Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System


Abstract
:1. Introduction
2. Molecular Properties of EAATs
3. Expression Patterns of EAATs
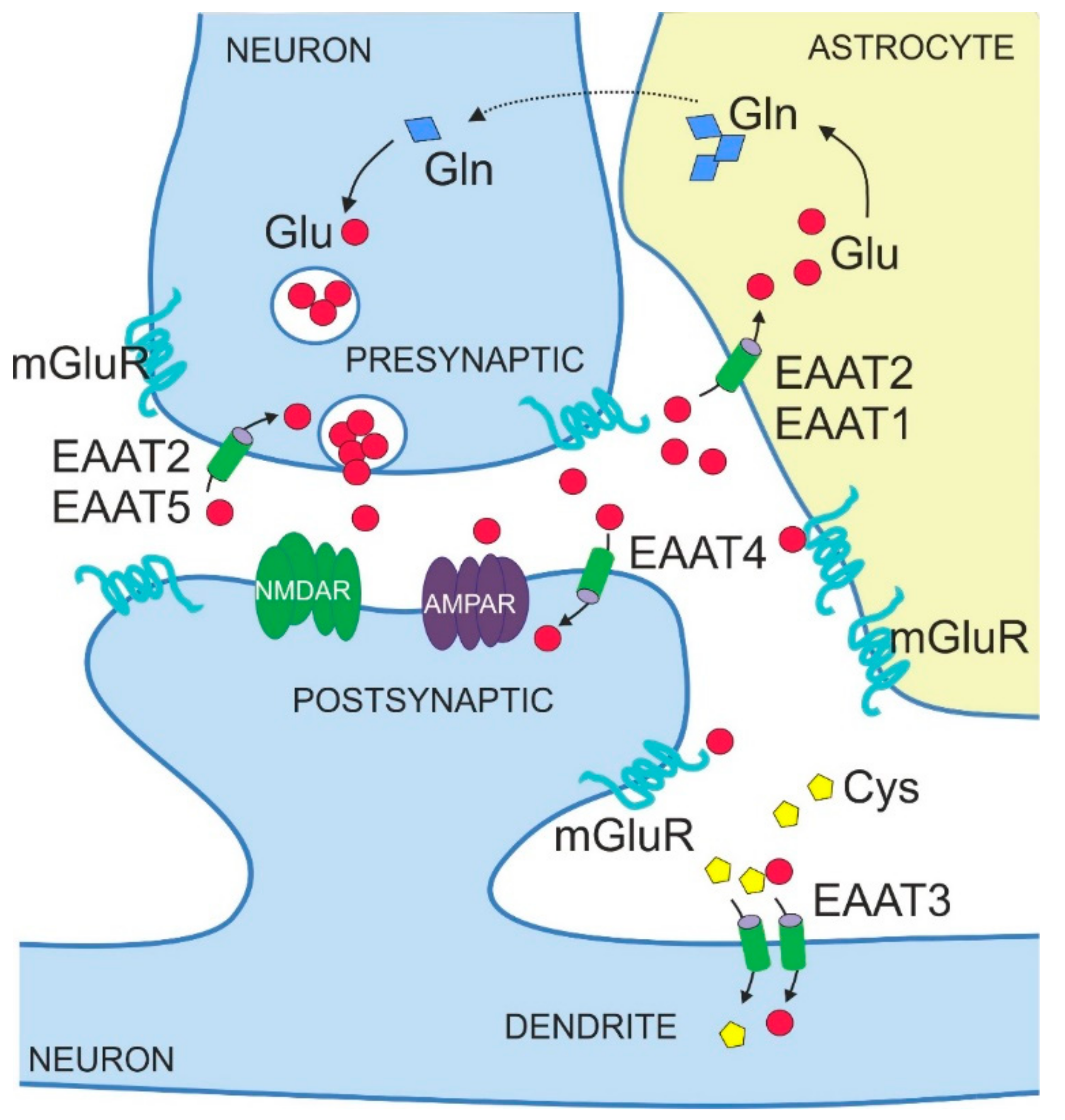
4. Physiological Functions of EAATs
5. Regulation of Expression Levels of EAATs
5.1. Regulation by Growth Factors, Polypeptides and Hormones
5.2. Regulation by Neuronal Activity
5.3. Regulation by Hypoxia and Oxidative Stress
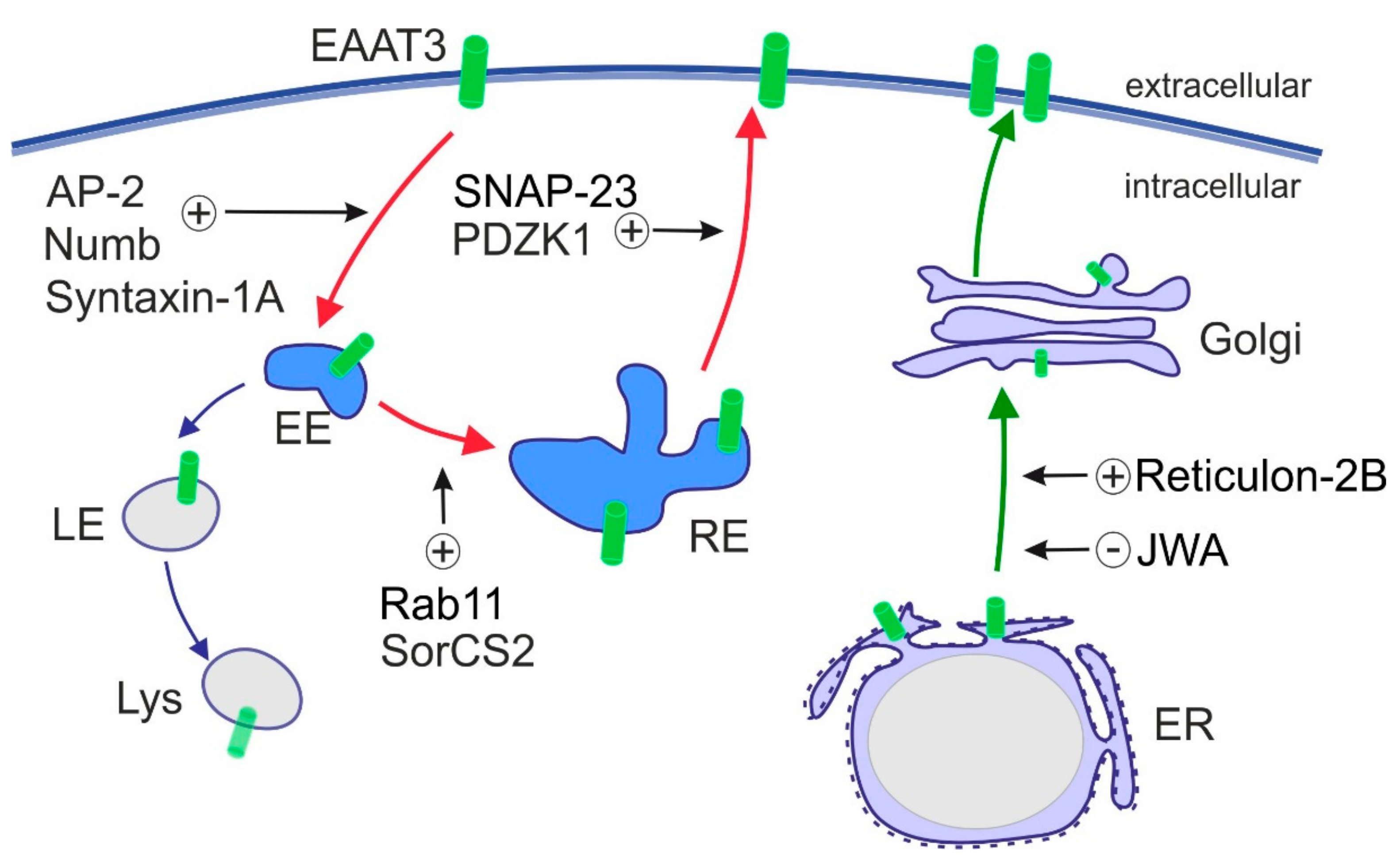
6. Regulation of Subcellular Localization of EAATs
7. EAATs in the CNS Pathology

{kind=link}
{kind=link}
{kind=link}
| Disease | EAAT | EAAT’s Activity Crucial for Preventing Pathology/Proposed Pathological Mechanism | Changes in Levels/Distribution in Course of the Disease | References |
|---|---|---|---|---|
| epilepsy | EAAT2 | EAAT2 prevents aberrant excitability and excitotoxic damage by removing excess glutamate | decreased levels | [64,163,164,165,166,167,168,169,170,171,172] |
| EAAT3 | EAAT3 has a protective role through involvement in GABA synthesis and in protection from oxidative damage | changes in subcellular distribution; increased levels in surviving neurons | [69,73,78,144,164,165,173,174,175,176] | |
| Alzheimer’s disease | EAAT2 | EAAT2 prevents excitotoxic damage by removing excess glutamate | decreased levels | [177,178,179,180,181,182,183,184,185,186,187] |
| Parkinson’s disease | EAAT3 | EAAT3 protects neurons from oxidative damage | shift to the plasma membrane in a mouse model of PD | [79,188] |
| Huntington’s disease | EAAT2 | EAAT2 prevents excitotoxic damage by removing excess glutamate | decreased levels | [189,190,191,192,193,194,195,196] |
| EAAT3 | EAAT3 protects neurons from oxidative damage | decreased levels; aberrant intracellular trafficking | [145,193,196] | |
| multiple sclerosis | EAAT2 | EAAT2 may be potentially protective by preventing excitotoxic damage by removing excess glutamate | inconsistent results | [111,197,198,199,200] |
| amyotrophic lateral sclerosis | EAAT2 | EAAT2 prevents excitotoxic damage by removing excess glutamate | decreased levels, aberrant splicing | [83,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215] |
| episodic ataxia (EA6) | EAAT1 | EAAT1 mutants show impaired glutamate uptake and alterations in anion conductance | mutations in EAAT1 coding gene identified in patients | [216,217,218,219,220,221] |
| spinocerebellar ataxias (SCA1, SCA5) | EAAT4 | Reduced EAAT4 activity impairs spontaneous activity of Purkinje cells and causes neuronal death | decreased levels in SCA1; aberrant intracellular trafficking and decreased levels in SCA5 | [148,222,223,224,225] |
| spinocerebellar ataxias (SCA5, SCA7) | EAAT1 | EAAT1 prevents excitotoxic damage by removing excess glutamate | decreased levels | [224,225,226,227,228] |
| ischemic stroke | EAAT2 | EAAT2 prevents excitotoxic damage by removing excess glutamate | decreased levels | [229,230,231,232,233] |
| EAAT3 | EAAT3 protects neurons from oxidative damage | increased levels | [231,234,235,236] |
7.1. Epilepsy
7.2. Alzheimer’s Disease
7.3. Parkinson’s Disease
7.4. Huntington’s Disease
7.5. Multiple Sclerosis
7.6. Amyotrophic Lateral Sclerosis
7.7. Cerebellar Diseases
7.8. Ischemic Stroke
8. Summary
Funding
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| ARE | antioxidant responsive element |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| EA | episodic ataxia |
| EAE | experimental autoimmune encephalomyelitis |
| EAAT | excitatory amino acid transporter |
| ET | essential tremor |
| GABA | γ-aminobutyric acid |
| GLAST | glutamate-aspartate transporter |
| GLT-1 | glutamate transporter 1 |
| GSH | glutathione |
| HD | Huntington’s disease |
| KO | knockout |
| mGluR | metabotropic glutamate receptor |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NAC | N-acetylcysteine |
| NMDAR | N-methyl-d-aspartate receptor |
| NPC | Niemann-Pick disease type C |
| Nrf2 | nuclear factor (erythroid-derived 2)-like 2 |
| OGD | oxygen-glucose deprivation |
| PC | Purkinje cell |
| PD | Parkinson’s disease |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PMA | phorbol 12-myristate 13-acetate |
| PPARγ | peroxisome proliferator-activated receptors γ |
| PTZ | pentylenetetrazol |
| SCA | spinocerebellar ataxia |
| SE | status epilepticus |
References
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S. Glutamate as a Neurotransmitter in the Brain: Review of Physiology and Pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [CrossRef] [PubMed]
- Moussawi, K.; Riegel, A.; Nair, S.; Kalivas, P.W. Extracellular Glutamate: Functional Compartments Operate in Different Concentration Ranges. Front. Syst. Neurosci. 2011, 5, 94. [Google Scholar] [CrossRef] [PubMed]
- Logan, W.J.; Snyder, S.H. Unique high affinity uptake systems for glycine, glutamic and aspartic acids in central nervous tissue of the rat. Nature 1971, 234, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Storck, T.; Schulte, S.; Hofmann, K.; Stoffel, W. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc. Natl. Acad. Sci. USA 1992, 89, 10955–10959. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Hediger, M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 1992, 360, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Fairman, W.A.; Vandenberg, R.J.; Arriza, J.L.; Kavanaugh, M.P.; Amara, S.G. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 1995, 375, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Eliasof, S.; Kavanaugh, M.P.; Amara, S.G. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. USA 1997, 94, 4155–4160. [Google Scholar] [CrossRef] [PubMed]
- Pines, G.; Danbolt, N.C.; Bjørås, M.; Zhang, Y.; Bendahan, A.; Eide, L.; Koepsell, H.; Storm-Mathisen, J.; Seeberg, E.; Kanner, B.I. Cloning and expression of a rat brain L-glutamate transporter. Nature 1992, 360, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Ziemens, D.; Untiet, V.; Fahlke, C. Molecular and cellular physiology of sodium-dependent glutamate transporters. Brain Res. Bull. 2018, 136, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Grewer, C.; Rauen, T. Electrogenic Glutamate Transporters in the CNS: Molecular Mechanism, Pre-steady-state Kinetics, and their Impact on Synaptic Signaling. J. Membr. Biol. 2005, 203, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, R.J.; Ryan, R.M. Mechanisms of Glutamate Transport. Physiol. Rev. 2013, 93, 1621–1657. [Google Scholar] [CrossRef] [PubMed]
- Barbour, B.; Brew, H.; Attwell, D. Electrogenic glutamate uptake in glial cells is activated by intracellular potassium. Nature 1988, 335, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Owe, S.G.; Marcaggi, P.; Attwell, D. The ionic stoichiometry of the GLAST glutamate transporter in salamander retinal glia. J. Physiol. 2006, 577, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Wadiche, J.I.; Amara, S.G.; Kavanaugh, M.P. Ion fluxes associated with excitatory amino acid transport. Neuron 1995, 15, 721–728. [Google Scholar] [CrossRef]
- Yernool, D.; Boudker, O.; Jin, Y.; Gouaux, E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 2004, 431, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Grewer, C.; Watzke, N.; Wiessner, M.; Rauen, T. Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc. Natl. Acad. Sci. USA 2000, 97, 9706–9711. [Google Scholar] [CrossRef] [PubMed]
- Wadiche, J.I.; Kavanaugh, M.P. Macroscopic and Microscopic Properties of a Cloned Glutamate Transporter/Chloride Channel. J. Neurosci. 1998, 18, 7650–7661. [Google Scholar] [CrossRef] [PubMed]
- Wadiche, J.I.; Arriza, J.L.; Amara, S.G.; Kavanaugh, M.P. Kinetics of a human glutamate transporter. Neuron 1995, 14, 1019–1027. [Google Scholar] [CrossRef]
- Mim, C.; Balani, P.; Rauen, T.; Grewer, C. The Glutamate Transporter Subtypes EAAT4 and EAATs 1-3 Transport Glutamate with Dramatically Different Kinetics and Voltage Dependence but Share a Common Uptake Mechanism. J. Gen. Physiol. 2005, 126, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, A.; Braams, S.; Rauen, T.; Grewer, C. The Discovery of Slowness: Low-Capacity Transport and Slow Anion Channel Gating by the Glutamate Transporter EAAT5. Biophys. J. 2011, 100, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Dehnes, Y.; Chaudhry, F.A.; Ullensvang, K.; Lehre, K.P.; Storm-Mathisen, J.; Danbolt, N.C. The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: A glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J. Neurosci. 1998, 18, 3606–3619. [Google Scholar] [CrossRef] [PubMed]
- Veruki, M.L.; Mørkve, S.H.; Hartveit, E. Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat. Neurosci. 2006, 9, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Cordeiro, S.; Machtens, J.-P.; Braams, S.; Rauen, T.; Fahlke, C. Functional properties of the retinal glutamate transporters GLT-1c and EAAT5. J. Biol. Chem. 2014, 289, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, M.; Bendahan, A.; Kanner, B.I. Biotinylation of Single Cysteine Mutants of the Glutamate Transporter GLT-1 from Rat Brain Reveals Its Unusual Topology. Neuron 1998, 21, 623–632. [Google Scholar] [CrossRef]
- Seal, R.P.; Leighton, B.H.; Amara, S.G. A Model for the Topology of Excitatory Amino Acid Transporters Determined by the Extracellular Accessibility of Substituted Cysteines. Neuron 2000, 25, 695–706. [Google Scholar] [CrossRef]
- Jiang, J.; Amara, S.G. New views of glutamate transporter structure and function: Advances and challenges. Neuropharmacology 2011, 60, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.K. Structure-Function Relationship of Transporters in the Glutamate–Glutamine Cycle of the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 1177. [Google Scholar] [CrossRef] [PubMed]
- Canul-Tec, J.C.; Assal, R.; Cirri, E.; Legrand, P.; Brier, S.; Chamot-Rooke, J.; Reyes, N. Structure and allosteric inhibition of excitatory amino acid transporter 1. Nature 2017, 544, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Haugeto, O.; Ullensvang, K.; Levy, L.M.; Chaudhry, F.A.; Honoré, T.; Nielsen, M.; Lehre, K.P.; Danbolt, N.C. Brain glutamate transporter proteins form homomultimers. J. Biol. Chem. 1996, 271, 27715–27722. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, S.; Kreman, M.; Kavanaugh, M.P.; Wright, E.M.; Zampighi, G.A. Pentameric assembly of a neuronal glutamate transporter. Proc. Natl. Acad. Sci. USA 2000, 97, 8641–8646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothmann, D.; Leinenweber, A.; Torres-Salazar, D.; Kovermann, P.; Hotzy, J.; Gameiro, A.; Grewer, C.; Fahlke, C. Hetero-oligomerization of neuronal glutamate transporters. J. Biol. Chem. 2011, 286, 3935–3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendreau, S.; Voswinkel, S.; Torres-Salazar, D.; Lang, N.; Heidtmann, H.; Detro-Dassen, S.; Schmalzing, G.; Hidalgo, P.; Fahlke, C. A trimeric quaternary structure is conserved in bacterial and human glutamate transporters. J. Biol. Chem. 2004, 279, 39505–39512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewer, C.; Balani, P.; Weidenfeller, C.; Bartusel, T.; Tao, Z.; Rauen, T. Individual subunits of the glutamate transporter EAAC1 homotrimer function independently of each other. Biochemistry 2005, 44, 11913–11923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, H.P.; Brown, R.L.; Larsson, H.P. The glutamate-activated anion conductance in excitatory amino acid transporters is gated independently by the individual subunits. J. Neurosci. 2007, 27, 2943–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leary, G.P.; Stone, E.F.; Holley, D.C.; Kavanaugh, M.P. The glutamate and chloride permeation pathways are colocalized in individual neuronal glutamate transporter subunits. J. Neurosci. 2007, 27, 2938–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, F.A.; Lehre, K.P.; van Lookeren Campagne, M.; Ottersen, O.P.; Danbolt, N.C.; Storm-Mathisen, J. Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 1995, 15, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Danbolt, N.C.; Storm-Mathisen, J.; Kanner, B.I. An [Na+ + K+]coupledL-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience 1992, 51, 295–310. [Google Scholar] [CrossRef]
- Lehre, K.P.; Levy, L.M.; Ottersen, O.P.; Storm-Mathisen, J.; Danbolt, N.C. Differential expression of two glial glutamate transporters in the rat brain: Quantitative and immunocytochemical observations. J. Neurosci. 1995, 15, 1835–1853. [Google Scholar] [CrossRef] [PubMed]
- Holmseth, S.; Dehnes, Y.; Huang, Y.H.; Follin-Arbelet, V.V.; Grutle, N.J.; Mylonakou, M.N.; Plachez, C.; Zhou, Y.; Furness, D.N.; Bergles, D.E.; et al. The Density of EAAC1 (EAAT3) Glutamate Transporters Expressed by Neurons in the Mammalian CNS. J. Neurosci. 2012, 32, 6000–6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pow, D.V.; Barnett, N.L. Developmental expression of excitatory amino acid transporter 5: A photoreceptor and bipolar cell glutamate transporter in rat retina. Neurosci. Lett. 2000, 280, 21–24. [Google Scholar] [CrossRef]
- Pitt, D.; Nagelmeier, I.E.; Wilson, H.C.; Raine, C.S. Glutamate uptake by oligodendrocytes: Implications for excitotoxicity in multiple sclerosis. Neurology 2003, 61, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- DeSilva, T.M.; Kabakov, A.Y.; Goldhoff, P.E.; Volpe, J.J.; Rosenberg, P.A. Regulation of Glutamate Transport in Developing Rat Oligodendrocytes. J. Neurosci. 2009, 29, 7898–7908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, M.R.; Huang, Y.H.; Kim, Y.S.; Dykes-Hoberg, M.I.; Jin, L.; Watkins, A.M.; Bergles, D.E.; Rothstein, J.D. Variations in Promoter Activity Reveal a Differential Expression and Physiology of Glutamate Transporters by Glia in the Developing and Mature CNS. J. Neurosci. 2007, 27, 6607–6619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, A.; Martin, L.J.; Lin, C.-L.G.; Dykes-Hoberg, M.; Rothstein, J.D. Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4. Neuroscience 1997, 81, 1031–1042. [Google Scholar] [CrossRef]
- Nagao, S.; Kwak, S.; Kanazawa, I. EAAT4, a glutamate transporter with properties of a chloride channel, is predominantly localized in Purkinje cell dendrites, and forms parasagittal compartments in rat cerebellum. Neuroscience 1997, 78, 929–933. [Google Scholar] [PubMed]
- Yamada, K.; Watanabe, M.; Shibata, T.; Tanaka, K.; Wada, K.; Inoue, Y. EAAT4 is a post-synaptic glutamate transporter at Purkinje cell synapses. Neuroreport 1996, 7, 2013–2017. [Google Scholar] [CrossRef] [PubMed]
- Rauen, T.; Rothstein, J.D.; Wässle, H. Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res. 1996, 286, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, A.E.; Durry, S.; Aida, T.; Stock, M.C.; Rüther, U.; Tanaka, K.; Rose, C.R.; Kafitz, K.W. Laminar and subcellular heterogeneity of GLAST and GLT-1 immunoreactivity in the developing postnatal mouse hippocampus. J. Comp. Neurol. 2014, 522, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C.; Furness, D.N.; Zhou, Y. Neuronal vs. glial glutamate uptake: Resolving the conundrum. Neurochem. Int. 2016, 98, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Furness, D.N.; Dehnes, Y.; Akhtar, A.Q.; Rossi, D.J.; Hamann, M.; Grutle, N.J.; Gundersen, V.; Holmseth, S.; Lehre, K.P.; Ullensvang, K.; et al. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: New insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 2008, 157, 80–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wersinger, E.; Schwab, Y.; Sahel, J.-A.; Rendon, A.; Pow, D.V.; Picaud, S.; Roux, M.J. The glutamate transporter EAAT5 works as a presynaptic receptor in mouse rod bipolar cells. J. Physiol. 2006, 577, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Furuta, A.; Rothstein, J.D.; Martin, L.J. Glutamate Transporter Protein Subtypes Are Expressed Differentially during Rat CNS Development. J. Neurosci. 1997, 17, 8363–8375. [Google Scholar] [CrossRef] [PubMed]
- Ullensvang, K.; Lehre, K.P.; Storm-Mathisen, J.; Danbolt, N.C. Differential Developmental Expression of the Two Rat Brain Glutamate Transporter Proteins GLAST and GLT. Eur. J. Neurosci. 1997, 9, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Kugler, P.; Schleyer, V. Developmental expression of glutamate transporters and glutamate dehydrogenase in astrocytes of the postnatal rat hippocampus. Hippocampus 2004, 14, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Arnth-Jensen, N.; Jabaudon, D.; Scanziani, M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat. Neurosci. 2002, 5, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Verbich, D.; Prenosil, G.A.; Chang, P.K.-Y.; Murai, K.K.; McKinney, R.A. Glial glutamate transport modulates dendritic spine head protrusions in the hippocampus. Glia 2012, 60, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Ullian, E.M.; Christopherson, K.S.; Barres, B.A. Role for glia in synaptogenesis. Glia 2004, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Raponi, E.; Agenes, F.; Delphin, C.; Assard, N.; Baudier, J.; Legraverend, C.; Deloulme, J.-C. S100B expression defines a state in which GFAP-expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia 2007, 55, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartfuss, E.; Galli, R.; Heins, N.; Götz, M. Characterization of CNS precursor subtypes and radial glia. Dev. Biol. 2001, 229, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, M.; Watanabe, Y.; Watanabe, M.; Tanaka, K.; Wada, K.; Takashima, S. Expression of a glutamate transporter subtype, EAAT4, in the developing human cerebellum. Brain Res. 1997, 767, 265–271. [Google Scholar] [CrossRef]
- Inage, Y.W.; Itoh, M.; Wada, K.; Takashima, S. Expression of Two Glutamate Transporters, GLAST and EAAT4, in the Human Cerebellum: Their Correlation in Development and Neonatal Hypoxic-Ischemic Damage. J. Neuropathol. Exp. Neurol. 1998, 57, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef] [PubMed]
- Watase, K.; Hashimoto, K.; Kano, M.; Yamada, K.; Watanabe, M.; Inoue, Y.; Okuyama, S.; Sakagawa, T.; Ogawa, S.-I.; Kawashima, N.; et al. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur. J. Neurosci. 1998, 10, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, R.-M.; Tanaka, K.; Heilig, M.; Holmes, A. Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: Rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biol. Psychiatry 2008, 64, 810–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, R.-M.; Tanaka, K.; Saksida, L.M.; Bussey, T.J.; Heilig, M.; Holmes, A. Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 2009, 34, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Perkins, E.M.; Clarkson, Y.L.; Suminaite, D.; Lyndon, A.R.; Tanaka, K.; Rothstein, J.D.; Skehel, P.A.; Wyllie, D.J.A.; Jackson, M. Loss of cerebellar glutamate transporters EAAT4 and GLAST differentially affects the spontaneous firing pattern and survival of Purkinje cells. Hum. Mol. Genet. 2018, 27, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Peghini, P.; Janzen, J.; Stoffel, W. Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J. 1997, 16, 3822–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, J.S. Neuronal Glutamate Transporters Limit Activation of NMDA Receptors by Neurotransmitter Spillover on CA1 Pyramidal Cells. J. Neurosci. 2001, 21, 8328–8338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scimemi, A.; Tian, H.; Diamond, J.S. Neuronal Transporters Regulate Glutamate Clearance, NMDA Receptor Activation, and Synaptic Plasticity in the Hippocampus. J. Neurosci. 2009, 29, 14581–14595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, F.; DeBiasi, S.; Minelli, A.; Rothstein, J.D.; Melone, M. EAAC1, a high-affinity glutamate tranporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb. Cortex 1998, 8, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepkuty, J.P.; Cohen, A.S.; Eccles, C.; Rafiq, A.; Behar, K.; Ganel, R.; Coulter, D.A.; Rothstein, J.D. A Neuronal Glutamate Transporter Contributes to Neurotransmitter GABA Synthesis and Epilepsy. J. Neurosci. 2002, 22, 6372–6379. [Google Scholar] [CrossRef] [PubMed]
- Watts, S.D.; Torres-Salazar, D.; Divito, C.B.; Amara, S.G. Cysteine transport through excitatory amino acid transporter 3 (EAAT3). PLoS ONE 2014, 9, e109245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerangue, N.; Kavanaugh, M.P. Interaction of L-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493 Pt 2, 419–423. [Google Scholar] [CrossRef]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Himi, T.; Ikeda, M.; Yasuhara, T.; Nishida, M.; Morita, I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J. Neural Transm. 2003, 110, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; Swanson, R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1-/- mouse. Ann. Neurol. 2011, 69, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Li, L.; Zuo, Z. N-acetylcysteine reverses existing cognitive impairment and increased oxidative stress in glutamate transporter type 3 deficient mice. Neuroscience 2012, 220, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.H.; Dykes-Hoberg, M.; Tanaka, K.; Rothstein, J.D.; Bergles, D.E. Climbing Fiber Activation of EAAT4 Transporters and Kainate Receptors in Cerebellar Purkinje Cells. J. Neurosci. 2004, 24, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Lozada, Z.; Guillem, A.M.; Robinson, M.B. Transcriptional Regulation of Glutamate Transporters: From Extracellular Signals to Transcription Factors. Adv. Pharmacol. 2016, 76, 103–145. [Google Scholar] [PubMed] [Green Version]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Hoberg, M.D.; Vidensky, S.; Chung, D.S.; et al. β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-G.; Su, Z.-Z.; Emdad, L.; Gupta, P.; Sarkar, D.; Borjabad, A.; Volsky, D.J.; Fisher, P.B. Mechanism of Ceftriaxone Induction of Excitatory Amino Acid Transporter-2 Expression and Glutamate Uptake in Primary Human Astrocytes. J. Biol. Chem. 2008, 283, 13116–13123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlag, B.D.; Vondrasek, J.R.; Munir, M.; Kalandadze, A.; Zelenaia, O.A.; Rothstein, J.D.; Robinson, M.B. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol. Pharmacol. 1998, 53, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Vanoni, C.; Bossi, M.; Massari, S.; Basudev, H.; Longhi, R.; Pietrini, G. The GLT-1 and GLAST Glutamate Transporters Are Expressed on Morphologically Distinct Astrocytes and Regulated by Neuronal Activity in Primary Hippocampal Cocultures. J. Neurochem. 2000, 75, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Figiel, M.; Engele, J. Pituitary adenylate cyclase-activating polypeptide (PACAP), a neuron-derived peptide regulating glial glutamate transport and metabolism. J. Neurosci. 2000, 20, 3596–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelenaia, O.; Schlag, B.D.; Gochenauer, G.E.; Ganel, R.; Song, W.; Beesley, J.S.; Grinspan, J.B.; Rothstein, J.D.; Robinson, M.B. Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Mol. Pharmacol. 2000, 57, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Figiel, M.; Maucher, T.; Rozyczka, J.; Bayatti, N.; Engele, J. Regulation of glial glutamate transporter expression by growth factors. Exp. Neurol. 2003, 183, 124–135. [Google Scholar] [CrossRef]
- Suzuki, K.; Ikegaya, Y.; Matsuura, S.; Kanai, Y.; Endou, H.; Matsuki, N. Transient upregulation of the glial glutamate transporter GLAST in response to fibroblast growth factor, insulin-like growth factor and epidermal growth factor in cultured astrocytes. J. Cell Sci. 2001, 114, 3717–3725. [Google Scholar] [PubMed]
- Aronica, E.; Gorter, J.A.; Ijlst-Keizers, H.; Rozemuller, A.J.; Yankaya, B.; Leenstra, S.; Troost, D. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: Opposite regulation of glutamate transporter proteins. Eur. J. Neurosci. 2003, 17, 2106–2118. [Google Scholar] [CrossRef]
- Bonde, C.; Sarup, A.; Schousboe, A.; Gegelashvili, G.; Noraberg, J.; Zimmer, J. GDNF pre-treatment aggravates neuronal cell loss in oxygen-glucose deprived hippocampal slice cultures: A possible effect of glutamate transporter up-regulation. Neurochem. Int. 2003, 43, 381–388. [Google Scholar] [CrossRef]
- Rodriguez-Kern, A.; Gegelashvili, M.; Schousboe, A.; Zhang, J.; Sung, L.; Gegelashvili, G. Beta-amyloid and brain-derived neurotrophic factor, BDNF, up-regulate the expression of glutamate transporter GLT-1/EAAT2 via different signaling pathways utilizing transcription factor NF-kappaB. Neurochem. Int. 2003, 43, 363–370. [Google Scholar] [CrossRef]
- Feng, D.; Guo, B.; Liu, G.; Wang, B.; Wang, W.; Gao, G.; Qin, H.; Wu, S. FGF2 alleviates PTSD symptoms in rats by restoring GLAST function in astrocytes via the JAK/STAT pathway. Eur. Neuropsychopharmacol. 2015, 25, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Webb, A.; Zerguine, A.; Choi, J.; Son, D.-S.; Lee, E. Mechanism of raloxifene-induced upregulation of glutamate transporters in rat primary astrocytes. Glia 2014, 62, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, J.; Brito, V.; Küppers, E.; Beyer, C. Regulation of glutamate transporter GLAST and GLT-1 expression in astrocytes by estrogen. Brain Res. Mol. Brain Res. 2005, 138, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Webb, A.; Smith, K.; Lee, K.; Son, D.-S.; Aschner, M.; Lee, E. cAMP response element-binding protein (CREB) and nuclear factor κB mediate the tamoxifen-induced up-regulation of glutamate transporter 1 (GLT-1) in rat astrocytes. J. Biol. Chem. 2013, 288, 28975–28986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Sidoryk-Wegrzynowicz, M.; Yin, Z.; Webb, A.; Son, D.-S.; Aschner, M. Transforming growth factor-α mediates estrogen-induced upregulation of glutamate transporter GLT-1 in rat primary astrocytes. Glia 2012, 60, 1024–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Sidoryk-Wêgrzynowicz, M.; Wang, N.; Webb, A.; Son, D.-S.; Lee, K.; Aschner, M. GPR30 Regulates Glutamate Transporter GLT-1 Expression in Rat Primary Astrocytes. J. Biol. Chem. 2012, 287, 26817–26828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Yuan, P.; Wu, J.; Huang, J. Estrogen regulates excitatory amino acid carrier 1 (EAAC1) expression through sphingosine kinase 1 (SphK1) transacting FGFR-mediated ERK signaling in rat C6 astroglial cells. Neuroscience 2016, 319, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Zschocke, J.; Bayatti, N.; Clement, A.M.; Witan, H.; Figiel, M.; Engele, J.; Behl, C. Differential promotion of glutamate transporter expression and function by glucocorticoids in astrocytes from various brain regions. J. Biol. Chem. 2005, 280, 34924–34932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autry, A.E.; Grillo, C.A.; Piroli, G.G.; Rothstein, J.D.; McEwen, B.S.; Reagan, L.P. Glucocorticoid regulation of GLT-1 glutamate transporter isoform expression in the rat hippocampus. Neuroendocrinology 2006, 83, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Kim, C.; Smith, K.; Son, D.-S.; Aschner, M.; Lee, E. Transcriptional regulation of the astrocytic excitatory amino acid transporter 1 (EAAT1) via NF-kB and Yin Yang 1 (YY1). J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitcheran, R.; Gupta, P.; Fisher, P.B.; Baldwin, A.S. Positive and negative regulation of EAAT2 by NF-kappaB: A role for N-myc in TNFalpha-controlled repression. EMBO J. 2005, 24, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.; Yang, Y.; Rothstein, J.D.; Robinson, M.B. Nuclear Factor-κB Contributes to Neuron-Dependent Induction of Glutamate Transporter-1 Expression in Astrocytes. J. Neurosci. 2011, 31, 9159–9169. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Magnus, T.; Jung, S. Autoantigen specific T cells inhibit glutamate uptake in astrocytes by decreasing expression of astrocytic glutamate transporter GLAST: A mechanism mediated by tumor necrosis factor-alpha. FASEB J. 2005, 19, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Rozyczka, J.; Figiel, M.; Engele, J. Endothelins negatively regulate glial glutamate transporter expression. Brain Pathol. 2004, 14, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Romera, C.; Hurtado, O.; Botella, S.H.; Lizasoain, I.; Cárdenas, A.; Fernández-Tomé, P.; Leza, J.C.; Lorenzo, P.; Moro, M.A. In Vitro Ischemic Tolerance Involves Upregulation of Glutamate Transport Partly Mediated by the TACE/ADAM17-Tumor Necrosis Factor-α Pathway. J. Neurosci. 2004, 24, 1350–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.-N.; Park, W.-K.; Nam, K.-H.; Song, D.-Y.; Kim, H.-S.; Baik, T.-K.; Woo, R.-S. Neuregulin 1 Controls Glutamate Uptake by Up-regulating Excitatory Amino Acid Carrier 1 (EAAC1). J. Biol. Chem. 2015, 290, 20233–20244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorlin, T.; Roginski, R.S.; Choudhury, K.; Nilsson, M.; Rönnbäck, L.; Hansson, E.; Eriksson, P.S. Regulation of the glial glutamate transporter GLT-1 by glutamate and delta-opioid receptor stimulation. FEBS Lett. 1998, 425, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Vallejo-Illarramendi, A.; Domercq, M.; Pérez-Cerdá, F.; Ravid, R.; Matute, C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol. Dis. 2006, 21, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gozen, O.; Watkins, A.; Lorenzini, I.; Lepore, A.; Gao, Y.; Vidensky, S.; Brennan, J.; Poulsen, D.; Won Park, J.; et al. Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron 2009, 61, 880–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, L.M.; Lehre, K.P.; Walaas, S.I.; Storm-Mathisen, J.; Danbolt, N.C. Down-regulation of Glial Glutamate Transporters after Glutamatergic Denervation in the Rat Brain. Eur. J. Neurosci. 1995, 7, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Genoud, C.; Quairiaux, C.; Steiner, P.; Hirling, H.; Welker, E.; Knott, G.W. Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol. 2006, 4, e343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-H.; You, J.-R.; Wei, K.-C.; Gean, P.-W. Stimulating ERK/PI3K/NFκB signaling pathways upon activation of mGluR2/3 restores OGD-induced impairment in glutamate clearance in astrocytes. Eur. J. Neurosci. 2014, 39, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Lyon, L.; Kew, J.N.C.; Corti, C.; Harrison, P.J.; Burnet, P.W.J. Altered hippocampal expression of glutamate receptors and transporters in GRM2 and GRM3 knockout mice. Synapse 2008, 62, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, L.; Rockenstein, E.; Mallory, M.; Hashimoto, M.; Masliah, E. Altered expression of glutamate transporters under hypoxic conditions in vitro. J. Neurosci. Res. 2001, 64, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Dallas, M.; Boycott, H.E.; Atkinson, L.; Miller, A.; Boyle, J.P.; Pearson, H.A.; Peers, C. Hypoxia suppresses glutamate transport in astrocytes. J. Neurosci. 2007, 27, 3946–3955. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.-F.; Zhou, L.; Xie, Y.-J.; Xu, S.-M.; Zhu, J.; Teng, P.; Shao, C.-Y.; Wang, Y.; Luo, J.-H.; Shen, Y. Upregulation of glutamate transporter GLT-1 by mTOR-Akt-NF-кB cascade in astrocytic oxygen-glucose deprivation. Glia 2013, 61, 1959–1975. [Google Scholar] [CrossRef] [PubMed]
- Llorente, I.L.; Landucci, E.; Pellegrini-Giampietro, D.E.; Fernández-López, A. Glutamate receptor and transporter modifications in rat organotypic hippocampal slice cultures exposed to oxygen–glucose deprivation: The contribution of cyclooxygenase-2. Neuroscience 2015, 292, 118–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, S.-J.; Chen, L.-Y.; Zhang, M.; Gong, J.-X.; Ma, Y.-X.; Zhang, J.-M.; Wang, Y.-J.; Hu, Y.-Y.; Sun, X.-C.; Li, W.-B.; et al. Intermittent Hypobaric Hypoxia Preconditioning Induced Brain Ischemic Tolerance by Up-Regulating Glial Glutamate Transporter-1 in Rats. Neurochem. Res. 2012, 37, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Li, P.; Mangin, J.-M.; Huntsman, M.; Gallo, V. Chronic perinatal hypoxia reduces glutamate-aspartate transporter function in astrocytes through the Janus kinase/signal transducer and activator of transcription pathway. J. Neurosci. 2011, 31, 17864–17871. [Google Scholar] [CrossRef] [PubMed]
- An, S.-J.; Kang, T.-C.; Park, S.-K.; Hwang, I.-K.; Cho, S.S.; Chung, M.-H.; Won, M.H. Oxidative DNA damage and alteration of glutamate transporter expressions in the hippocampal Ca1 area immediately after ischemic insult. Mol. Cells 2002, 13, 476–480. [Google Scholar] [PubMed]
- Escartin, C.; Won, S.J.; Malgorn, C.; Auregan, G.; Berman, A.E.; Chen, P.-C.; Déglon, N.; Johnson, J.A.; Suh, S.W.; Swanson, R.A. Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J. Neurosci. 2011, 31, 7392–7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butchbach, M.E.R.; Tian, G.; Guo, H.; Lin, C.-L.G. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: Importance for excitatory amino acid transporter localization and function. J. Biol. Chem. 2004, 279, 34388–34396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy-Royal, C.; Dupuis, J.P.; Varela, J.A.; Panatier, A.; Pinson, B.; Baufreton, J.; Groc, L.; Oliet, S.H.R. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 2015, 18, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kalandadze, A.; Wu, Y.; Robinson, M.B. Protein kinase C activation decreases cell surface expression of the GLT-1 subtype of glutamate transporter. Requirement of a carboxyl-terminal domain and partial dependence on serine 486. J. Biol. Chem. 2002, 277, 45741–45750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Sutherland, M.L. Glutamate transporter cluster formation in astrocytic processes regulates glutamate uptake activity. J. Neurosci. 2004, 24, 6301–6306. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Otsubo, Y.; Yatani, Y.; Shirakawa, H.; Kaneko, S. Mechanisms of substrate transport-induced clustering of a glial glutamate transporter GLT-1 in astroglial-neuronal cultures. Eur. J. Neurosci. 2008, 28, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Brouillet, E.; Gubellini, P.; Trioulier, Y.; Jacquard, C.; Smadja, C.; Knott, G.W.; Kerkerian-Le Goff, L.; Déglon, N.; Hantraye, P.; et al. Ciliary neurotrophic factor activates astrocytes, redistributes their glutamate transporters GLAST and GLT-1 to raft microdomains, and improves glutamate handling in vivo. J. Neurosci. 2006, 26, 5978–5989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Awabdh, S.; Gupta-Agarwal, S.; Sheehan, D.F.; Muir, J.; Norkett, R.; Twelvetrees, A.E.; Griffin, L.D.; Kittler, J.T. Neuronal activity mediated regulation of glutamate transporter GLT-1 surface diffusion in rat astrocytes in dissociated and slice cultures. Glia 2016, 64, 1252–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, A.; Noda, M.; Suzuki, S.O.; Goto, Y.; Kanahori, Y.; Rothstein, J.D.; Iwaki, T. Translocation of glutamate transporter subtype excitatory amino acid carrier 1 protein in kainic acid-induced rat epilepsy. Am. J. Pathol. 2003, 163, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.-X.; Shen, L.; Xia, P.; Tang, Y.-W.; Bao, L.; Pei, G. Syntaxin 1A promotes the endocytic sorting of EAAC1 leading to inhibition of glutamate transport. J. Cell Sci. 2006, 119, 3776–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, K.M.; Gonzalez, M.I.; Robinson, M.B. Rapid Trafficking of the Neuronal Glutamate Transporter, Eaac1: Evidence for Distinct Trafficking Pathways Differentially Regulated by Protein Kinase C and Platelet-Derived Growth Factor. J. Biol. Chem. 2004, 279, 34505–34513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, K.E.; Straff, D.J.; Weinstein, E.A.; Bannerman, P.G.; Correale, D.M.; Rothstein, J.D.; Robinson, M.B. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J. Neurosci. 1998, 18, 2475–2485. [Google Scholar] [CrossRef] [PubMed]
- Guillet, B.A.; Velly, L.J.; Canolle, B.; Masmejean, F.M.; Nieoullon, A.L.; Pisano, P. Differential regulation by protein kinases of activity and cell surface expression of glutamate transporters in neuron-enriched cultures. Neurochem. Int. 2005, 46, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Sims, K.D.; Straff, D.J.; Robinson, M.B. Platelet-derived growth factor rapidly increases activity and cell surface expression of the EAAC1 subtype of glutamate transporter through activation of phosphatidylinositol 3-kinase. J. Biol. Chem. 2000, 275, 5228–5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Vidensky, S.; Ruggiero, A.M.; Maier, S.; Sitte, H.H.; Rothstein, J.D. Reticulon RTN2B Regulates Trafficking and Function of Neuronal Glutamate Transporter EAAC1. J. Biol. Chem. 2008, 283, 6561–6571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggiero, A.M.; Liu, Y.; Vidensky, S.; Maier, S.; Jung, E.; Farhan, H.; Robinson, M.B.; Sitte, H.H.; Rothstein, J.D. The Endoplasmic Reticulum Exit of Glutamate Transporter Is Regulated by the Inducible Mammalian Yip6b/GTRAP3-18 Protein. J. Biol. Chem. 2008, 283, 6175–6183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, M.I.; Susarla, B.T.S.; Fournier, K.M.; Sheldon, A.L.; Robinson, M.B. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J. Neurochem. 2007, 103, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Fournier, K.M.; Robinson, M.B. A dominant-negative variant of SNAP-23 decreases the cell surface expression of the neuronal glutamate transporter EAAC1 by slowing constitutive delivery. Neurochem. Int. 2006, 48, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-F.; Wei, J.; Li, P.-S.; Miao, H.-H.; Ma, Y.-C.; Qu, Y.-X.; Xu, J.; Qin, J.; Li, B.-L.; Song, B.-L.; et al. Numb directs the subcellular localization of EAAT3 through binding the YxNxxF motif. J. Cell Sci. 2016, 129, 3104–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’ Amico, A.; Soragna, A.; Di Cairano, E.; Panzeri, N.; Anzai, N.; Vellea Sacchi, F.; Perego, C. The Surface Density of the Glutamate Transporter EAAC1 is Controlled by Interactions with PDZK1 and AP2 Adaptor Complexes. Traffic 2010, 11, 1455–1470. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.R.; Szydlowska, K.; Nizinska, K.; Asaro, A.; van Vliet, E.A.; Popp, O.; Dittmar, G.; Fritsche-Guenther, R.; Kirwan, J.A.; Nykjaer, A.; et al. SorCS2 Controls Functional Expression of Amino Acid Transporter EAAT3 and Protects Neurons from Oxidative Stress and Epilepsy-Induced Pathology. Cell Rep. 2019, 26, 2792–2804.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Valencia, A.; Sapp, E.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Aronin, N.; DiFiglia, M. Aberrant Rab11-Dependent Trafficking of the Neuronal Glutamate Transporter EAAC1 Causes Oxidative Stress and Cell Death in Huntington’s Disease. J. Neurosci. 2010, 30, 4552–4561. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Song, W.; Liu, M.Y.; Jin, L.; Dykes-Hoberg, M.; Lin, C.I.; Bowers, W.J.; Federoff, H.J.; Sternweis, P.C.; Rothstein, J.D. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature 2001, 410, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Dick, K.A.; Weatherspoon, M.R.; Gincel, D.; Armbrust, K.R.; Dalton, J.C.; Stevanin, G.; Dürr, A.; Zühlke, C.; Bürk, K.; et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet. 2006, 38, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, Y.L.; Gillespie, T.; Perkins, E.M.; Lyndon, A.R.; Jackson, M. Beta-III spectrin mutation L253P associated with spinocerebellar ataxia type 5 interferes with binding to Arp1 and protein trafficking from the Golgi. Hum. Mol. Genet. 2010, 19, 3634–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkin, G.M.; Udawela, M.; Gibbons, A.; Dean, B. Glutamate transporters, EAAT1 and EAAT2, are potentially important in the pathophysiology and treatment of schizophrenia and affective disorders. World J. Psychiatry 2018, 8, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kaneko, S. SLC1 glutamate transporters and diseases: Psychiatric diseases and pathological pain. Curr. Mol. Pharmacol. 2013, 6, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.-Y.; Wu, D.C.; Zhou, N. Astrocytic Regulation of Glutamate Transmission in Schizophrenia. Front. Psychiatry 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donovan, S.M.; Sullivan, C.R.; McCullumsmith, R.E. The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders. NPJ Schizophr. 2017, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Barker-Haliski, M.; White, H.S. Glutamatergic Mechanisms Associated with Seizures and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandini, F.; Porter, R.H.; Greenamyre, J.T. Glutamate and Parkinson’s disease. Mol. Neurobiol. 1996, 12, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Alford, M.; DeTeresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W.; Rothman, S.M. The Role of Glutamate Neurotoxicity in Hypoxic-Ischemic Neuronal Death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Domercq, M.; Sánchez-Gómez, M.-V. Glutamate-mediated glial injury: Mechanisms and clinical importance. Glia 2006, 53, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.; Zivkovic, N.; Stojanovic, I. Multiple sclerosis and glutamate excitotoxicity. Rev. Neurosci. 2013, 24, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C.; Tovar-y-Romo, L.B.; Tapia, R. Glutamate excitotoxicity and therapeutic targets for amyotrophic lateral sclerosis. Expert Opin. Ther. Targets 2007, 11, 1415–1428. [Google Scholar] [CrossRef] [PubMed]
- Obrenovitch, T.P.; Urenjak, J.; Zilkha, E.; Jay, T.M. Excitotoxicity in neurological disorders--the glutamate paradox. Int. J. Dev. Neurosci. 2000, 18, 281–287. [Google Scholar] [CrossRef]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, J.A.; Szu, J.I.; Yonan, J.M.; Binder, D.K. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp. Neurol. 2016, 283, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, T.; Ueda, Y.; Nagatomo, K.; Willmore, L.J. Role of Glutamate and GABA Transporters in Development of Pentylenetetrazol-Kindling. Neurochem. Res. 2009, 34, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Proper, E.A.; Hoogland, G.; Kappen, S.M.; Jansen, G.H.; Rensen, M.G.A.; Schrama, L.H.; van Veelen, C.W.M.; van Rijen, P.C.; van Nieuwenhuizen, O.; Gispen, W.H.; et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain J. Neurol. 2002, 125, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, G.S.; Kabakov, A.Y.; Hameed, M.Q.; Dhamne, S.C.; Rosenberg, P.A.; Rotenberg, A. Ceftriaxone Treatment after Traumatic Brain Injury Restores Expression of the Glutamate Transporter, GLT-1, Reduces Regional Gliosis, and Reduces Post-Traumatic Seizures in the Rat. J. Neurotrauma 2013, 30, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Ess, K.C.; Uhlmann, E.J.; Jansen, L.A.; Li, W.; Crino, P.B.; Mennerick, S.; Yamada, K.A.; Gutmann, D.H. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann. Neurol. 2003, 54, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.-H.; Ouyang, Y.; Gazit, V.; Cirrito, J.R.; Jansen, L.A.; Ess, K.C.; Yamada, K.A.; Wozniak, D.F.; Holtzman, D.M.; Gutmann, D.H.; et al. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol. Dis. 2007, 28, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petr, G.T.; Sun, Y.; Frederick, N.M.; Zhou, Y.; Dhamne, S.C.; Hameed, M.Q.; Miranda, C.; Bedoya, E.A.; Fischer, K.D.; Armsen, W.; et al. Conditional Deletion of the Glutamate Transporter GLT-1 Reveals That Astrocytic GLT-1 Protects against Fatal Epilepsy While Neuronal GLT-1 Contributes Significantly to Glutamate Uptake into Synaptosomes. J. Neurosci. 2015, 35, 5187–5201. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Takahashi, K.; Schulte, D.; Stouffer, N.; Lin, Y.; Lin, C.-L.G. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol. Dis. 2012, 47, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.-H.; Bero, A.W.; Zhang, B.; Holtzman, D.M.; Wong, M. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of Tuberous Sclerosis Complex. Neurobiol. Dis. 2010, 37, 764–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, L.; Wang, X.; Li, J.; Shi, X.; Wu, L.; Shen, Y.; Xu, Q. Pharmacologic inhibition of Hsp90 to prevent GLT-1 degradation as an effective therapy for epilepsy. J. Exp. Med. 2017, 214, 547–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crino, P.B.; Jin, H.; Shumate, M.D.; Robinson, M.B.; Coulter, D.A.; Brooks-Kayal, A.R. Increased Expression of the Neuronal Glutamate Transporter (EAAT3/EAAC1) in Hippocampal and Neocortical Epilepsy. Epilepsia 2002, 43, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, H.P.; Levey, A.I.; Rothstein, J.D.; Tzingounis, A.V.; Conn, P.J. Alterations in Glutamate Transporter Protein Levels in Kindling-Induced Epilepsy. J. Neurochem. 1997, 68, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Voutsinos-Porche, B.; Koning, E.; Cléent, Y.; Kaplan, H.; Ferrandon, A.; Motte, J.; Nehlig, A. EAAC1 Glutamate Transporter Expression in the Rat Lithium-Pilocarpine Model of Temporal Lobe Epilepsy. J. Cereb. Blood Flow Metab. 2006, 26, 1419–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.R.; Porter, B.E.; Buckley, P.T.; Eberwine, J.H.; Robinson, M.B. mRNA for the EAAC1 subtype of glutamate transporter is present in neuronal dendrites in vitro and dramatically increases in vivo after a seizure. Neurochem. Int. 2011, 58, 366–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, C.P.; Koutsilieri, E.; Bartl, J.; Neuen-Jacob, E.; Arzberger, T.; Zander, N.; Ravid, R.; Roggendorf, W.; Riederer, P.; Grünblatt, E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J. Alzheimers Dis. 2007, 11, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, A.; Tsunoda, A.; Yamamoto, T.; Tada, M.; Kakita, A.; Ugawa, Y. Altered expression of glutamate transporter-1 and water channel protein aquaporin-4 in human temporal cortex with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2018, 44, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Nakano, M.; Kubota, K.; Himuro, N.; Mizoguchi, S.; Chikenji, T.; Otani, M.; Mizue, Y.; Nagaishi, K.; Fujimiya, M. Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 2018, 8, 1712. [Google Scholar] [CrossRef] [PubMed]
- Audrain, M.; Fol, R.; Dutar, P.; Potier, B.; Billard, J.-M.; Flament, J.; Alves, S.; Burlot, M.-A.; Dufayet-Chaffaud, G.; Bemelmans, A.-P.; et al. Alzheimer’s disease-like APP processing in wild-type mice identifies synaptic defects as initial steps of disease progression. Mol. Neurodegener. 2016, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Serviddio, G.; Gaetani, S.; Romano, A.; Dipasquale, P.; Cianci, S.; Bellanti, F.; Laconca, L.; Romano, A.D.; Padalino, I.; et al. Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol. Aging 2012, 33, 1121.e1–1121.e12. [Google Scholar] [CrossRef] [PubMed]
- Zumkehr, J.; Rodriguez-Ortiz, C.J.; Cheng, D.; Kieu, Z.; Wai, T.; Hawkins, C.; Kilian, J.; Lim, S.L.; Medeiros, R.; Kitazawa, M. Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2260–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schallier, A.; Smolders, I.; van Dam, D.; Loyens, E.; De Deyn, P.P.; Michotte, A.; Michotte, Y.; Massie, A. Region- and age-specific changes in glutamate transport in the AβPP23 mouse model for Alzheimer’s disease. J. Alzheimers Dis. 2011, 24, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Tong, H.; Lei, M.; Zhou, M.; Guo, W.; Li, G.; Tang, X.; Li, Z.; Mo, M.; Zhang, X.; et al. Astrocytic glutamatergic transporters are involved in Aβ-induced synaptic dysfunction. Brain Res. 2018, 1678, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid-β1–42 Slows Clearance of Synaptically Released Glutamate by Mislocalizing Astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, P.; Green, P.S.; Watson, G.S.; Marques, M.A.; Tanaka, K.; Meeker, K.D.; Meabon, J.S.; Li, N.; Zhu, P.; Olson, V.G.; et al. GLT-1 Loss Accelerates Cognitive Deficit Onset in an Alzheimer’s Disease Animal Model. J. Alzheimers Dis. 2011, 26, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Xian, X.; Li, L.; Yao, X.; Hu, Y.; Zhang, M.; Li, W. Ceftriaxone Improves Cognitive Function and Upregulates GLT-1-Related Glutamate-Glutamine Cycle in APP/PS1 Mice. J. Alzheimers Dis. 2018, 66, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Matsumura, N.; Watabe, M.; Nakaki, T. Oxidative stress on EAAC1 is involved in MPTP-induced glutathione depletion and motor dysfunction. Eur. J. Neurosci. 2008, 27, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Arzberger, T.; Krampfl, K.; Leimgruber, S.; Weindl, A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease--an in situ hybridization study. J. Neuropathol. Exp. Neurol. 1997, 56, 440–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liévens, J.C.; Woodman, B.; Mahal, A.; Spasic-Boscovic, O.; Samuel, D.; Kerkerian-Le Goff, L.; Bates, G.P. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol. Dis. 2001, 8, 807–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, P.F.; Franz, P.; Woodman, B.; Lindenberg, K.S.; Landwehrmeyer, G.B. Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the Huntington mutation. Brain J. Neurol. 2002, 125, 1908–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.-Y.; Fang, Z.-H.; Yu, Z.-X.; Wang, C.-E.; Li, S.-H.; Li, X.-J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Petr, G.T.; Schultheis, L.A.; Hussey, K.C.; Sun, Y.; Dubinsky, J.M.; Aoki, C.; Rosenberg, P.A. Decreased expression of GLT-1 in the R6/2 model of Huntington’s disease does not worsen disease progression. Eur. J. Neurosci. 2013, 38, 2477–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, J.; Shin, J.-Y.; Roberts, M.; Wang, C.-E.; Li, X.-J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. USA 2009, 106, 22480–22485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.R.; Dorner, J.L.; Shou, M.; Sari, Y.; Barton, S.J.; Sengelaub, D.R.; Kennedy, R.T.; Rebec, G.V. Up-regulation of GLT1 Expression Increases Glutamate Uptake and Attenuates the Huntington’s Disease Phenotype in the R6/2 Mouse. Neuroscience 2008, 153, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgoh, M.; Hanada, T.; Smith, T.; Hashimoto, T.; Ueno, M.; Yamanishi, Y.; Watanabe, M.; Nishizawa, Y. Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002, 125, 170–178. [Google Scholar] [CrossRef]
- Sulkowski, G.; Dąbrowska-Bouta, B.; Salińska, E.; Strużyńska, L. Modulation of Glutamate Transport and Receptor Binding by Glutamate Receptor Antagonists in EAE Rat Brain. PLoS ONE 2014, 9, e113954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitosek-Szewczyk, K.; Sulkowski, G.; Stelmasiak, Z.; Struzyńska, L. Expression of glutamate transporters GLT-1 and GLAST in different regions of rat brain during the course of experimental autoimmune encephalomyelitis. Neuroscience 2008, 155, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, M.; Merola, A.; Piacentino, C.; Votta, B.; Capello, E.; Mancardi, G.L.; Mutani, R.; Giordana, M.T.; Cavalla, P. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: Correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J. Neuropathol. Exp. Neurol. 2007, 66, 732–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Komori, T.; Iwata, M. Excitatory amino acid transporter 1 and 2 immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol. 2000, 100, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Lin, C.L.; Rothstein, J.D.; Geller, B.A.; Hosler, B.A.; Munsat, T.L.; Horvitz, H.R.; Brown, R.H. Mutations in the glutamate transporter EAAT2 gene do not cause abnormal EAAT2 transcripts in amyotrophic lateral sclerosis. Ann. Neurol. 1998, 43, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Aoki, M.; Pasinelli, P.; Berger, U.V.; Danbolt, N.C.; Brown, R.H.; Hediger, M.A. Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity. J. Biol. Chem. 2001, 276, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.L.; Bristol, L.A.; Jin, L.; Dykes-Hoberg, M.; Crawford, T.; Clawson, L.; Rothstein, J.D. Aberrant RNA processing in a neurodegenerative disease: The cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 1998, 20, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Bendotti, C.; Tortarolo, M.; Suchak, S.K.; Calvaresi, N.; Carvelli, L.; Bastone, A.; Rizzi, M.; Rattray, M.; Mennini, T. Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J. Neurochem. 2001, 79, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Duan, W.; Li, Z.; Huang, J.; Yin, Y.; Zhang, K.; Wang, Q.; Zhang, Z.; Li, C. Decreased GLT-1 and increased SOD1 and HO-1 expression in astrocytes contribute to lumbar spinal cord vulnerability of SOD1-G93A transgenic mice. FEBS Lett. 2010, 584, 1615–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, A.C.; Wong, V.; Benson, L.M.; Dykes, M.; Tanaka, K.; Rothstein, J.D.; Maragakis, N.J. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1G93A mice. Exp. Neurol. 2006, 201, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, C.; Massari, S.; Losa, M.; Carrega, P.; Perego, C.; Conforti, L.; Pietrini, G. Increased internalisation and degradation of GLT-1 glial glutamate transporter in a cell model for familial amyotrophic lateral sclerosis (ALS). J. Cell Sci. 2004, 117, 5417–5426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotti, D.; Rolfs, A.; Danbolt, N.C.; Brown, R.H.; Hediger, M.A. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat. Neurosci. 1999, 2, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.-G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Lai, L.; Butchbach, M.E.R.; Stockinger, M.P.; Shan, X.; Bishop, G.A.; Lin, C.G. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum. Mol. Genet. 2003, 12, 2519–2532. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Hala, T.J.; Seetharam, S.; Poulsen, D.J.; Wright, M.C.; Lepore, A.C. GLT1 overexpression in SOD1(G93A) mouse cervical spinal cord does not preserve diaphragm function or extend disease. Neurobiol. Dis. 2015, 78, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Cudkowicz, M.E.; Titus, S.; Kearney, M.; Yu, H.; Sherman, A.; Schoenfeld, D.; Hayden, D.; Shui, A.; Brooks, B.; Conwit, R.; et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: A multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014, 13, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Jen, J.C.; Wan, J.; Palos, T.P.; Howard, B.D.; Baloh, R.W. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 2005, 65, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Vries, B.; de Mamsa, H.; Stam, A.H.; Wan, J.; Bakker, S.L.M.; Vanmolkot, K.R.J.; Haan, J.; Terwindt, G.M.; Boon, E.M.J.; Howard, B.D.; et al. Episodic Ataxia Associated with EAAT1 Mutation C186S Affecting Glutamate Reuptake. Arch. Neurol. 2009, 66, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Winter, N.; Kovermann, P.; Fahlke, C. A point mutation associated with episodic ataxia 6 increases glutamate transporter anion currents. Brain J. Neurol. 2012, 135, 3416–3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parinejad, N.; Peco, E.; Ferreira, T.; Stacey, S.M.; van Meyel, D.J. Disruption of an EAAT-Mediated Chloride Channel in a Drosophila Model of Ataxia. J. Neurosci. 2016, 36, 7640–7647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.-D.; Jen, J.C.; Choi, S.Y.; Shin, J.-H.; Kim, H.-S.; Kim, H.-J.; Kim, J.-S.; Choi, J.-H. Late-onset episodic ataxia associated with SLC1A3 mutation. J. Hum. Genet. 2017, 62, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Smertenko, T.; Bargiela, D.; Griffin, H.; Duff, J.; Appleton, M.; Douroudis, K.; Pfeffer, G.; Santibanez-Koref, M.; Eglon, G.; et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 2015, 138, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Antalffy, B.; Kang, D.; Orr, H.T.; Zoghbi, H.Y. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat. Neurosci. 2000, 3, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Serra, H.G.; Byam, C.E.; Lande, J.D.; Tousey, S.K.; Zoghbi, H.Y.; Orr, H.T. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum. Mol. Genet. 2004, 13, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Perkins, E.M.; Clarkson, Y.L.; Sabatier, N.; Longhurst, D.M.; Millward, C.P.; Jack, J.; Toraiwa, J.; Watanabe, M.; Rothstein, J.D.; Lyndon, A.R.; et al. Loss of beta-III spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J. Neurosci. 2010, 30, 4857–4867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankewich, M.C.; Gwynn, B.; Ardito, T.; Ji, L.; Kim, J.; Robledo, R.F.; Lux, S.E.; Peters, L.L.; Morrow, J.S. Targeted deletion of betaIII spectrin impairs synaptogenesis and generates ataxic and seizure phenotypes. Proc. Natl. Acad. Sci. USA 2010, 107, 6022–6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, E.M.; Suminaite, D.; Clarkson, Y.L.; Lee, S.K.; Lyndon, A.R.; Rothstein, J.D.; Wyllie, D.J.A.; Tanaka, K.; Jackson, M. Posterior cerebellar Purkinje cells in an SCA5/SPARCA1 mouse model are especially vulnerable to the synergistic effect of loss of β-III spectrin and GLAST. Hum. Mol. Genet. 2016, 25, 4448–4461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvetanovic, M. Decreased Expression of Glutamate Transporter GLAST in Bergmann Glia Is Associated with the Loss of Purkinje Neurons in the Spinocerebellar Ataxia Type 1. The Cerebellum 2015, 14, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Custer, S.K.; Garden, G.A.; Gill, N.; Rueb, U.; Libby, R.T.; Schultz, C.; Guyenet, S.J.; Deller, T.; Westrum, L.E.; Sopher, B.L.; et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat. Neurosci. 2006, 9, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Mallolas, J.; Hurtado, O.; Castellanos, M.; Blanco, M.; Sobrino, T.; Serena, J.; Vivancos, J.; Castillo, J.; Lizasoain, I.; Moro, M.A.; et al. A polymorphism in the EAAT2 promoter is associated with higher glutamate concentrations and higher frequency of progressing stroke. J. Exp. Med. 2006, 203, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-C.; Hsu-Chou, H.; Lu, J.-L.; Chiang, Y.-C.; Huang, H.-M.; Wang, H.-L.; Wu, T.; Liao, J.-J.; Yeh, T.-S. Down-regulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology 2005, 49, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.L.; Dogan, A.; Todd, K.G.; Bowen, K.K.; Kim, B.T.; Rothstein, J.D.; Dempsey, R.J. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J. Neurosci. 2001, 21, 1876–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzyzanowska, W.; Pomierny, B.; Budziszewska, B.; Filip, M.; Pera, J. N-Acetylcysteine and Ceftriaxone as Preconditioning Strategies in Focal Brain Ischemia: Influence on Glutamate Transporters Expression. Neurotox. Res. 2016, 29, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romera, C.; Hurtado, O.; Mallolas, J.; Pereira, M.P.; Morales, J.R.; Romera, A.; Serena, J.; Vivancos, J.; Nombela, F.; Lorenzo, P.; et al. Ischemic Preconditioning Reveals that GLT1/EAAT2 Glutamate Transporter is a Novel PPARγ Target Gene Involved in Neuroprotection. J. Cereb. Blood Flow Metab. 2007, 27, 1327–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, S.J.; Yoo, B.H.; Brennan, A.M.; Shin, B.S.; Kauppinen, T.M.; Berman, A.E.; Swanson, R.A.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J. Neurosci. 2010, 30, 15409–15418. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zuo, Z. Glutamate transporter type 3 knockout reduces brain tolerance to focal brain ischemia in mice. J. Cereb. Blood Flow Metab. 2011, 31, 1283–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.Y.; Won, S.J.; Kim, J.H.; Sohn, M.; Song, H.K.; Chung, T.N.; Kim, T.Y.; Suh, S.W. EAAC1 gene deletion reduces adult hippocampal neurogenesis after transient cerebral ischemia. Sci. Rep. 2018, 8, 6903. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE Official Report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- During, M.J.; Spencer, D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet Lond. Engl. 1993, 341, 1607–1610. [Google Scholar] [CrossRef]
- Bailey, C.G.; Ryan, R.M.; Thoeng, A.D.; Ng, C.; King, K.; Vanslambrouck, J.M.; Auray-Blais, C.; Vandenberg, R.J.; Bröer, S.; Rasko, J.E.J. Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J. Clin. Investig. 2011, 121, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinc, B.; Grabnar, I.; Vovk, T. Antioxidants as a Preventive Treatment for Epileptic Process: A Review of the Current Status. Curr. Neuropharmacol. 2014, 12, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.-J.; Jeong, J.H.; Chung, Y.H.; Kim, W.-K.; Ko, K.-H.; Bach, J.-H.; Hong, J.-S.; Yoneda, Y.; Kim, H.-C. Role of oxidative stress in epileptic seizures. Neurochem. Int. 2011, 59, 122–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, J.; González, M.E.; Lorigados, L.; Morales, L.; Riverón, G.; Bauzá, J.Y. Oxidative stress markers in surgically treated patients with refractory epilepsy. Clin. Biochem. 2007, 40, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Tampellini, D. Synaptic activity and Alzheimer’s disease: A critical update. Front. Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, H.S.; Dodd, P.R. Glutamate–glutamine cycling in Alzheimer’s disease. Neurochem. Int. 2007, 50, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, R.; Hardy, J.; Roberts, P.; Briggs, R. Presynaptic and postsynaptic glutamatergic function in Alzheimer’s disease. Neurosci. Lett. 1988, 86, 109–113. [Google Scholar] [CrossRef]
- Liang, Z.; Valla, J.; Sefidvash-Hockley, S.; Rogers, J.; Li, R. Effects of estrogen treatment on glutamate uptake in cultured human astrocytes derived from cortex of Alzheimer’s disease patients. J. Neurochem. 2002, 80, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Esparcia, P.; Diaz-Lucena, D.; Ainciburu, M.; Torrejón-Escribano, B.; Carmona, M.; Llorens, F.; Ferrer, I. Glutamate Transporter GLT1 Expression in Alzheimer Disease and Dementia with Lewy Bodies. Front. Aging Neurosci. 2018, 10, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerson, K.; Woltjer, R.L.; Mookherjee, P.; Leverenz, J.B.; Montine, T.J.; Bird, T.D.; Pow, D.V.; Rauen, T.; Cook, D.G. Detergent-Insoluble EAAC1/EAAT3 Aberrantly Accumulates in Hippocampal Neurons of Alzheimer’s Disease Patients. Brain Pathol. Zurich Switz. 2009, 19, 267–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F.; Armentero, M.-T. Animal models of Parkinson’s disease. FEBS J. 2012, 279, 1156–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assous, M.; Had-Aissouni, L.; Gubellini, P.; Melon, C.; Nafia, I.; Salin, P.; Kerkerian-Le-Goff, L.; Kachidian, P. Progressive Parkinsonism by acute dysfunction of excitatory amino acid transporters in the rat substantia nigra. Neurobiol. Dis. 2014, 65, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Chotibut, T.; Davis, R.W.; Arnold, J.C.; Frenchek, Z.; Gurwara, S.; Bondada, V.; Geddes, J.W.; Salvatore, M.F. Ceftriaxone increases glutamate uptake and reduces striatal tyrosine hydroxylase loss in 6-OHDA Parkinson’s model. Mol. Neurobiol. 2014, 49, 1282–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD Gene with an Expanded CAG Repeat Is Sufficient to Cause a Progressive Neurological Phenotype in Transgenic Mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Slow, E.J.; van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.-Z.; et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Hassel, B.; Tessler, S.; Faull, R.L.M.; Emson, P.C. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem. Res. 2008, 33, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Kang, M.H.; Askew, C.; Kang, R.; Sanders, S.S.; Wan, J.; Davis, N.G.; Hayden, M.R. Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2010, 40, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Nylander, A.; Hafler, D.A. Multiple sclerosis. J. Clin. Investig. 2012, 122, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.P.; Richards, M.H.; Miller, S.D. Mouse models of multiple sclerosis: Experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol. Biol. 2012, 900, 381–401. [Google Scholar] [PubMed] [Green Version]
- Brambilla, R. The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2019, 137, 757–783. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Farez, M.F. The Role of Astrocytes in Multiple Sclerosis Progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef] [PubMed]
- Groom, A.J.; Smith, T.; Turski, L. Multiple Sclerosis and Glutamate. Ann. N. Y. Acad. Sci. 2003, 993, 229–275. [Google Scholar] [CrossRef] [PubMed]
- Werner, P.; Pitt, D.; Raine, C.S. Multiple sclerosis: Altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann. Neurol. 2001, 50, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.N.; Lim, J.L.; Nijland, P.G.; Witte, M.E.; van Horssen, J. Glutathione in multiple sclerosis: More than just an antioxidant? Mult. Scler. J. 2014, 20, 1425–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linker, R.A.; Lee, D.-H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain J. Neurol. 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowicka, N.; Juranek, J.; Juranek, J.K.; Wojtkiewicz, J. Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2019, 20, 2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The Glutamate Hypothesis in ALS: Pathophysiology and Drug Development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaitakis, A.; Caroscio, J.T. Abnormal glutamate metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 1987, 22, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Tsai, G.; Kuncl, R.W.; Clawson, L.; Cornblath, D.R.; Drachman, D.B.; Pestronk, A.; Stauch, B.L.; Coyle, J.T. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 1990, 28, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Wuolikainen, A.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Disease-Related Changes in the Cerebrospinal Fluid Metabolome in Amyotrophic Lateral Sclerosis Detected by GC/TOFMS. PLoS ONE 2011, 6, e17947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Andreadou, E.; Kapaki, E.; Kokotis, P.; Paraskevas, G.P.; Katsaros, N.; Libitaki, G.; Zis, V.; Sfagos, C.; Vassilopoulos, D. Plasma glutamate and glycine levels in patients with amyotrophic lateral sclerosis: The effect of riluzole treatment. Clin. Neurol. Neurosurg. 2008, 110, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Župunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, E.; Suminaite, D.; Jackson, M. Cerebellar ataxias: β-III spectrin’s interactions suggest common pathogenic pathways. J. Physiol. 2016, 594, 4661–4676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, K.A.; Ikeda, Y.; Day, J.W.; Ranum, L.P.W. Spinocerebellar ataxia type 5. Handb. Clin. Neurol. 2012, 103, 451–459. [Google Scholar] [PubMed]
- Burright, E.N.; Brent Clark, H.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Clark, H.B.; Burright, E.N.; Yunis, W.S.; Larson, S.; Wilcox, C.; Hartman, B.; Matilla, A.; Zoghbi, H.Y.; Orr, H.T. Purkinje Cell Expression of a Mutant Allele of SCA1in Transgenic Mice Leads to Disparate Effects on Motor Behaviors, Followed by a Progressive Cerebellar Dysfunction and Histological Alterations. J. Neurosci. 1997, 17, 7385–7395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretto, D.I.; Kumar, M.; Cao, Z.; Cunningham, C.L.; Durbin-Johnson, B.; Qi, L.; Berman, R.; Noctor, S.C.; Hagerman, R.J.; Pessah, I.N.; et al. Reduced excitatory amino acid transporter 1 and metabotropic glutamate receptor 5 expression in the cerebellum of fragile X mental retardation gene 1 premutation carriers with fragile X-associated tremor/ataxia syndrome. Neurobiol. Aging 2014, 35, 1189–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zesiewicz, T.A.; Chari, A.; Jahan, I.; Miller, A.M.; Sullivan, K.L. Overview of essential tremor. Neuropsychiatr. Dis. Treat. 2010, 6, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filip, P.; Lungu, O.V.; Manto, M.-U.; Bareš, M. Linking Essential Tremor to the Cerebellum: Physiological Evidence. Cerebellum 2016, 15, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Thier, S.; Lorenz, D.; Nothnagel, M.; Poremba, C.; Papengut, F.; Appenzeller, S.; Paschen, S.; Hofschulte, F.; Hussl, A.-C.; Hering, S.; et al. Polymorphisms in the glial glutamate transporter SLC1A2 are associated with essential tremor. Neurology 2012, 79, 243–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, S.H.; Girard, S.L.; Hopfner, F.; Merner, N.D.; Bourassa, C.V.; Lorenz, D.; Clark, L.N.; Tittmann, L.; Soto-Ortolaza, A.I.; Klebe, S.; et al. Genome-wide association study in essential tremor identifies three new loci. Brain 2016, 139, 3163–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.P.; Rayaprolu, S.; Bernales, C.Q.; Soto-Ortolaza, A.I.; van Gerpen, J.; Uitti, R.J.; Wszolek, Z.K.; Rajput, A.; Rajput, A.H.; Rajput, M.L.; et al. SLC1A2 rs3794087 does not associate with essential tremor. Neurobiol. Aging 2014, 35, 935.e9–935.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, E.-K.; Foo, J.-N.; Tan, L.; Au, W.-L.; Prakash, K.M.; Ng, E.; Ikram, M.K.; Wong, T.-Y.; Liu, J.-J.; Zhao, Y. SLC1A2 variant associated with essential tremor but not Parkinson disease in Chinese subjects. Neurology 2013, 80, 1618–1619. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. SLC1A2 rs3794087 variant and risk for essential tremor: A systematic review and meta-analysis. Pharmacogenet. Genom. 2015, 25, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Cheng, M.M.; Lin, C.-Y.; Louis, E.D.; Faust, P.L.; Kuo, S.-H. Decreased EAAT2 protein expression in the essential tremor cerebellar cortex. Acta Neuropathol. Commun. 2014, 2, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Kelly, G.C.; Tate, W.J.; Li, Y.-S.; Lee, M.; Gutierrez, J.; Louis, E.D.; Faust, P.L.; Kuo, S.-H. Excitatory Amino acid transporter expression in the essential tremor dentate nucleus and cerebellar cortex: A postmortem study. Parkinsonism Relat. Disord. 2016, 32, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bräuer, A.U.; Kuhla, A.; Holzmann, C.; Wree, A.; Witt, M. Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann-Pick Disease Type C1. Int. J. Mol. Sci. 2019, 20, 4392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporali, P.; Bruno, F.; Palladino, G.; Dragotto, J.; Petrosini, L.; Mangia, F.; Erickson, R.P.; Canterini, S.; Fiorenza, M.T. Developmental delay in motor skill acquisition in Niemann-Pick C1 mice reveals abnormal cerebellar morphogenesis. Acta Neuropathol. Commun. 2016, 4, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabenstein, M.; Peter, F.; Rolfs, A.; Frech, M.J. Impact of Reduced Cerebellar EAAT Expression on Purkinje Cell Firing Pattern of NPC1-deficient Mice. Sci. Rep. 2018, 8, 3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Fontana, A.C.K. Current approaches to enhance glutamate transporter function and expression. J. Neurochem. 2015, 134, 982–1007. [Google Scholar] [CrossRef] [PubMed]
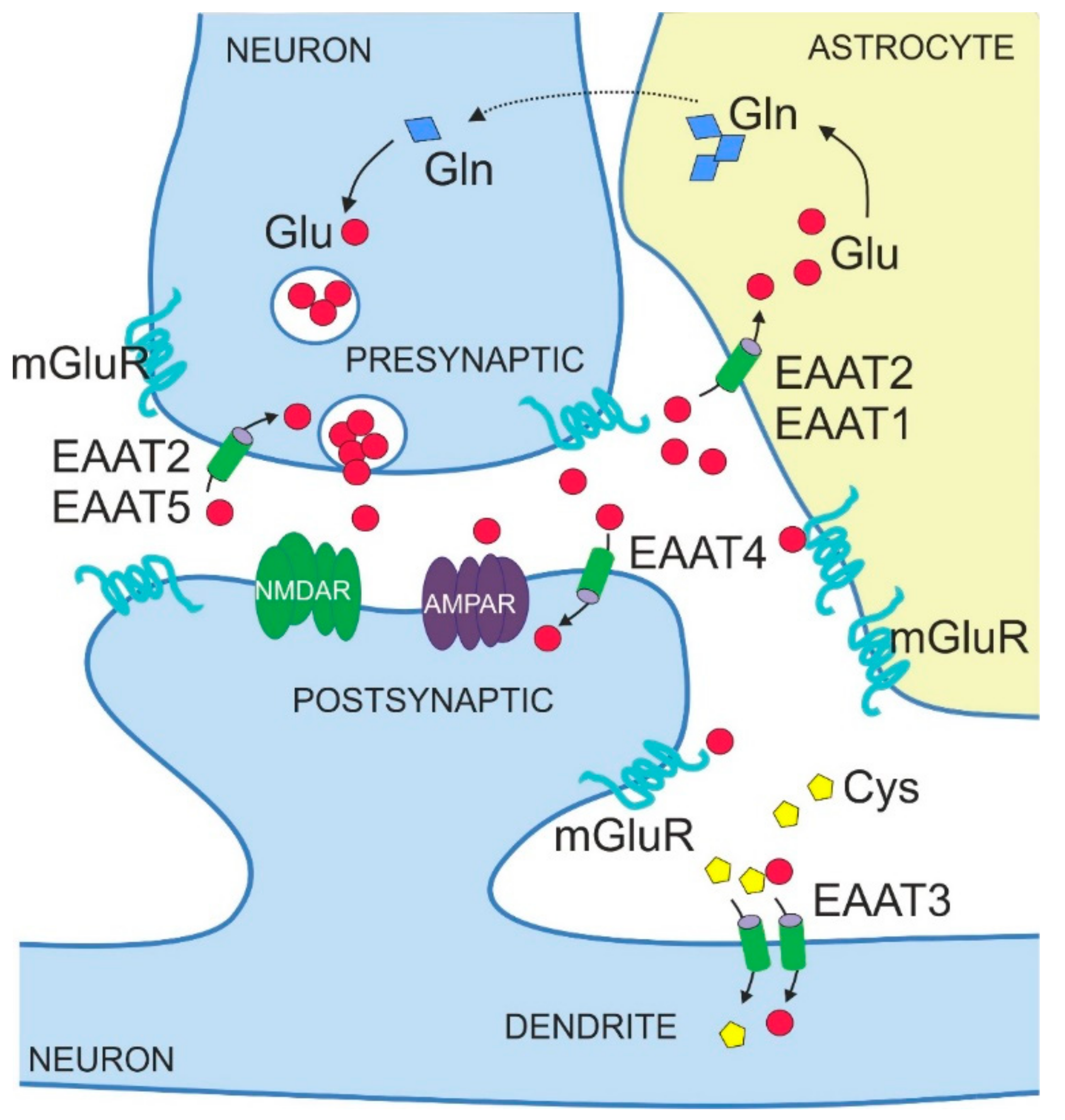
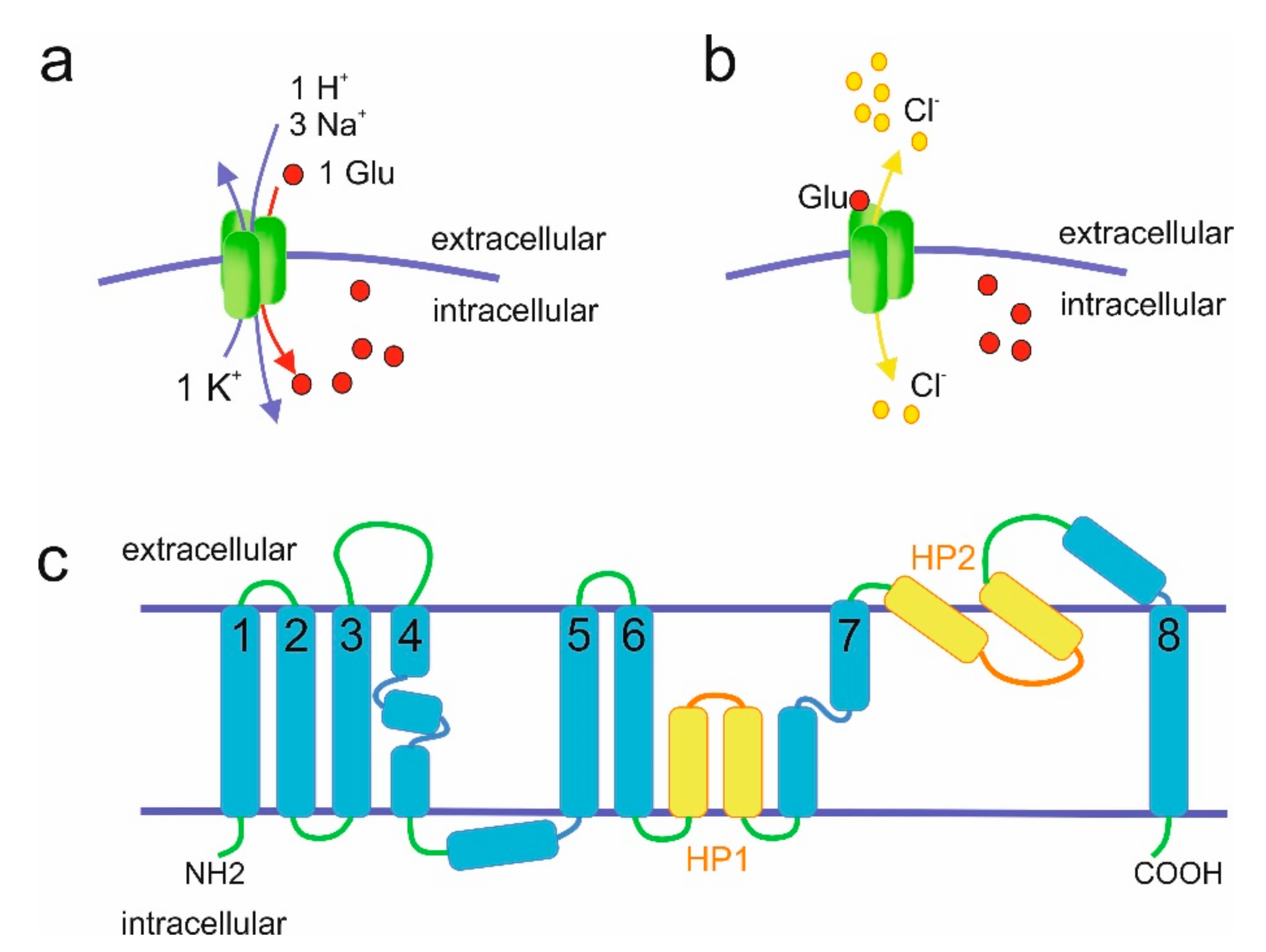
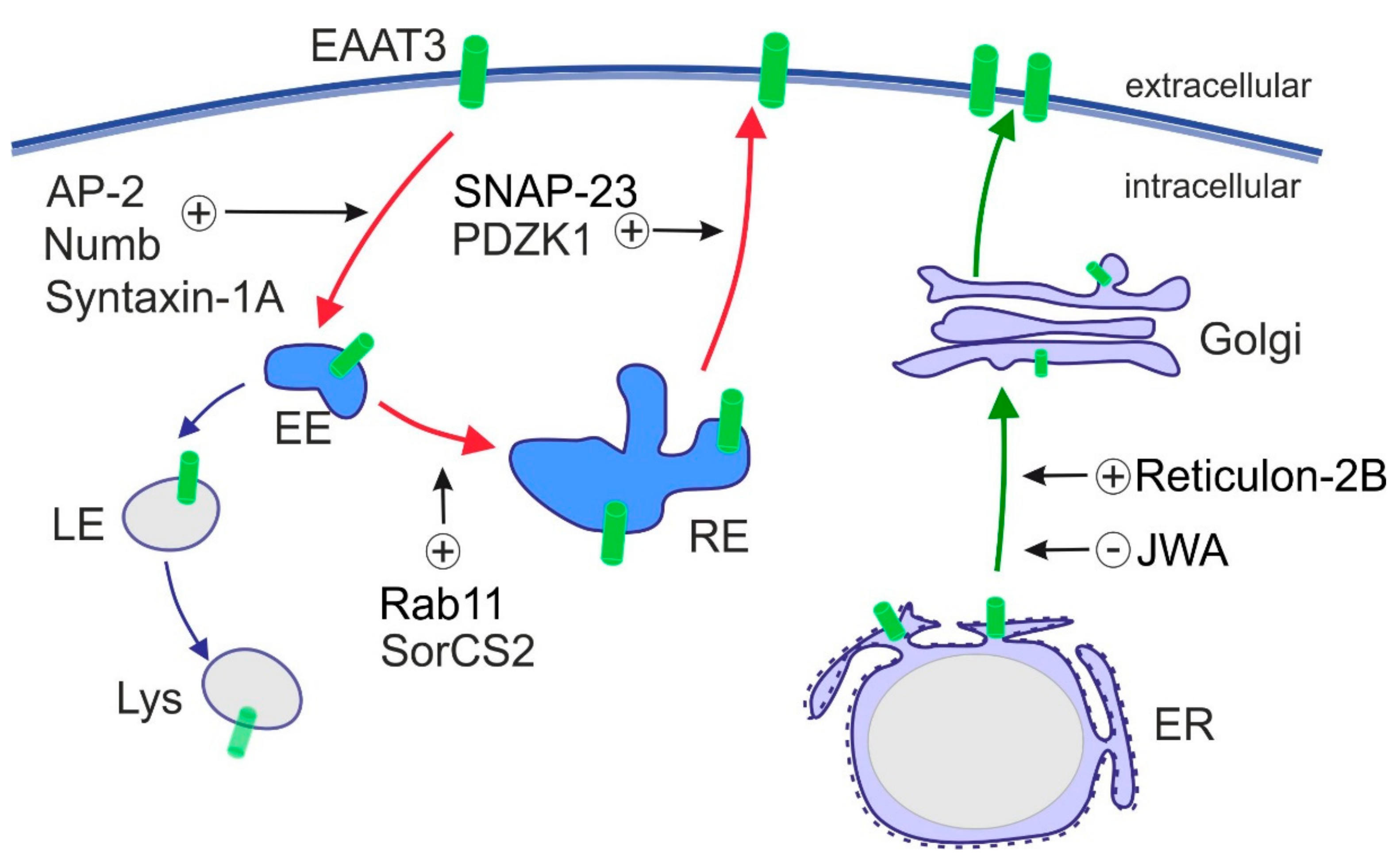
| Protein Name (Human) | Protein Name (Rodent) | Gene | Main Biological Activity | Predominant Expression Pattern in the Mature Brain | |
|---|---|---|---|---|---|
| Brain Regions | Cell Type and Subcellular Localization | ||||
| EAAT1 | GLAST | SLC1A3 | glutamate transporter | cerebellum | astrocytes (perisynaptic) |
| EAAT2 | GLT-1 | SLC1A2 | glutamate transporter | whole brain | astrocytes (perisynaptic); axon terminals (presynaptic) |
| EAAT3 | EAAC1 | SLC1A1 | glutamate and cysteine transporter | whole brain | neurons (postsynaptic, cell soma and dendrites) |
| EAAT4 | EAAT4 | SLC1A6 | glutamate transporter; glutamate-gated chloride channel | cerebellum | neurons (postsynaptic, dendritic spines) |
| EAAT5 | EAAT5 | SLC1A7 | glutamate-gated chloride channel | retina | neurons (presynaptic) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. https://doi.org/10.3390/ijms20225671
Malik AR, Willnow TE. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. International Journal of Molecular Sciences. 2019; 20(22):5671. https://doi.org/10.3390/ijms20225671
Chicago/Turabian StyleMalik, Anna R., and Thomas E. Willnow. 2019. "Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System" International Journal of Molecular Sciences 20, no. 22: 5671. https://doi.org/10.3390/ijms20225671
APA StyleMalik, A. R., & Willnow, T. E. (2019). Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. International Journal of Molecular Sciences, 20(22), 5671. https://doi.org/10.3390/ijms20225671