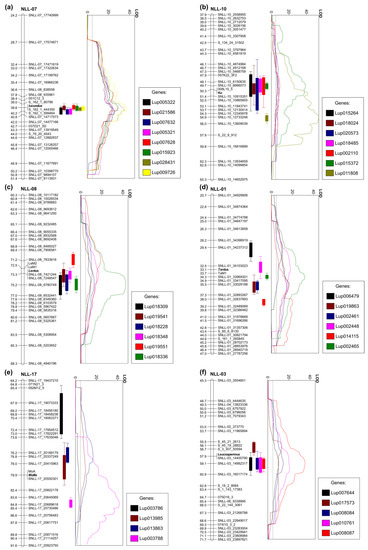
Figure 2.
Major expression quantitative trait loci (eQTLs) revealed for narrow-leafed lupin domestication trait loci: (a) main alkaloid content iucundus locus, (b) vernalization responsiveness Ku locus, (c) pod shattering lentus locus, (d) pod shattering tardus locus, (e) soft seededness mollis locus, and (f) white flower color leucospermus locus. Linear plots show LOD values (threshold 4.8), whereas vertical bar graphs visualize eQTL ranges (outer, LODmax-2 and inner, LODmax-1) on corresponding linkage group fragments. Linkage groups are drawn to scale indicated by ticks and labels.
In addition to Lup007628 and Lup007632, nine other cis-regulated genes revealed eQTLs localized at iucundus locus, including three hypothetical components of alkaloid biosynthesis pathways, Lup007706 (OIW07664.1), Lup017658 (OIW07643.1), and Lup032669 (OIW21355.1). Lup007706 encodes a representative of a S-adenosyl-l-methionine-dependent methyltransferases superfamily protein. N-methylation of quinolizidine alkaloids was confirmed to occur in crude protein extracts from Laburnum anagyroides carrying S-adenosyl-l-methionine: cytisine N-methyltransferase [67]. Moreover, a homolog of S-adenosyl-l-methionine-dependent N methyltransferase catalyzes a nitrogen methylation involved in vindoline alkaloid biosynthesis in Madagascar periwinkle (Catharanthus roseus) [68]. Lup017658 encodes a 4-HYDROXY-TETRAHYDRODIPICOLINATE SYNTHASE gene which is generally involved in biosynthesis of l-lysine, a precursor of quinolizidine alkaloids. This gene revealed considerably elevated expression in bitter accessions of L. angustifolius, indicating its hypothetical involvement in alkaloid biosynthesis pathway [59]. Lup032669 encodes the CARBOXYLESTERASE 1 gene. CARBOXYLESTERASE 1 was evidenced to be involved in one of the final three steps of noscapine alkaloid biosynthesis [69]. To summarize, this study highlighted a relatively high number of alkaloid biosynthesis genes with gene expression orchestrated by a regulatory agent(s) localized at iucundus locus. This study provided novel evidences supporting the concept that RAP2.7 may control low-alkaloid iucundus phenotype, however, further evidence would require cis-trans tests. Nonetheless, such studies are hampered by very low transformation efficiency in narrow-leafed lupin [70].
2.5. Genes Revealed for Vernalization Independence Ku Locus
The P27255 parent is late flowering and requires vernalization for flowering induction, whereas the 83A:476 parent is early flowering and vernalization independent. In this study, seeds were subjected to vernalization procedure to ensure transition from vegetative to generative phase in all analyzed RILs. Such an approach could result in diminishing of some differences in expression profiles of vernalization-responsive genes between early and late flowering RILs. However, despite this partial pre-sowing vernalization, relatively a large number of genes revealed to have their eQTL peaks closely localized to Ku locus (Table 4, Figure 2b). Contrary to iucundus, the same ratio of cis- and trans-regulation for major eQTL loci was observed (44%). Genes showing the highest gene expression association and eQTL peak co-localization (≤2 cM) with Ku included Lup011808 (OIW03171.1, LOD 41.0) and Lup015372 (OIW20567.1, LOD 38.9). Lup015372 encodes hypothetical uncharacterized protein, whereas Lup011808 a homolog of A. thaliana CALCIUM/CALMODULIN-REGULATED RECEPTOR-LIKE KINASE 1, CRLK1. CRLK1 confers cold responsiveness in plants via C-repeat binding factors (CBF) pathway [71]. Overexpression of the CBF in Arabidopsis delays flowering by promoting the expression of FLOWERING LOCUS C (FLC), indicating a link between cold signaling and flowering time regulation [72]. One of the downstream genes in this pathway is INDUCER OF CBF EXPRESSION 1 (ICE1) which integrates cold signals into FLC-mediated flowering pathway [73]. Another cross-talk between cold response and flowering initiation pathways, involving a SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1) gene was also identified [74]. A recent study confirmed that CBF pathway affects flowering time but does not affect vernalization response in Arabidopsis [75]. Moreover, a calcium and calmodulin-binding protein kinase (NtCBK1) from N. tabacum functions as a negative regulator of flowering; high levels of NtCBK1 in the shoot apical meristem extended the vegetative phase of growth [76]. Indeed, CRLK1, in this study, was revealed to be positively associated with late flowering phenotype. These observations bring attention to the hypothetical involvement of calcium and calmodulin link with FLC pathway in flowering time regulation in L. angustifolius. Legume genomes do not contain FLC homologs but other genes from this pathway, including activators and repressors of FLC, are present [77].
Table 4.
Genes showing the highest gene expression association and eQTL peak LOD co-localization (≤2 cM) with vernalization independence Ku locus.
Other Ku eQTLs include Lup011781 (OIW03144.1), Lup011739 (OIV96743.1), Lup011836 (OIW03199.1), and Lup002110 (OIW20134.1) sequences. Lup011781 has been identified as a homolog of MHM17-10 (AT5G56980) gene. It is pathogen-associated molecular pattern-induced gene with unknown function, putatively participating in jasmonic acid pathway [78]. Lup011739 encodes a homolog of GALACTURONOSYLTRANSFERASE-LIKE 10, which is involved in cell wall organization and its expression is regulated by FLAVIN-BINDING KELCH REPEAT, F-BOX 1 (FKF1), blue light receptor, and well-known photoperiodic flowering time regulator [79]. Lup011836 encodes general transcription factor IIH subunit 2 (GTF2H2), performing basic functions in transcription and nucleotide excision repair of damaged DNA. Lup002110 has been annotated as a representative of the UDP-glycosyltransferases protein family. One of the A. thaliana UDP-glycosyltransferases, UGT87A2, was revealed to be involved in the regulation of flowering in vernalization and gibberellin pathways via the flowering repressor FLC [80]. UGT87A2 vs. OIW20134.1 protein alignment revealed 96% coverage, 31% identical sites, and 46% positive sites.
As plants in this study were subjected to a moderate vernalization procedure, differences in gene expression resulting from variation in vernalization responsiveness should be reduced as compared with those expected for nonvernalized plants. This reduction might be highlighted by a relatively low LOD value (as compared with other eQTLs from this region) of the major L. angustifolius gene underlying vernalization responsiveness, a homolog of A. thaliana FLOWERING LOCUS T (FT), LanFTc1 (Lup015264, OIW03334.1, LOD 7.0) [49,81]. As expected, LanFTc1 revealed negative association with late flowering phenotype. Similar LOD values were also revealed for Lup018024 (OIV92673.1), Lup020573 (OIW03269.1), and Lup018485 (OIW19675.1) genes. Lup018024 encodes a homolog of bZIP transcription factor, FD, which mediates signals from the FT gene at the shoot apex and promotes plant flowering in general [82]. However, in this study, FD expression revealed a positive association with late flowering phenotype. In Arabidopsis, the FT-FD complex induces the transcription of several floral-promoting genes, such as SOC1 and FRUITFULL (FUL), which accelerate flowering, as well as APETALA1 (AP1) and LEAFY (LFY), which control floral meristem identity [83]. Indeed, Lup018485 revealed similarity to AGAMOUS-LIKE 8 (AGL8)/ FUL and AP1 MADS-box transcription factors and was found to be negatively associated with late flowering phenotype. Lup020573 was annotated as MYB60 transcription factor. MYB transcription factors perform various regulatory functions in plants in responses to biotic and abiotic stresses, development, differentiation, metabolism, defense, etc. [84].
To summarize, analysis of Ku eQTLs shed light on vernalization pathway in L. angustifolius, providing transcriptomic evidence for the contribution of several genes acting upstream of FLC in CBF and UDP-glycosyltransferases pathways. The study also revealed transcriptomic contribution of conserved mechanism of FT-FD regulon on transition from vegetative to generative development phase in L. angustifolius.
2.6. Genes Profiled for Lentus and Tardus Pod Shattering Loci
The recessive lentus (le) allele changes the orientation of the sclerified endocarp in the pod, substantially reducing torsional forces after drying [12]. In this study, the following three genes highly associated with lentus were identified to have major eQTL peaks localized in the proximity of this locus: Lup018336 (OIW06948.1, LOD 43.0), Lup018348 (OIW06960.1, LOD 17.4), and Lup018228 (OIW06846.1, LOD 13.0) (Table 5, Figure 2c). All these genes originated from the same region at chromosome NLL-08, carrying lentus. Lup018336 encoded a homolog of A. thaliana fiber protein carrying DUF1218 domain. The genome of A. thaliana contained 15 members of DUF1218 genes. Members of the DEAL subfamily of the DUF1218 confer bilateral symmetry of Arabidopsis leaves by controlling proper coordination of cell proliferation between different domains of the leaf lamina margin [85]. Another group of DUF1218 genes has been related to secondary cell wall biosynthesis and includes AtUNKA (At4g27435), MODIFYING WALL LIGNIN-1, and MODIFYING WALL LIGNIN-2 (At1g31720/MWL-1 and At4g19370/MWL-2) [86,87,88]. Lup018348 encodes a homolog of DETOXIFICATION14, a member of the multidrug and toxic compound extrusion (MATE efflux) family [89]. MATE transporters perform various functions including phytohormone transport, secondary metabolite transport, xenobiotic detoxification, aluminium tolerance, disease resistance, tip growth processes, and senescence [90]. Some MATE proteins have been involved in the transport of anthocyanins or proanthocyanidins to vacuoles and in the flavonoid metabolism pathways [91,92]. Anthocyanins are accumulated in cell vacuoles and are responsible for diverse pigmentation from orange to red, purple, and blue [93]. Interestingly, Lup018336 revealed positive gene expression association with pod shattering phenotype, whereas Lup018348 was positively associated with nonshattering pods. These results are in line with the general observation that le allele affects a pod pigmentation, resulting in a purplish hue of young pods and a bright yellowish-brown color on the internal surface of mature pods. Lup018348 may be responsible for this pigmentation, whereas Lup018336 for pod shattering in L. angustifolius.
Table 5.
Genes showing the highest gene expression association and eQTL peak LOD co-localization (≤2 cM) with anthracnose resistance Lanr1, white flower color leucospermus, soft seededness mollis, and pod shattering tardus and lentus loci.
The recessive tardus (ta) allele affects the sclerenchyma strips of the dorsal and ventral pod seams, greatly increasing the fusion of two pod halves and moderately hampering their separation when drying [12]. Two genes revealed high association and eQTL peak co-localization with tardus, namely Lup002465 (OIW17837.1) and Lup002448 (OIW17820.1) (Table 5, Figure 2d). Lup002465 encodes BolA-like family protein with unknown function. Lup002448 is a G family ATP-binding ABC transporter. Such a transporter in rice (RCN1) is required for hypodermal suberization of roots [94]. Similarly, ABCG1 confers suberin formation in potato tuber periderm [95]. Some ABCG transporters are involved in sclerenchyma fiber development via monolignol transport in lignin biosynthesis pathway [96,97]. Moreover, one of ABCG transporters has been revealed to be involved in the silicon-induced formation of Casparian bands in the exodermis of rice [98]. ABCG transporters also perform other diverse functions, including abiotic and biotic stress responses, however, these examples provide non-negligible support to select Lup002448 as a candidate gene involved in tardus trait.
2.7. Gene Related to the Soft Seededness Mollis Allele
Recessive allele mollis provides water permeable testa at maturity [14,99]. Seed dormancy in legumes is related to the deposition of phenolics and, hypothetically, development of suberin-impregnated layers of palisade cells as observed in pea and soybean [100,101]. Only one highly associated gene was revealed by eQTL analysis, Lup013985 (OIW15058.1), annotated as a protein FAM32A/7-dehydrocholesterol reductase (homolog of A. thaliana DWARF5 gene) (Table 5, Figure 2e). Because the sequence homology of these genes between L. angustifolius and Arabidopsis is quite low, it is difficult to elucidate a particular function by comparative analysis. It can be concluded that it is putatively a gene involved in plant sterol metabolism. Plant sterols are essential structural components that influence biophysical properties of membranes such as permeability and fluidity [102]. Mutation in one of enzymes contributing to steryl glycoside biosynthesis pathway, UDP-Glc:sterol glycosyltransferase, alters embryonic development, seed suberin accumulation, and cutin formation in the seed coat, resulting in abnormal permeability [103]. Recently, it has been evidenced that a maternally deposited endosperm cuticle underlies this seed coat permeability in A. thaliana [104]. Mollis is also maternally determined and as such is considered to be the most difficult L. angustifolius domestication gene for selection by phenotype observation. Lup013985 cannot be considered to be a candidate gene conferring mollis allele, because it is located in different chromosome than mollis locus, however, it might be considered to be a hypothetical trans-regulated component eventually contributing to mollis phenotype.
2.8. Genes with eQTL Loci Matching White Flower Color Leucospermus Allele
Recessive leucospermus allele confers white flower and bright seed pigmentation in L. angustifolius. A similar trait in pea was conferred by a basic-helix-loop-helix (bHLH) transcription factor [105] but eQTL analysis did not highlight any bHLH transcription factor with LOD peak close to leucospermus locus. Two genes with expression positively associated with recessive allele revealed eQTL peaks close to leucospermus, namely Lup008087 (OIW21684.1) and (Lup017573) OIV97389.1 (Table 5, Figure 2f). Lup008087 encodes ubiquitin-60S ribosomal protein L40 (RPL40A) isoform and is localized in Scaffold_168_4 mapped in this study in linkage group NLL-03 close to leucospermus, however, a particular biological function of RPL40 gene is unknown. Lup017573, from the chromosome NLL-15, revealed similarity to the NRT1 and PTR family proteins. Three eQTLs revealed a negative association with recessive allele, including Lup008084 (OIW15287.1) annotated as F-BOX/WD repeat-containing protein. Interestingly, a single mutation in an F-BOX domain-containing protein, OsFBX310, confers brown hull phenotype in rice resulting from a high content of total flavonoids and anthocyanins [106], however, putatively due to the large evolutionary distance between monocots and dicots, sequence alignment reveled very limited similarity between the OsFBX310 and OIW15287.1 protein sequences.
2.9. Applicability of MACE for Gene-Based Studies
This study is the first report on exploitation of the MACE for L. angustifolius genome and transcriptome analysis. As a method for gene expression analysis, the MACE was first used in chronic kidney disease survey [35] and in de novo transcriptome analysis of Calliphora vicina pupae [37]. The MACE was also exploited for stem rust transcriptomic response in perennial ryegrass (Lolium perenne), highlighting a candidate LpPg1 resistance gene and yielding numerous SNPs which were further transformed into PCR-based molecular markers [36]. The MACE protocol was also applied in pea (Pisum sativum) providing single nucleotide variants subsequently converted into CAPS markers [38]. Furthermore, MACE-based studies in pea resulted in the identification of a new mutant allele of the key nodulation gene Sym33 [107]. The MACE was also used for transcriptomic profiling of Phaseolus vulgaris seeds and Solanum lycopersicum pollen [39,40]. The MACE was also exploited for GWAS, tagging several candidate genes for salt stress tolerance in Triticum aestivum [41].
Several previous L. angustifolius genotyping approaches were based on diversity arrays technology (DArT) profiling. DArT studies have highlighted low genetic diversity in narrow-leafed lupin breeding material as compared with primitive and wild germplasm [108]. This domestication bottleneck resulted from narrow genetic variability of exploited resources and significantly limited adaptation range in this crop [109,110]. The DArT-seq has also been exploited for genome-wide association studies (GWAS) targeting several narrow-leafed lupin phenology and yield traits, but it did not provide any candidate gene with significant associations between a marker and a quantitative trait [111,112].
In this study, the MACE was revealed to be an advantageous technique for marker development and gene expression profiling. The eQTL mapping highlighted numerous genes involved in the vernalization response and alkaloid biosynthesis, providing a valuable contribution for further advancement of knowledge on the complexity of molecular networks controlling these two biological processes. Taking into consideration the recent improvements in deciphering the molecular basis underlying early flowering and low-alkaloid phenotypes, as well as addressing results reported here, L. angustifolius can serve as a reference model for such studies across the whole genus. Moreover, information about candidate genes identified in L. angustifolius can be translated to other legume species as these processes are generally conserved.
2.10. Recommendations for Improving Narrow-Leafed Lupin As a Crop
During the process of L. angustifolius domestication several agronomic traits were identified and transferred into improved germplasm by classical selection approaches. Current breeding materials and cultivars usually carry desired alleles of all major domestication traits in homozygous state (Ku, iucundus, lentus, tardus, mollis, leucospermus and Lanr1). However, domestication process was highly focused on these traits and resulted in approximately threefold reduction in genome-wide diversity across domesticated accessions as compared with their wild relatives [112]. Further improvement of this species as a crop will require harnessing of primitive germplasm and subsequent reselection of domesticated alleles in the progenies. One of the most challenging issue is related to the influence of global warming on temperature and rainfall patterns in all major areas where lupins are currently cultivated. This issue could be partially resolved by SNP-based selection of wild accessions of narrow-leafed lupin with well-established local adaptation to warm and dry climate of the eastern Mediterranean basin [111]. Novel opportunities for reducing the time required for transition between phenological phases could also be uncovered by exploitation of natural variability in genes from vernalization and cold pathways highlighted in this study, particularly LanFTc1, CRLK1, FD, UGT85A2, GAUT10, and MYB60. Moreover, a common issue related to dry and warm weather patterns, which are expected to occur more frequently due to changing climate, is pod dehiscence. Identified candidate genes for lentus (a homolog carrying DUF1218 domain) and tardus (an ABCG5 transporter) await further genotypic and phenotypic exploration in wide genetic background because mapping population represents only a small fraction of diversity existing in L. angustifolius germplasm.
3. Materials and Methods
3.1. Plant Material
The reference 83A:476 × P27255 recombinant inbred line (RIL) population (n = 89, F8) of L. angustifolius [27] delivered by the Department of Agriculture and Food Western Australia was used in the study. This population was developed from a cross between a domesticated Australian breeding line (83A:476) and a wild accession from Morocco (P27255). The line P27255 is late flowering and vernalization-responsive (recessive allele ku), pod shattering (dominant alleles Tardus and Lentus), hard seeded (Mollis), blue flower and dark seed color Leucospermus), high alkaloid (Iucundus) and anthracnose susceptible (lanr1). 83A:476 has an opposite allele combination (Ku, tardus, lentus, mollis, leucospermus, iucundus, Lanr1). Both parental lines are homozygous in relation to these alleles.
3.2. Controlled Environment Experiment
Seeds of mapping population and parental lines (83A:476 × P27255) were vernalized for 16 days at 4 °C in darkness on Petri dishes with moist filter paper. Filter paper (Chemland, Stargard, Poland) was changed every four days to maintain phytosanitary conditions. Following vernalization, plants were transferred to pots (2 plants per 11 cm × 11 cm pot, about 8 cm between plants) and grown in controlled conditions (photoperiod 16 h, temperature +25 °C day and +18 °C night) at the Wielkopolska Center of Advanced Technologies in Poznań, Poland. Tissue was sampled from young leaves two times a day, 4 h after beginning of photoperiod and 1 h before the end of photoperiod on the 28th, 36th, and 44th day from sowing. Five biological replicates were collected.
3.3. Massive Analysis of cDNA Ends
Frozen plant tissue (50 mg, −80 °C) was homogenized using TissueLyser II (Qiagen, Hilden, Germany) and two stainless steel beads (ø 5 mm) placed in a 2 mL tube (Eppendorf, Hamburg, Germany). RNA isolation was performed using SV Total RNA Isolation System (Promega, Madison, WI, USA) according to the protocol. The concentration of RNA was measured using NanoDrop 2000 (ThermoFisher Scientific, Waltham, MA, USA) and A260/A280 ratio. RNA quality was visualized by 1% agarose gel electrophoresis (1X TAE) of denaturated samples. RNA concentration was equalized to 400 ng/µL in nuclease-free water. Samples from particular line (representing 5 terms and 5 biological replicates) were bulked together in equal aliquots. 10 µL of mixture (4 µg of RNA) was subjected to the MACE protocol. The MACE profiling and SNP calling was outsourced (GenXPro, Frankfurt, Germany). The MACE reads were aligned to the L. angustifolius genome assembly [34] (http://www.lupinexpress.org). Normalization procedure was as follows [113]: The average raw count of each gene within a library was divided by the geometric mean of all counts in all samples and the median of the quotients was calculated per library. Each raw count was then divided by the library-specific median value.
3.4. Molecular Markers and Linkage Mapping
The 83A:476-like scores were assigned as “a”, the P27255-like scores as “b”, and the heterozygotes as “h”. If several cosegregating MACE markers in particular gene were identified, the marker with the lowest percentage of missing data was chosen to infer consensus segregation representing each cluster. To provide a mapping file, heterozygote scores were removed. Accepted missing data threshold was 11%. Chi-square (χ2) values for Mendelian segregation were estimated using the expected 1:1 ratio. The calculation of probability was based on χ2 and 2 degrees of freedom. Based on the segregation distortion observed in recently published L. angustifolius linkage map versions [29,30,34], χ2 p-value threshold of 1 × 10−7 was applied.
Segregation data for domestication traits and tightly linked SSR-derived markers were included in the study [13,21,22,23,25,52,53]. Moreover, to provide landmarks for chromosome map integration, recently published BAC-derived markers were incorporated [43,44,45,46,47,49,51]. Additionally, novel markers were developed using BAC-end sequences. PCR primers were designed using Primer3′lus [114]. Amplification was performed using DNA isolated from the parental lines of the L. angustifolius mapping population, 83A:476 and P27255. Amplicons were extracted directly from the post-reaction mixtures (QIAquick PCR Purification Kit; Qiagen) and sequenced using ABI PRISM 3130 Genetic Analyzer XL (Applied Biosystems, Hitachi, Tokyo, Japan). Allele-specific PCR (AS-PCR) polymorphisms were visualized by 1% agarose gel electrophoresis, whereas nucleotide substitution polymorphisms were revealed by the cleaved amplified polymorphic sequence (CAPS) and dCAPS approaches [115,116]. Restriction sites were identified using dCAPS Finder 2.0 and SNP2dCAPS [117,118]. Digestion products were separated by 2% agarose gel electrophoresis. Multipoint mapping (JoinMap 5, Kyazma, Wageningen, Netherlands) was performed after grouping under independence LOD of 9.5. Some inconsistency in segregation patterns were observed between previously published and newly developed marker sets. In such cases, marker segregation was tested using current DNA isolates (if possible) or questionable data was deleted from segregation file. Linkage group optimization was performed according to the procedure previously applied for white lupin by [54].
3.5. Expression Quantitative Trait Loci Mapping
Normalized gene expression values (continuous traits) obtained for RILs and mapping population parental lines were associated with Ku, tardus, lentus, mollis, leucospermus, iucundus, and Lanr1 segregation data (binary trait) by t-Student test in two classes of polymorphism. Obtained p-values were false discovery rate (FDR) corrected [119]. Genes with corrected p-value ≤ 0.01 were subjected to composite interval mapping performed in Windows QTL Cartographer V2.5 (North Carolina State University, Raleigh, NC, USA) using 5 background control markers, window size 10 cM, walk speed 0.5 cM, and backward regression method. LOD threshold for QTL calling was established by permutation test (N = 1000) using the same parameters. Linkage groups and LOD graphs were drawn in MapChart [120]. Moreover, sets of genes with corrected p-value ≤ 0.01 were analyzed for gene ontology (GO) term enrichment by hypergeometric test with FDR correction in Bingo [121] using GO annotation of L. angustifolius genes obtained from Ensembl Plants Genes database (rel. 45, genome assembly LupAngTanjil v1.0). Whole-genome annotation was used as reference set. Results were provided as −log10 (corrected p-value).
4. Conclusions
- The massive analysis of cDNA ends was revealed to be applicable for molecular marker development and linkage map construction, as well as for gene expression evaluation and expression quantitative trait loci mapping.
- The analysis of vernalization independence Ku locus shed light on vernalization response via FLOWERING LOCUS T and FD regulon, providing transcriptomic evidence for contribution of several genes acting in C-repeat binding factor (CBF) cold responsiveness and in UDP-glycosyltransferases pathways. This information can be relevant to decipher vernalization pathway in legumes, because legume genomes do not contain a major vernalization-responsive gene FLOWERING LOCUS C (FLC) but other genes from this pathway, including activators and repressors of FLC, are present.
- The study of low-alkaloid iucundus locus highlighted a high number of cis- and trans-regulated alkaloid biosynthesis genes with gene expression orchestrated by a regulatory agent localized at iucundus locus, supporting the concept that the ETHYLENE RESPONSIVE TRANSCRIPTION FACTOR RAP2-7 gene may control low-alkaloid phenotype in narrow-leafed lupin.
- Research on reduced pod shattering lentus locus selected a DUF1218 domain homolog as a candidate gene controlling the orientation of the sclerified endocarp and a DETOXIFICATION14 homolog for purplish hue of young pods.
- An ABCG transporter gene was identified as a hypothetical contributor to sclerenchyma fortification underlying reduced pod shattering tardus locus.
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/20/22/5670/s1.
Author Contributions
Conceptualization, M.K. and B.W.; methodology, M.K, B.W., and P.P.; software, P.P. and M.K.; validation, M.K. and P.P.; formal analysis, M.K. and P.P.; investigation, S.R.-B., E.R., P.P., and M.K.; resources, S.R.-B., E.R., M.K., and P.P.; data curation, M.K and P.P.; writing—original draft preparation, M.K.; visualization, M.K.; supervision, B.W.; project administration, B.W. and M.K.; funding acquisition, B.W.
Funding
This research was funded by the Polish National Centre for Research and Development (Narodowe Centrum Badań i Rozwoju) grant number PBS3/A8/28/2015, SEGENMAS. The APC was funded by the Institute of Plant Genetics, Polish Academy of Sciences.
Acknowledgments
Authors thank Krzysztof Pudełko from the Wielkopolska Center of Advanced Technologies in Poznań, Poland for assistance in plant experiment. Moreover, authors acknowledge Magdalena Tomaszewska from the Department of Genomics of the Institute of Plant Genetics, Polish Academy of Sciences for help in plant sowing and leaf sampling, and Paweł Krajewski from the Department of Biometry and Bioinformatics, Institute of Plant Genetics, Polish Academy of Sciences, for help with MACE data processing and GO analyses. Authors would also like to thank Björn Rotter and Peter Winter from GenXPro GmBH, Frankfurt Main, Germany for executing MACE protocol, MACE marker identification, and production of annotated sequence read count and haplotype datasets. Part of the computations were performed at the Poznań Supercomputing and Networking Center (www.man.poznan.pl).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 2OG | DMR6-like OXYGENASE 2 |
| AAP | Amino acid permease |
| ABCG5 | G-family ATP-binding ABC transporter 5 |
| AGL8 | AGAMOUS-LIKE 8 |
| AK1 | Aspartate kinase 1 |
| AP1 | APETALA1 |
| ASDH | Aspartate-semialdehyde dehydrogenase |
| BAC | Bacterial artificial chromosome |
| BAN | Anthocyanidin reductase |
| BolA2 | BolA-like family protein 2 |
| CBF | C-repeat binding factor |
| CCR1 | Cinnamoyl-CoA reductase 1 |
| CRLK1 | CALCIUM/CALMODULIN-REGULATED RECEPTOR-LIKE KINASE 1 |
| CXE1 | Carboxylesterase 1 |
| DAPDC1 | Diaminopimelate decarboxylase 1 |
| DapL | LL-diaminopimelate aminotransferase |
| DArT | Diversity Arrays Technology |
| DFR | Dihydroflavonol 4-reductase |
| DHDPS | 4-Hydroxy-tetrahydrodipicolinate synthase |
| DTX14 | MATE efflux family protein DETOXIFICATION14 |
| DUF1218 | Fiber protein Fb34, domain of unknown function 1218 |
| eQTL | Expression quantitative trait locus |
| FKF1 | FLAVIN-BINDING KELCH REPEAT, F-BOX 1 |
| FLC | FLOWERING LOCUS C |
| FSD2 | Fe superoxide dismutase 2 |
| FT | FLOWERING LOCUS T |
| FUL | FRUITFULL |
| GAUT10 | Galacturonosyltransferase 10-like |
| GLT1 | Glutamate synthase 1 |
| GO | GENE Gene ontology |
| GTF2H2 | General transcription factor IIH subunit 2 |
| GWAS | Genome-wide association study |
| ICE1 | INDUCER OF CBF EXPRESSION 1 |
| KAB1 | Voltage-gated potassium channel subunit beta 1 |
| KNAT1 | Homeobox protein knotted-1-like |
| LaAT | HXXXD-type ACYL-TRANSFERASE |
| LanFTc1 | L. angustifolius FLOWERING LOCUS T c1 |
| LDC | Lysine/ornithine decarboxylase |
| LFY | LEAFY |
| MACE | Massive analysis of cDNA ends |
| MATE | Multidrug and toxic compound extrusion |
| MFLP | Molecular fragment length polymorphism |
| MHK10.21 | Copper amine oxidase |
| MLP31 | MAJOR LATEX PROTEIN 31 |
| MLP423 | MAJOR LATEX PROTEIN 423 |
| MWL-1 | MODIFYING WALL LIGNIN-1 |
| MWL-2 | MODIFYING WALL LIGNIN-2 |
| MYB60 | MYB transcription factor 60 |
| NACA2 | Nascent polypeptide-associated complex subunit alpha 2 |
| NPF3.1 | Protein NRT1/ PTR FAMILY 3.1-like |
| PGPS2 | CDP-diacylglycerol–glycerol-3-phosphate 3-phosphatidyltransferase 2 |
| PUP | Purine permease transporter |
| RAP2-7 | ETHYLENE RESPONSIVE TRANSCRIPTION FACTOR RAP2-7 |
| RIL | Recombinant inbred line |
| RPL40A | Ubiquitin-60S ribosomal protein L40A |
| SDR | Short-chain dehydrogenase reductase |
| SNP | Single nucleotide polymorphism |
| SOC1 | SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 |
| UGT85A2 | UDP-Glycosyltransferase 85A2 |
| UGT87A2 | UDP-Glycosyltransferase 87A2 |
References
- Święcicki, W.; Święcicki, W.K. Domestication and breeding improvement of narrow-leafed lupin (L. angustifolius L.). J. Appl. Genet. 1995, 36, 155–167. [Google Scholar]
- Landers, K.F. Vernalization responses in narrow-leafed lupin (Lupinus angustifolius) genotypes. Aust. J. Agric. Res. 1995, 46, 1011–1025. [Google Scholar] [CrossRef]
- Adhikari, K.N.; Buirchell, B.J.; Sweetingham, M.W. Length of vernalization period affects flowering time in three lupin species. Plant Breed. 2012, 131, 631–636. [Google Scholar] [CrossRef]
- Gladstones, J.; Hill, G. Selection for economic characters in Lupinus angustifolius and L. digitatus. 2. Time of flowering. Aust. J. Exp. Agric. 1969, 9, 213–220. [Google Scholar] [CrossRef]
- Wink, M.; Meißner, C.; Witte, L. Patterns of quinolizidine alkaloids in 56 species of the genus Lupinus. Phytochemistry 1995, 38, 139–153. [Google Scholar] [CrossRef]
- Bassoli, A.; Borgonovo, G.; Busnelli, G. Alkaloids and the bitter taste. In Modern Alkaloids, Fattorusso, E., Taglialatela-Scafati, O.; Wiley-VCH: Weinheim, Germany, 2007; pp. 53–72. [Google Scholar]
- Matsuura, H.N.; Fett-Neto, A.G. Plant alkaloids: Main features, toxicity, and mechanisms of action. In Plant Toxins; Gopalakrishnakone, P., Carlini, C.R., Ligabue-Braun, R., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 1–15. [Google Scholar]
- Gladstones, J.S. Lupins as crop plants. Field Crop Abstracts 1970, 23, 26. [Google Scholar]
- Von Sengbusch, R. Süßlupinen und Öllupinen. Die Entstehungsgeschichte einiger neuer Kulturpflanzen. Landwirtsch. Jahrbücher 1942, 91, 719–880. [Google Scholar]
- Kamel, K.A.; Święcicki, W.; Kaczmarek, Z.; Barzyk, P. Quantitative and qualitative content of alkaloids in seeds of a narrow-leafed lupin (Lupinus angustifolius L.) collection. Genet. Resour. Crop Evol. 2016, 63, 711–719. [Google Scholar] [CrossRef]
- Frick, K.M.; Kamphuis, L.G.; Siddique, K.H.; Singh, K.B.; Foley, R.C. Quinolizidine alkaloid biosynthesis in lupins and prospects for grain quality improvement. Front. Plant Sci. 2017, 8, 87. [Google Scholar] [CrossRef]
- Gladstones, J. Selection for economic characters in Lupinus angustifolius and L. digitatus. Aust. J. Exp. Agric. 1967, 7, 360–366. [Google Scholar] [CrossRef]
- Boersma, J.G.; Buirchell, B.J.; Sivasithamparam, K.; Yang, H. Development of two sequence-specific PCR markers linked to the le gene that reduces pod shattering in narrow-leafed lupin (Lupinus angustifolius L.). Genet. Mol. Biol. 2007, 30, 623–629. [Google Scholar] [CrossRef]
- Mikolajczyk, J. Genetic studies in Lupinus angustifolius. 2. Inheritance of some morphological characters in blue lupine. Genet. Pol. 1966, 7, 153–180. [Google Scholar]
- Li, X.; Buirchell, B.; Yan, G.; Yang, H. A molecular marker linked to the mollis gene conferring soft-seediness for marker-assisted selection applicable to a wide range of crosses in lupin (Lupinus angustifolius L.) breeding. Mol. Breed. 2012, 29, 361–370. [Google Scholar] [CrossRef]
- Nirenberg, H.I.; Feiler, U.; Hagedorn, G. Description of Colletotrichum lupini comb. nov. in modern terms. Mycologia 2002, 94, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Boersma, J.G.; You, M.; Buirchell, B.J.; Sweetingham, M.W. Development and implementation of a sequence-specific PCR marker linked to a gene conferring resistance to anthracnose disease in narrow-leafed lupin (Lupinus angustifolius L.). Mol. Breed. 2004, 14, 145–151. [Google Scholar] [CrossRef]
- Yang, H.; Renshaw, D.; Thomas, G.; Buirchell, B.; Sweetingham, M. A strategy to develop molecular markers applicable to a wide range of crosses for marker assisted selection in plant breeding: A case study on anthracnose disease resistance in lupin (Lupinus angustifolius L.). Mol. Breed. 2008, 21, 473–483. [Google Scholar] [CrossRef]
- Fischer, K.; Dieterich, R.; Nelson, M.N.; Kamphuis, L.G.; Singh, K.B.; Rotter, B.; Krezdorn, N.; Winter, P.; Wehling, P.; Ruge-Wehling, B. Characterization and mapping of LanrBo: A locus conferring anthracnose resistance in narrow-leafed lupin (Lupinus angustifolius L.). Theor. Appl. Genet. 2015, 128, 2121–2130. [Google Scholar] [CrossRef]
- Yang, H.; Sweetingham, M.W.; Cowling, W.A.; Smith, P.M.C. DNA fingerprinting based on microsatellite-anchored fragment length polymorphisms, and isolation of sequence-specific PCR markers in lupin (Lupinus angustifolius L.). Mol. Breed. 2001, 7, 203–209. [Google Scholar] [CrossRef]
- Boersma, J.G.; Pallotta, M.; Li, C.; Buirchell, B.J.; Sivasithamparam, K.; Yang, H. Construction of a genetic linkage map using MFLP and identification of molecular markers linked to domestication genes in narrow-leafed lupin (Lupinus angustifolius L.). Cell. Mol. Biol. Lett. 2005, 10, 331–344. [Google Scholar]
- Li, X.; Yang, H.; Buirchell, B.; Yan, G. Development of a DNA marker tightly linked to low-alkaloid gene iucundus in narrow-leafed lupin (Lupinus angustifolius L.) for marker-assisted selection. Crop Pasture Sci. 2011, 62, 218–224. [Google Scholar] [CrossRef]
- Boersma, J.G.; Buirchell, B.J.; Sivasithamparam, K.; Yang, H. Development of a sequence-specific PCR marker linked to the Ku gene which removes the vernalization requirement in narrow-leafed lupin. Plant Breed. 2007, 126, 306–309. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.; Yan, G. Development of a co-dominant DNA marker linked to the gene lentus conferring reduced pod shattering for marker-assisted selection in narrow-leafed lupin (Lupinus angustifolius) breeding. Plant Breed. 2012, 131, 540–544. [Google Scholar] [CrossRef]
- Boersma, J.G.; Nelson, M.N.; Sivasithamparam, K.; Yang, H.A. Development of sequence-specific PCR markers linked to the Tardus gene that reduces pod shattering in narrow-leafed lupin (Lupinus angustifolius L.). Mol. Breed. 2009, 23, 259–267. [Google Scholar] [CrossRef]
- Li, X.; Renshaw, D.; Yang, H.; Yan, G. Development of a co-dominant DNA marker tightly linked to gene tardus conferring reduced pod shattering in narrow-leafed lupin (Lupinus angustifolius L.). Euphytica 2010, 176, 49–58. [Google Scholar] [CrossRef]
- Nelson, M.N.; Phan, H.T.T.; Ellwood, S.R.; Moolhuijzen, P.M.; Hane, J.; Williams, A.; O’Lone, C.E.; Fosu-Nyarko, J.; Scobie, M.; Cakir, M.; et al. The first gene-based map of Lupinus angustifolius L.-location of domestication genes and conserved synteny with Medicago truncatula. Theor. Appl. Genet. 2006, 113, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.N.; Moolhuijzen, P.M.; Boersma, J.G.; Chudy, M.; Lesniewska, K.; Bellgard, M.; Oliver, R.P.; Swiecicki, W.; Wolko, B.; Cowling, W.A.; et al. Aligning a new reference genetic map of Lupinus angustifolius with the genome sequence of the model legume, Lotus japonicus. DNA Res. 2010, 17, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, L.G.; Hane, J.K.; Nelson, M.N.; Gao, L.; Atkins, C.A.; Singh, K.B. Transcriptome sequencing of different narrow-leafed lupin tissue types provides a comprehensive uni-gene assembly and extensive gene-based molecular markers. Plant Biotechnol. J. 2015, 13, 14–25. [Google Scholar] [CrossRef]
- Zhou, G.; Jian, J.; Wang, P.; Li, C.; Tao, Y.; Li, X.; Renshaw, D.; Clements, J.; Sweetingham, M.; Yang, H. Construction of an ultra-high density consensus genetic map, and enhancement of the physical map from genome sequencing in Lupinus angustifolius. Theor. Appl. Genet. 2018, 131, 209–223. [Google Scholar] [CrossRef]
- Kasprzak, A.; Safár, J.; Janda, J.; Dolezel, J.; Wolko, B.; Naganowska, B. The bacterial artificial chromosome (BAC) library of the narrow-leafed lupin (Lupinus angustifolius L.). Cell. Mol. Biol. Lett. 2006, 11, 396–407. [Google Scholar] [CrossRef]
- Gao, L.-L.; Hane, J.K.; Kamphuis, L.G.; Foley, R.; Shi, B.-J.; Atkins, C.A.; Singh, K.B. Development of genomic resources for the narrow-leafed lupin (Lupinus angustifolius): Construction of a bacterial artificial chromosome (BAC) library and BAC-end sequencing. BMC Genom. 2011, 12, 521. [Google Scholar] [CrossRef]
- Yang, H.; Tao, Y.; Zheng, Z.; Zhang, Q.; Zhou, G.; Sweetingham, M.W.; Howieson, J.G.; Li, C. Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L. PLoS ONE 2013, 8, e64799. [Google Scholar] [CrossRef] [PubMed]
- Hane, J.K.; Ming, Y.; Kamphuis, L.G.; Nelson, M.N.; Garg, G.; Atkins, C.A.; Bayer, P.E.; Bravo, A.; Bringans, S.; Cannon, S.; et al. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: Insights into plant-microbe interactions and legume evolution. Plant Biotechnol. J. 2017, 15, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.M.; Rogacev, K.S.; Muller, S.; Rotter, B.; Winter, P.; Fliser, D.; Heine, G.H. Massive analysis of cDNA Ends (MACE) and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease. Epigenetics 2014, 9, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Bojahr, J.; Nhengiwa, O.; Krezdorn, N.; Rotter, B.; Saal, B.; Ruge-Wehling, B.; Struck, C.; Winter, P. Massive analysis of cDNA ends (MACE) reveals a co-segregating candidate gene for LpPg1 stem rust resistance in perennial ryegrass (Lolium perenne). Theor. Appl. Genet. 2016, 129, 1915–1932. [Google Scholar] [CrossRef]
- Zajac, B.K.; Amendt, J.; Horres, R.; Verhoff, M.A.; Zehner, R. De novo transcriptome analysis and highly sensitive digital gene expression profiling of Calliphora vicina (Diptera: Calliphoridae) pupae using MACE (Massive Analysis of cDNA Ends). Forensic Science International: Genetics 2015, 15, 137–146. [Google Scholar] [CrossRef]
- Zhernakov, A.; Rotter, B.; Winter, P.; Borisov, A.; Tikhonovich, I.; Zhukov, V. Massive Analysis of cDNA Ends (MACE) for transcript-based marker design in pea (Pisum sativum L.). Genom. Data 2017, 11, 75–76. [Google Scholar] [CrossRef]
- Keller, M.; Consortium, S.-I.; Simm, S. The coupling of transcriptome and proteome adaptation during development and heat stress response of tomato pollen. BMC Genom. 2018, 19, 447. [Google Scholar] [CrossRef]
- Parreira, J.R.; Balestrazzi, A.; Fevereiro, P.; Araujo, S.S. Maintaining genome integrity during seed development in Phaseolus vulgaris L.: Evidence from a transcriptomic profiling study. Genes 2018, 9, 463. [Google Scholar] [CrossRef]
- Oyiga, B.C.; Sharma, R.C.; Baum, M.; Ogbonnaya, F.C.; Léon, J.; Ballvora, A. Allelic variations and differential expressions detected at quantitative trait loci for salt stress tolerance in wheat. Plant Cell Environ. 2018, 41, 919–935. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Naganowska, B.; Wolko, B. Karyotyping of the narrow-leafed lupin (Lupinus angustifolius L.) by using FISH, PRINS and computer measurements of chromosomes. J. Appl. Genet. 2009, 50, 77–82. [Google Scholar] [CrossRef]
- Wyrwa, K.; Książkiewicz, M.; Szczepaniak, A.; Susek, K.; Podkowiński, J.; Naganowska, B. Integration of Lupinus angustifolius L. (narrow-leafed lupin) genome maps and comparative mapping within legumes. Chromosome Res. 2016, 24, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Przysiecka, Ł.; Książkiewicz, M.; Wolko, B.; Naganowska, B. Structure, expression profile and phylogenetic inference of chalcone isomerase-like genes from the narrow-leafed lupin (Lupinus angustifolius L.) genome. Front. Plant Sci. 2015, 6, 268. [Google Scholar] [CrossRef] [PubMed]
- Książkiewicz, M.; Zielezinski, A.; Wyrwa, K.; Szczepaniak, A.; Rychel, S.; Karlowski, W.; Wolko, B.; Naganowska, B. Remnants of the legume ancestral genome preserved in gene-rich regions: Insights from Lupinus angustifolius physical, genetic, and comparative mapping. Plant Mol. Biol. Rep. 2015, 33, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Książkiewicz, M.; Wyrwa, K.; Szczepaniak, A.; Rychel, S.; Majcherkiewicz, K.; Przysiecka, Ł.; Karlowski, W.; Wolko, B.; Naganowska, B. Comparative genomics of Lupinus angustifolius gene-rich regions: BAC library exploration, genetic mapping and cytogenetics. BMC Genom. 2013, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Leśniewska, K.; Książkiewicz, M.; Nelson, M.N.; Mahé, F.; Aïnouche, A.; Wolko, B.; Naganowska, B. Assignment of 3 genetic linkage groups to 3 chromosomes of narrow-leafed lupin. J. Hered. 2011, 102, 228–236. [Google Scholar] [CrossRef]
- Szczepaniak, A.; Książkiewicz, M.; Podkowiński, J.; Czyż, K.B.; Figlerowicz, M.; Naganowska, B. Legume cytosolic and plastid acetyl-coenzyme-A carboxylase genes differ by evolutionary patterns and selection pressure schemes acting before and after whole-genome duplications. Genes 2018, 9, 563. [Google Scholar] [CrossRef]
- Nelson, M.N.; Książkiewicz, M.; Rychel, S.; Besharat, N.; Taylor, C.M.; Wyrwa, K.; Jost, R.; Erskine, W.; Cowling, W.A.; Berger, J.D.; et al. The loss of vernalization requirement in narrow-leafed lupin is associated with a deletion in the promoter and de-repressed expression of a Flowering Locus T (FT) homologue. New Phytol. 2017, 213, 220–232. [Google Scholar] [CrossRef]
- Narożna, D.; Książkiewicz, M.; Przysiecka, Ł.; Króliczak, J.; Wolko, B.; Naganowska, B.; Mądrzak, C.J. Legume isoflavone synthase genes have evolved by whole-genome and local duplications yielding transcriptionally active paralogs. Plant Sci. 2017, 264, 149–167. [Google Scholar] [CrossRef]
- Książkiewicz, M.; Rychel, S.; Nelson, M.N.; Wyrwa, K.; Naganowska, B.; Wolko, B. Expansion of the phosphatidylethanolamine binding protein family in legumes: A case study of Lupinus angustifolius L. FLOWERING LOCUS T homologs, LanFTc1 and LanFTc2. BMC Genom. 2016, 17, 820. [Google Scholar]
- Boersma, J.G.; Buirchell, B.J.; Sivasithamparam, K.; Yang, H. Development of a PCR marker tightly linked to mollis, the gene that controls seed dormancy in Lupinus angustifolius L. Plant Breed. 2007, 126, 612–616. [Google Scholar] [CrossRef]
- You, M.; Boersma, J.G.; Buirchell, B.J.; Sweetingham, M.W.; Siddique, K.H.M.; Yang, H. A PCR-based molecular marker applicable for marker-assisted selection for anthracnose disease resistance in lupin breeding. Cell. Mol. Biol. Lett. 2005, 10, 123–134. [Google Scholar] [PubMed]
- Książkiewicz, M.; Nazzicari, N.; Yang, H.A.; Nelson, M.N.; Renshaw, D.; Rychel, S.; Ferrari, B.; Carelli, M.; Tomaszewska, M.; Stawiński, S.; et al. A high-density consensus linkage map of white lupin highlights synteny with narrow-leafed lupin and provides markers tagging key agronomic traits. Sci. Rep. 2017, 7, 15335. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.M.; Huynh, M.; Udall, J.A.; Kilian, A.; Adhikari, K.N.; Berger, J.D.; Erskine, W.; Nelson, M.N. The first genetic map for yellow lupin enables genetic dissection of adaptation traits in an orphan grain legume crop. BMC Genet. 2019, 20, 68. [Google Scholar] [CrossRef] [PubMed]
- Rychel, S.; Książkiewicz, M. Development of gene-based molecular markers tagging low alkaloid pauper locus in white lupin (Lupinus albus L.). J. Appl. Genet. 2019, 60, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Bunsupa, S.; Katayama, K.; Ikeura, E.; Oikawa, A.; Toyooka, K.; Saito, K.; Yamazaki, M. Lysine decarboxylase catalyzes the first step of quinolizidine alkaloid biosynthesis and coevolved with alkaloid production in Leguminosae. Plant Cell 2012, 24, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Frick, K.M.; Foley, R.C.; Kamphuis, L.G.; Siddique, K.H.M.; Garg, G.; Singh, K.B. Characterization of the genetic factors affecting quinolizidine alkaloid biosynthesis and its response to abiotic stress in narrow-leafed lupin (Lupinus angustifolius L.). Plant Cell Environ. 2018, 41, 2155–2168. [Google Scholar] [CrossRef]
- Kroc, M.; Koczyk, G.; Kamel, K.A.; Czepiel, K.; Fedorowicz-Strońska, O.; Krajewski, P.; Kosińska, J.; Podkowiński, J.; Wilczura, P.; Święcicki, W. Transcriptome-derived investigation of biosynthesis of quinolizidine alkaloids in narrow-leafed lupin (Lupinus angustifolius L.) highlights candidate genes linked to iucundus locus. Sci. Rep. 2019, 9, 2231. [Google Scholar] [CrossRef]
- Kroc, M.; Czepiel, K.; Wilczura, P.; Mokrzycka, M.; Swiecicki, W. Development and validation of a gene-targeted dCAPS marker for marker-assisted selection of low-alkaloid content in seeds of narrow-leafed lupin (Lupinus angustifolius L.). Genes 2019, 10, 428. [Google Scholar] [CrossRef]
- Kato, K.; Shoji, T.; Hashimoto, T. Tobacco nicotine uptake permease regulates the expression of a key transcription factor gene in the nicotine biosynthesis pathway. Plant Physiol. 2014, 166, 2195–2204. [Google Scholar] [CrossRef]
- Hildreth, S.B.; Gehman, E.A.; Yang, H.; Lu, R.-H.; Ritesh, K.C.; Harich, K.C.; Yu, S.; Lin, J.; Sandoe, J.L.; Okumoto, S.; et al. Tobacco nicotine uptake permease (NUP1) affects alkaloid metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, 18179–18184. [Google Scholar] [CrossRef]
- Yang, T.; Nagy, I.; Mancinotti, D.; Otterbach, S.L.; Andersen, T.B.; Motawia, M.S.; Asp, T.; Geu-Flores, F. Transcript profiling of a bitter variety of narrow-leafed lupin to discover alkaloid biosynthetic genes. J. Exp. Bot. 2017, 68, 5527–5537. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Kang, Y.-G.; Lee, T.; Kim, M.; Yoon, M.Y.; Lee, E.; Yang, X.; Kim, D.; Kim, Y.-J.; Lee, T.R.; et al. Comprehensive RNA sequencing and co-expression network analysis to complete the biosynthetic pathway of coumestrol, a phytoestrogen. Sci. Rep. 2019, 9, 1934. [Google Scholar] [CrossRef] [PubMed]
- Goldsbrough, A.; Belzile, F.; Yoder, J.I. Complementation of the tomato anthocyanin without (aw) mutant using the dihydroflavonol 4-reductase gene. Plant Physiol. 1994, 105, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Bunsupa, S.; Okada, T.; Saito, K.; Yamazaki, M. An acyltransferase-like gene obtained by differential gene expression profiles of quinolizidine alkaloid-producing and nonproducing cultivars of Lupinus angustifolius. Plant Biotechnol. 2011, 28, 89–94. [Google Scholar] [CrossRef]
- Wink, M. N-Methylation of quinolizidine alkaloids: An S-adenosyl-L-methionine: Cytisine N-methyltransferase from Laburnum anagyroides plants and cell cultures of L. alpinum and Cytisus canariensis. Planta 1984, 161, 339–344. [Google Scholar] [CrossRef]
- Liscombe, D.K.; Usera, A.R.; O’Connor, S.E. Homolog of tocopherol C methyltransferases catalyzes N methylation in anticancer alkaloid biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18793–18798. [Google Scholar] [CrossRef]
- Li, Y.; Smolke, C.D. Engineering biosynthesis of the anticancer alkaloid noscapine in yeast. Nat. Commun. 2016, 7, 12137. [Google Scholar] [CrossRef]
- Barker, S.J.; Si, P.; Hodgson, L.; Ferguson-Hunt, M.; Khentry, Y.; Krishnamurthy, P.; Averis, S.; Mebus, K.; O’Lone, C.; Dalugoda, D.; et al. Regeneration selection improves transformation efficiency in narrow-leaf lupin. Plant Cell Tissue Organ Cult. 2016, 126, 219–228. [Google Scholar] [CrossRef]
- Yang, T.; Chaudhuri, S.; Yang, L.; Du, L.; Poovaiah, B.W. A calcium/calmodulin-regulated member of the receptor-like kinase family confers cold tolerance in plants. J. Biol. Chem. 2010, 285, 7119–7126. [Google Scholar] [CrossRef]
- Kim, H.-J.; Hyun, Y.; Park, J.-Y.; Park, M.-J.; Park, M.-K.; Kim, M.D.; Kim, H.-J.; Lee, M.H.; Moon, J.; Lee, I.; et al. A genetic link between cold responses and flowering time through FVE in Arabidopsis thaliana. Nat. Genet. 2004, 36, 167–171. [Google Scholar] [CrossRef]
- Lee, J.-H.; Jung, J.-H.; Park, C.-M. INDUCER OF CBF EXPRESSION 1 integrates cold signals into FLOWERING LOCUS C-mediated flowering pathways in Arabidopsis. Plant J. 2015, 84, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Lee, H.; Jeon, J.; Park, H.; Kim, J.; Noh, Y.S.; Lee, I. Crosstalk between cold response and flowering in Arabidopsis is mediated through the flowering-time gene SOC1 and its upstream negative regulator FLC. Plant Cell 2009, 21, 3185–3197. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Gilmour, S.J.; Grumet, R.; Thomashow, M.F. CBF-dependent and CBF-independent regulatory pathways contribute to the differences in freezing tolerance and cold-regulated gene expression of two Arabidopsis ecotypes locally adapted to sites in Sweden and Italy. PLoS ONE 2018, 13, e0207723. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Zhang, L.; Liang, S.; Jones, R.L.; Lu, Y.T. A tobacco calcium/calmodulin-binding protein kinase functions as a negative regulator of flowering. J. Biol. Chem. 2004, 279, 31483–31494. [Google Scholar] [CrossRef]
- Hecht, V.; Foucher, F.; Ferrandiz, C.; Macknight, R.; Navarro, C.; Morin, J.; Vardy, M.E.; Ellis, N.; Beltran, J.P.; Rameau, C.; et al. Conservation of Arabidopsis flowering genes in model legumes. Plant Physiol. 2005, 137, 1420–1434. [Google Scholar] [CrossRef]
- Akiyama, K.; Kurotani, A.; Iida, K.; Kuromori, T.; Shinozaki, K.; Sakurai, T. RARGE II: An integrated phenotype database of Arabidopsis mutant traits using a controlled vocabulary. Plant Cell Physiol. 2014, 55, e4. [Google Scholar] [CrossRef]
- Yuan, N.; Balasubramanian, V.K.; Chopra, R.; Mendu, V. The photoperiodic flowering time regulator FKF1 negatively regulates cellulose biosynthesis. Plant Physiol. 2019, 180, 2240–2253. [Google Scholar] [CrossRef]
- Wang, B.; Jin, S.-H.; Hu, H.-Q.; Sun, Y.-G.; Wang, Y.-W.; Han, P.; Hou, B.-K. UGT87A2, an Arabidopsis glycosyltransferase, regulates flowering time via FLOWERING LOCUS C. New Phytol. 2012, 194, 666–675. [Google Scholar] [CrossRef]
- Taylor, C.M.; Kamphuis, L.G.; Zhang, W.; Garg, G.; Berger, J.D.; Mousavi-Derazmahalleh, M.; Bayer, P.E.; Edwards, D.; Singh, K.B.; Cowling, W.A.; et al. INDEL variation in the regulatory region of the major flowering time gene LanFTc1 is associated with vernalization response and flowering time in narrow-leafed lupin (Lupinus angustifolius L.). Plant Cell Environ. 2019, 42, 174–187. [Google Scholar] [CrossRef]
- Abe, M.; Kobayashi, Y.; Yamamoto, S.; Daimon, Y.; Yamaguchi, A.; Ikeda, Y.; Ichinoki, H.; Notaguchi, M.; Goto, K.; Araki, T. FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 2005, 309, 1052–1056. [Google Scholar] [CrossRef]
- Andrés, F.; Romera-Branchat, M.; Martínez-Gallegos, R.; Patel, V.; Schneeberger, K.; Jang, S.; Altmüller, J.; Nürnberg, P.; Coupland, G. Floral induction in Arabidopsis by FLOWERING LOCUS T requires direct repression of BLADE-ON-PETIOLE genes by the homeodomain protein PENNYWISE. Plant Physiol. 2015, 169, 2187–2199. [Google Scholar] [CrossRef]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Sánchez, D.; Martínez-López, S.; Navarro-Cartagena, S.; Jover-Gil, S.; Micol, J.L. Members of the DEAL subfamily of the DUF1218 gene family are required for bilateral symmetry but not for dorsoventrality in Arabidopsis leaves. New Phytol. 2018, 217, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Persson, S.; Wei, H.; Milne, J.; Page, G.P.; Somerville, C.R. Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. Proc. Natl. Acad. Sci. USA 2005, 102, 8633–8638. [Google Scholar] [CrossRef] [PubMed]
- Ubeda-Tomas, S.; Edvardsson, E.; Eland, C.; Singh, S.K.; Zadik, D.; Aspeborg, H.; Gorzsàs, A.; Teeri, T.T.; Sundberg, B.; Persson, P.; et al. Genomic-assisted identification of genes involved in secondary growth in Arabidopsis utilising transcript profiling of poplar wood-forming tissues. Physiol. Plant. 2007, 129, 415–428. [Google Scholar] [CrossRef]
- Mewalal, R.; Mizrachi, E.; Coetzee, B.; Mansfield, S.D.; Myburg, A.A. The Arabidopsis Domain of Unknown Function 1218 (DUF1218) containing proteins, MODIFYING WALL LIGNIN-1 and 2 (At1g31720/MWL-1 and At4g19370/MWL-2) function redundantly to alter secondary cell wall lignin content. PLoS ONE 2016, 11, e0150254. [Google Scholar] [CrossRef]
- Miyauchi, H.; Moriyama, S.; Kusakizako, T.; Kumazaki, K.; Nakane, T.; Yamashita, K.; Hirata, K.; Dohmae, N.; Nishizawa, T.; Ito, K.; et al. Structural basis for xenobiotic extrusion by eukaryotic MATE transporter. Nat. Commun. 2017, 8, 1633. [Google Scholar] [CrossRef]
- Upadhyay, N.; Kar, D.; Deepak Mahajan, B.; Nanda, S.; Rahiman, R.; Panchakshari, N.A.; Bhagavatula, L.; Datta, S. The multitasking abilities of MATE transporters in plants. J. Exp. Bot. 2019, 70, 4643–4656. [Google Scholar] [CrossRef]
- Yang, X.; Xia, X.; Zhang, Z.; Nong, B.; Zeng, Y.; Wu, Y.; Xiong, F.; Zhang, Y.; Liang, H.; Pan, Y.; et al. Identification of anthocyanin biosynthesis genes in rice pericarp using PCAMP. Plant Biotechnol. J. 2019, 17, 1700–1702. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.; Liu, H.; Kang, L.; Geng, J.; Gai, Y.; Ding, Y.; Sun, H.; Li, Y. Identification and expression analysis of MATE genes involved in flavonoid transport in blueberry plants. PLoS ONE 2015, 10, e0118578. [Google Scholar] [CrossRef]
- Ellis, T.J.; Field, D.L. Repeated gains in yellow and anthocyanin pigmentation in flower colour transitions in the Antirrhineae. Ann. Bot. 2016, 117, 1133–1140. [Google Scholar] [CrossRef]
- Shiono, K.; Ando, M.; Nishiuchi, S.; Takahashi, H.; Watanabe, K.; Nakamura, M.; Matsuo, Y.; Yasuno, N.; Yamanouchi, U.; Fujimoto, M.; et al. RCN1/OsABCG5, an ATP-binding cassette (ABC) transporter, is required for hypodermal suberization of roots in rice (Oryza sativa). Plant J. 2014, 80, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, R.; Smolka, U.; Altmann, S.; Eschen-Lippold, L.; Senning, M.; Sonnewald, S.; Weigel, B.; Frolova, N.; Strehmel, N.; Hause, G.; et al. The ABC transporter ABCG1 is required for suberin formation in potato tuber periderm. Plant cell 2014, 26, 3403–3415. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, S.; Lee, Y.; Tohge, T.; Sudre, D.; Osorio, S.; Park, J.; Bovet, L.; Lee, Y.; Geldner, N.; Fernie, A.R.; et al. AtABCG29 is a monolignol transporter involved in lignin biosynthesis. Curr. Biol. 2012, 22, 1207–1212. [Google Scholar] [CrossRef]
- Sibout, R.; Höfte, H. Plant Cell Biology: The ABC of monolignol transport. Curr. Biol. 2012, 22, R533–R535. [Google Scholar] [CrossRef]
- Hinrichs, M.; Fleck, A.T.; Biedermann, E.; Ngo, N.S.; Schreiber, L.; Schenk, M.K. An ABC transporter is involved in the silicon-induced formation of Casparian Bands in the exodermis of rice. Front. Plant Sci. 2017, 8, 671. [Google Scholar] [CrossRef]
- Forbes, I.; Wells, H.D. Hard and soft seededness in blue lupine, Lupinus angustifolius L.: Inheritance and phenotype classification. Crop Sci. 1968, 8, 195–197. [Google Scholar] [CrossRef]
- Miao, Z.H.; Fortune, J.A.; Gallagher, J. Anatomical structure and nutritive value of lupin seed coats. Aust. J. Agric. Res. 2001, 52, 985–993. [Google Scholar] [CrossRef]
- Smýkal, P.; Vernoud, V.; Blair, M.W.; Soukup, A.; Thompson, R.D. The role of the testa during development and in establishment of dormancy of the legume seed. Front. Plant Sci. 2014, 5, 351. [Google Scholar]
- Silvestro, D.; Andersen, T.G.; Schaller, H.; Jensen, P.E. Plant sterol metabolism. Δ(7)-Sterol-C5-desaturase (STE1/DWARF7), Δ(5,7)-sterol-Δ(7)-reductase (DWARF5) and Δ(24)-sterol-Δ(24)-reductase (DIMINUTO/DWARF1) show multiple subcellular localizations in Arabidopsis thaliana (Heynh) L. PLoS ONE 2013, 8, e56429. [Google Scholar] [CrossRef] [PubMed]
- DeBolt, S.; Scheible, W.-R.; Schrick, K.; Auer, M.; Beisson, F.; Bischoff, V.; Bouvier-Navé, P.; Carroll, A.; Hematy, K.; Li, Y.; et al. Mutations in UDP-Glucose:sterol glucosyltransferase in Arabidopsis cause transparent testa phenotype and suberization defect in seeds. Plant Physiol. 2009, 151, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Loubéry, S.; De Giorgi, J.; Utz-Pugin, A.; Demonsais, L.; Lopez-Molina, L. A maternally deposited endosperm cuticle contributes to the physiological defects of transparent testa seeds. Plant Physiol. 2018, 177, 1218–1233. [Google Scholar] [CrossRef] [PubMed]
- Hellens, R.P.; Moreau, C.; Lin-Wang, K.; Schwinn, K.E.; Thomson, S.J.; Fiers, M.W.E.J.; Frew, T.J.; Murray, S.R.; Hofer, J.M.I.; Jacobs, J.M.E.; et al. Identification of Mendel’s white flower character. PLoS ONE 2010, 5, e13230. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, X.-B.; Shi, Y.-F.; Wang, H.-M.; Feng, B.-H.; Li, X.-H.; Huang, Q.-N.; Song, L.-X.; Guo, D.; He, Y.; et al. A point mutation in an F-Box domain-containing protein is responsible for brown hull phenotype in rice. Rice Sci. 2016, 23, 1–8. [Google Scholar] [CrossRef]
- Zhernakov, A.I.; Shtark, O.Y.; Kulaeva, O.A.; Fedorina, J.V.; Afonin, A.M.; Kitaeva, A.B.; Tsyganov, V.E.; Afonso-Grunz, F.; Hoffmeier, K.; Rotter, B.; et al. Mapping-by-sequencing using NGS-based 3’-MACE-Seq reveals a new mutant allele of the essential nodulation gene Sym33 (IPD3) in pea (Pisum sativum L.). Peer J. 2019, 7, e6662. [Google Scholar] [CrossRef]
- Berger, J.D.; Buirchell, B.J.; Luckett, D.J.; Nelson, M.N. Domestication bottlenecks limit genetic diversity and constrain adaptation in narrow-leafed lupin (Lupinus angustifolius L.). Theor. Appl. Genet. 2012, 124, 637–652. [Google Scholar] [CrossRef]
- Cowling, W.A. Pedigrees and characteristics of narrow-leafed lupin cultivars released in Australia from 1967 to 1998. Bull. Agric. West. Aust. 1999, 4365, 4–11. [Google Scholar]
- Cowling, W.A.; Buirchell, B.J.; Falk, D.E. A model for incorporating novel alleles from the primary gene pool into elite crop breeding programs while reselecting major genes for domestication or adaptation. Crop Pasture Sci. 2009, 60, 1009–1015. [Google Scholar] [CrossRef]
- Mousavi-Derazmahalleh, M.; Bayer, P.E.; Nevado, B.; Hurgobin, B.; Filatov, D.; Kilian, A.; Kamphuis, L.G.; Singh, K.B.; Berger, J.D.; Hane, J.K.; et al. Exploring the genetic and adaptive diversity of a pan-Mediterranean crop wild relative: Narrow-leafed lupin. Theor. Appl. Genet. 2018, 131, 887–901. [Google Scholar] [CrossRef]
- Mousavi-Derazmahalleh, M.; Nevado, B.; Bayer, P.E.; Filatov, D.A.; Hane, J.K.; Edwards, D.; Erskine, W.; Nelson, M.N. The western Mediterranean region provided the founder population of domesticated narrow-leafed lupin. Theor. Appl. Genet. 2018, 131, 2543–2554. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, A.; Ausubel, F.M. A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 1993, 4, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Neff, M.M.; Neff, J.D.; Chory, J.; Pepper, A.E. dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: Experimental applications in Arabidopsis thaliana genetics. Plant J. 1998, 14, 387–392. [Google Scholar] [CrossRef]
- Neff, M.M.; Turk, E.; Kalishman, M. Web-based primer design for single nucleotide polymorphism analysis. Trends Genet. 2002, 18, 613–615. [Google Scholar] [CrossRef]
- Thiel, T.; Kota, R.; Grosse, I.; Stein, N.; Graner, A. SNP2CAPS: A SNP and INDEL analysis tool for CAPS marker development. Nucleic Acids Res. 2004, 32, e5. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).