Abstract
Centromeres contain specialized nucleosomes at which histone H3 is partially replaced by the centromeric histone H3 variant cenH3 that is required for the assembly, maintenance, and proper function of kinetochores during mitotic and meiotic divisions. Previously, we identified a KINETOCHORE NULL 2 (KNL2) of Arabidopsis thaliana that is involved in the licensing of centromeres for the cenH3 recruitment. We also demonstrated that a knockout mutant for KNL2 shows mitotic and meiotic defects, slower development, reduced growth rate, and fertility. To analyze an effect of KNL2 mutation on global gene transcription of Arabidopsis, we performed RNA-sequencing experiments using seedling and flower bud tissues of knl2 and wild-type plants. The transcriptome data analysis revealed a high number of differentially expressed genes (DEGs) in knl2 plants. The set was enriched in genes involved in the regulation of the cell cycle, transcription, development, and DNA damage repair. In addition to comprehensive information regarding the effects of KNL2 mutation on the global gene expression, physiological changes in plants are also presented, which provides an integrated understanding of the critical role played by KNL2 in plant growth and development.
1. Introduction
The centromeres are specialized chromosomal domains that are required for proper separation of chromosomes during mitosis and meiosis. The centromere is composed of centromeric DNA, often enriched in satellite repeats, and the large protein complex “kinetochore”. In centromeric nucleosomes of most eukaryotes, histone H3 is partially replaced by the centromeric histone H3 variant cenH3 (also known as CENP-A in mammals, CID in Drosophila, Cse4 in Saccharomyces cerevisiae, and Cnp1 in Schizosaccharomyces pombe [1]). Deposition of cenH3 at the centromeric region is a prerequisite of the correct assembly and function of the kinetochore complex. It depends on different cenH3 assembly factors and chaperones [2], the transcription of the centromeric repeats [3,4,5], and the epigenetic status of centromeric chromatin [6,7]. In mammals, the Mis18 complex composed of Mis18α, Mis18β, and Mis18-binding protein 1 (also known as KNL2) plays an important role in the licensing of centromeres for cenH3 recruitment [8,9]. The human Mis18 protein complex localizes to centromeres during late telophase and remains associated with the centromere during early G1 phase when new CENP-A is deposited [2,9,10]. It mediates the recruitment of the cenH3 chaperone Holliday junction recognition protein (HJURP) to endogenous centromeres [11,12]. Knockout of murine Mis18a is embryo lethal [7]. Cultured homozygous mutant embryos showed misaligned chromosomes, anaphase bridges, and lagging chromosomes [7].
Up to now, only the cenH3 assembly factor KNL2 has been identified and characterized in plants [13,14]. In contrast to the mammalian cenH3 assembly factor Mis18BP1, the Arabidopsis KNL2 protein is present at centromeres during all stages of the mitotic cell cycle, except from metaphase to mid-anaphase [13]. Knockout of KNL2 in Arabidopsis resulted in a reduced amount of cenH3 at centromeres, mitotic and meiotic defects, decreased DNA methylation degree, and lowered growth rate and fertility [13].
All homologs of Mis18BP1 (KNL2) identified up to now contain the conserved SANTA domain [15] at the N-terminus. However, the functional role of this domain still remains obscure. It was suggested that it might be involved in protein–protein interactions due to the presence of many conserved hydrophobic residues. It was shown that an absence of the SANTA domain in Arabidopsis KNL2 does not disturb its centromeric localization. Recently, the conserved C-terminal CENPC-k motif [14,16] required for the targeting of Mis18BP1 (KNL2) to centromeres was identified [14,17,18]. It presents in the Mis18BP1 (KNL2) proteins of most eukaryotes excluding therian mammals and Caenorhabditis elegans [14,16]. Arabidopsis KNL2 binds centromeric repeat pAL1 and non-centromeric DNA sequences in vitro, whereas in vivo it associates preferentially with the centromeric repeat pAL1. The level and function of the Mis18BP1 protein in human cell culture is regulated by SUMOylation [19], and its centromeric localization is controlled by the phosphorylation in a cell cycle-dependent manner [20]. Whether the KNL2 of plants is regulated in the similar way remains to be elucidated.
Although the Mis18 protein complex is important for the deposition of cenH3 to centromeres in different organisms, its mechanism of function remains to be elucidated in detail. For mammals, it was shown that an interaction of the Mis18 complex with the de novo DNA methyltransferases DNMT3A and DNMT3B is required for the regulation of the epigenetic status of centromeric DNA and subsequently the transcription of centromeric repeats [7]. A knockout of mammalian Mis18α resulted in reduced DNA methylation, altered histone modifications, and increased centromeric transcripts in cultured embryos [7]. However, it was not tested whether a knockout of Mis18 complex components has an effect on the methylation status of non-centromeric chromatin and on the expression of other repetitive or gene-coding chromosomal regions. For instance, knockout of KNL2 resulted in decreased DNA methylation of the marker regions MEA-ISR and the At-SN1 in Arabidopsis [13].
In the current study, we used an RNA-sequencing (RNA-seq) approach to address the question of whether the inactivation of KNL2 influences the genome-wide gene expression during seedling and flower bud stages in Arabidopsis. This analysis allowed the identification of highly differentially expressed genes (DEGs) in flower buds (n = 1861) and seedlings (n = 459) of the knl2 mutant. Gene Ontology (GO) term enrichment analysis links the activity of KNL2 to centromere function, DNA repair, DNA methylation as well as regulation of transcription. The specific pattern of gene expression in response to the inactivation of the KNL2 gene provides a resource for future functional studies to unravel the role of KNL2 in kinetochore assembly and function.
2. Results and Discussion
2.1. Loss of KNL2 Function Leads to Massive Transcriptional Changes
To understand the role of KNL2 for the kinetochore function, mitotic and meiotic divisions, and subsequently for plant growth and development, we compared the transcriptomes from two tissue types, namely seedlings and flower buds, of wild-type and the knl2 plants. The experiments in the current study were designed in a way to weaken factors that could introduce experimental noise and would diminish the biological relevance of the data. For the RNA-seq study, we compared control and mutant plants with the same genetic background (Columbia ecotype) and physical age by performing experiments under the same plant growth conditions. This allowed the identification of DEGs and enriched biological processes. At the same time, the information obtained is limited to a single snapshot of gene expression reflecting the physical age of plants. Further comparative experiments including more time points to study temporal gene expression profiles in the knl2 mutant line would be desirable, such as the comparison of the transcriptomes of knl2 and wild-type plants not of the same “physical” but “biological age”, since the knl2 mutant plants showed a delay in growth and development [13].
RNA-seq analysis based on DeSeq2 [21] identified 3261 genes in seedlings (Table S1, Supplementary Materials) that were altered in the knl2 mutant line (adjusted p-value < 0.05). Among them, 459 genes (Figure 1) were highly differentially expressed (fold change (FC) cutoffs of ≥ 2).

Figure 1.
Differential gene expression. Venn diagram showing the number of highly differentially expressed genes (DEGs) (2-fold cutoff, false discovery rate (FDR) corrected p-value < 0.05) obtained by comparing knl2 versus wild-type ecotype Columbia-0 (Col-0) genes in seedlings and knl2 versus Col-0 genes in flower buds.
In flower buds, more DEGs were identified in the knl2 (4750 in total). Out of 1858 highly expressed genes (FC cutoffs of ≥ 2) (Figure 1), 1194 were highly upregulated and 664 were highly downregulated in the knl2 mutant (adjusted p-value < 0.05). In both tissues, more genes were significantly upregulated than downregulated (Table S1).
In agreement with previously published data [13], which showed absence of the full-length KNL2 transcript in the knl2 plants, the mRNA level of KNL2 was greatly reduced (log2 FC of −2.63 and −4.57) in seedling and flower bud tissues, respectively, of knl2 plants, suggesting that the observed transcription responses are the result of KNL2 depletion.
To explore the distribution of DEGs and to identify the biological consequences of the KNL2 inactivation, we used the GO::TermFinder [22]. Gene Ontology (GO) enrichment analysis for the “Biological process” was compared for downregulated and upregulated DEGs (adjusted p-value < 0.05) represented in seedlings and, separately, in flower buds. Detailed information about all overrepresented processes is given in a supporting information dataset (Dataset S1, Supplementary Materials). In this paper, we consider in more detail the categories associated with cell division, chromatin status, and plant growth and development. The results of the GO enrichment analysis can shed new light on how KNL2 depletion leads to plant growth and development alterations.
2.2. Knockout of KNL2 Impairs Expression of Genes Involved in Kinetochore Function
Kinetochores are responsible for the accurate segregation of chromosomes during mitosis and meiosis. Our RNA-seq analysis revealed GO terms related to physical events associated with both types of cell division, such as the “chromosome segregation”, “meiosis I”, “meiotic chromosome separation”, “mitotic metaphase plate congression”, “mitotic recombination”, and “attachment of mitotic spindle microtubules to kinetochore” in seedlings (Table 1). All DEGs representing these categories were downregulated in the knl2 plants (Dataset S1, Tables S2 and S3).

Table 1.
Assignment of differentially expressed downregulated genes in knl2 seedlings to different functional categories related to cell processes. Gene Ontologies were analyzed for term enrichment using the Generic Gene Ontology GO::TermFinder tool (cutoff of 5% false discovery rate (FDR)).
GO terms characterizing the functions of kinetochores, like microtubule binding, chromosome movement, and checkpoint signaling were overrepresented in the downregulated gene list of knl2 flower bud samples (Table 2).
Table 2.
Assignment of differentially expressed downregulated genes in knl2 flower buds to different functional categories related to cell processes. Gene Ontologies were analyzed for term enrichment using the Generic Gene Ontology GO::TermFinder tool (cutoff of 5% FDR).
For instance, reduced transcript levels were found for genes with known microtubule-associated functions [23] such as KINESIN 5 (ATK5; AT4G05190), TITAN1 (TTN1; AT3G60740), and HINKEL (AT1G18370). The CELL DIVISION CYCLE 20.1 (CDC20.1; AT4G33270), known for its critical role in the spindle assembly checkpoint-dependent meiotic chromosome segregation [24], was also significantly suppressed in the flower buds of knl2 plants. Another example is the reduced expression of BUB1 (AT2G20635), a protein kinase containing the Mad3-BUB1-I domain. Being together with CDC20.1 in the same protein–protein interaction network, BUB1 plays an important role in the assembly of the checkpoint proteins at the kinetochore [25]. In our RNA-seq study, it was highly downregulated in both types of tissue. Several other kinetochore associated genes, like GAMMA-TUBULIN (TUBG1; AT3G61650), PLEIADE (PLE; AT5G51600), and AURORA KINASE 1 (AT4G32830) were downregulated in knl2 flower buds. The expression of the inner kinetochore CENP-C (AT1G15660) was decreased in both seedlings and flower buds. Knockout of KNL2 results in a reduced amount of cenH3 transcripts of Arabidopsis [13]. This result was confirmed by RT-qPCR analysis of the samples used for the current RNA-seq study.
2.3. Reduced DNA Methylation in the knl2 Mutant Might Be Responsible for the Activation of a High Number of Transposons
Knockout of Arabidopsis KNL2 leads to a reduced level of DNA methylation [13]. In our RNA-seq study, a number of genes related to the DNA and histone methylation showed repressed differential expression in the knl2 line (in both tissues). For example, histone H3 lysine 9 di-methyltransferase SUVH4 (AT5G13960), CHROMOMETHYLASE 2 (CMT2; AT4G19020), and ARGONAUTE 6 (AGO6; AT2G32940) were found among differentially downregulated genes in the flower buds (adjusted p-value < 0.05), whereas DEMETER-LIKE 1 (DML1; AT2G36490) showed reduced expression in both tissues used for analysis.
It is known that the activity of transposons is controlled epigenetically through DNA methylation and repressive histone marks (reviewed in [26,27,28]). It is likely that a reduced level of DNA methylation in the knl2 mutant results in an altered expression of transposons. Indeed, a considerable number of transposable elements were differentially expressed in knl2 seedlings (n = 52) and flower buds (n = 89). These elements belonged to the DNA transposons (CACTA-like, hAT-like, Mutator-like) and LTR retrotransposons (Copia-like, Gypsy-like). In both tissues, more transposons were upregulated in knl2 plants (Figure 2).
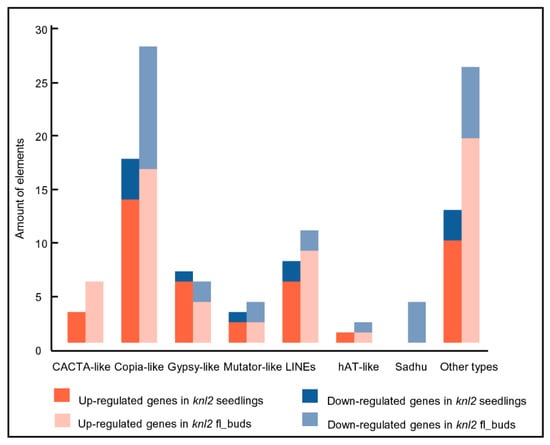
Figure 2.
Transposable elements and their amount identified in the knl2 mutant line. Information is given about transposons up- and downregulated in the knl2 seedlings and knl2 flower buds.
In contrast, the non-LTR retrotransposons Sadhu 1-3 (AT3G44042), Sadhu 3-1 (AT3G44042), Sadhu 4-1 (AT5G28913), and Sadhu 5-1 (AT4G01525) had decreased expression level in the knl2 flower buds. Depression of Sadhu 3-1 retroelement was also observed in the epigenetic mutant suvh4 [29].
2.4. Altered Expression of the Root-related Genes Explains the Slow Root Growth of knl2
Root length was compared between seven-day-old control and the knl2 mutant seedlings. The average root length of mutant seedlings (0.66 cm) was about 3 times shorter than that of the wild-type (1.82 cm) (Figure 3), and GO terms related to the root development such as “regulation of root development” and “primary root development” were highly overrepresented among the genes downregulated in the knl2 seedlings (Table 1).
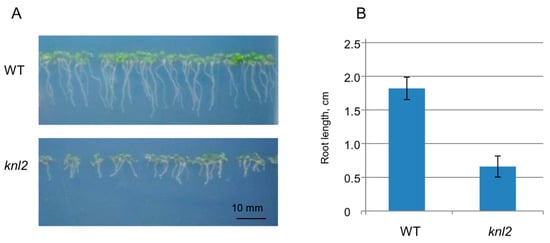
Figure 3.
Effect of KNL2 depletion on the root development. (A) Representative phenotypes of wild-type (WT) and knl2 plants germinated and grown for seven days on ½ Murashige and Skoog (MS) medium. Bar = 10 mm. (B) Quantitative data for plants described in (A). Data are the means ± standard errors, n = 20. Student’s t-test, p < 0.01.
We additionally tested all root-related genes available in TAIR for their overlap with the differentially expressed genes in the knl2 seedlings. As a result, 119 differentially expressed genes were identified (Table S4, Supplementary Materials). Among them, 14 were highly suppressed (FC cutoffs of ≥ 2). The reduced transcript level was found for SHORT AND SWOLLEN ROOT 1 (SSR1; AT5G02130). The ssr1-2 plants show reduced root growth [30]. Other downregulated genes encoding the chloroplast/plastid localized GAPDH isoforms are GAPCp1 (AT1G16300) and GAPCp2 (AT1G79530). gapcp double mutants are characterized by arrested root development, dwarfism, and sterility [31]. The transcription factor AGAMOUS-like 14/XAL2 (AT4G11880) preferentially expressed in roots is highly suppressed in knl2 seedlings (log2 FC of −1.05, adjusted p-value 0.0001). Several studies demonstrated that xal2 mutants have short roots [32,33]. In addition, we observed the reduced transcript level of EMBRYO DEFECTIVE 2757 (EMB2757; AT4G29860) in knl2 seedlings. Genetic analysis of EMB2757 in Arabidopsis has demonstrated that mutations in this gene cause defects in both embryo and seedling development [34]. Upregulated differentially expressed genes in knl2 seedlings can be further exemplified with RAPID ALKALINIZATION FACTOR 23 (RALF23; AT3G16570). Overexpression of RALF23 leads to slower growing seedlings with roots that have reduced capacity to acidify the rhizosphere [35].
2.5. DNA Damage Repair Genes are Downregulated in the knl2 Plants
The genome stability is maintained by DNA damage responses. The failure to repair DNA damage leads to negative processes related to plant growth, reproduction, and even lethality. We found that genes representing the GO term “DNA repair” (26 genes) and related categories, such as “double-strand break repair via homologous recombination”, “non-recombinational repair”, and “DNA ligation involved in DNA repair” were overrepresented among knl2 DEGs (Table 1). All genes of these categories were downregulated in the mutant. Among downregulated genes were the key players participating in the canonical nonhomologous end joining, such as KU70 (AT1G16970), KU80 (AT1G48050), and LIGASE 4 (AT5G57160). It is known that mutations in these genes lead to increased sensitivity to double-strand DNA breaks (DSB)-inducing factors (reviewed in [36]). AT5G20850, encoding a DNA recombination and repair protein RAD51, is another example of a gene with a decreased expression level in knl2 seedlings (log2 FC of −0.99, adjusted p-value 1.47 × 10−6). Loss of RAD51 function does not affect the vegetative development of Arabidopsis, probably due to functional redundancy with other genes of the RAD51 family, but is essential for meiotic repair of DSBs caused by AtSPO11−1 [37]. In addition, the RAD51-like gene, DISRUPTION OF MEIOTIC CONTROL 1 (DMC1; AT3G22880), known to promote interhomolog recombination was strongly downregulated in the knl2 flower buds (log2 FC of −1.20, adjusted p-value 1.25 × 10−8). Interestingly, the mammalian kinetochore protein BUB1, playing an important role in chromosome segregation, is also known to participate in the DNA damage response [38]. In human and mouse cells, cenH3/CENP-A accumulates at DSBs together with CENP-N, CENP-T, and CENP-U [39]. Since the induction of DNA damage by radiation resulted in an increased expression of cenH3, the authors proposed a cenH3 function in DNA repair. Further research is needed to unravel the interrelationships between kinetochore genes and the DNA damage response.
To test whether the altered mRNA levels are associated with changed capacity of knl2 plants for DNA damage repair, we exposed wild-type and mutant plants to 10 µM DNA inter-strand crosslink inducer mitomycin C (MMC), 50 nM DNA strand breaker bleomycin, and 10 nM enzymatic DNA-protein crosslinker camptothecin (Figure S1, Supplementary Materials). This pilot experiment indicated sensitivity of knl2 plants to MMC, but not other tested drugs (Figure 4).
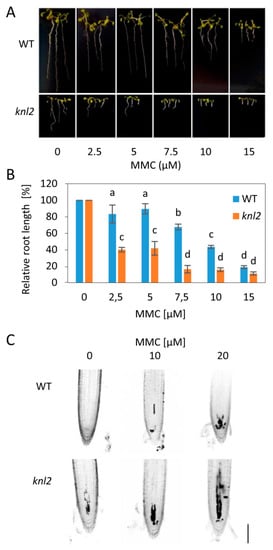
Figure 4.
Sensitivity of knl2 plants to DNA damage. (A) Representative phenotypes of wild-type (WT) and knl2 plants grown for 14 days on mitomycin C (MMC)-containing media. Bar = 10 mm. (B) Quantitative data for plants grown as described in (A). Error bars represent the standard deviation between the means of three biological replicates, each represented by 15 to 20 plants. Letters above the bars indicate statistically significantly different groups in ANOVA (p-value ≤ 0.05) and post hoc Tukey‘s test. (C) Analysis of propidium iodide (PI) uptake (dark sectors) in roots of WT and knl2 plants. Four-day-old plants were treated with the specified concentration of MMC for 24 h prior to analysis. Bar = 50 µm.
To validate this result, we treated knl2 plants with a series of MMC concentrations (2.5 to 15 µM) and found significant reduction in their root growth compared to wild-type plants (Figure 4A,B). Next, we performed propidium iodide (PI) staining of root apices from plants treated for 24 h with 0, 10, and 20 µM MMC. Intense PI staining inside the cells indicates damaged plasma membranes, typical for stressed and dead cells. Under mock conditions, wild-type plant root apical meristems were intact, whereas the meristems of knl2 plants showed few intensely stained cells. MMC treatment caused dose-dependent accumulation of intensely stained cells in the meristems of wild-type plants, but the increase in the meristem of knl2 plants was more prominent.
Based on this, we conclude that knl2 plants fail to properly activate specific DNA damage repair factors, have reduced capacity for repair of DNA inter-strand crosslinks, and suffer from increased cellular damage.
2.6. Knockout of KNL2 Results in Deregulated Expression of a High Number of Transcription Factors
We hypothesized that a large number of DEGs in the knl2 mutant may be the result of deregulated expression of transcription factors. Therefore, the knl2 mutant transcriptome was further screened for the presence of differentially expressed transcription factors (TFs). TFs were retrieved from The Plant TF database v.4 [40]. In knl2 seedlings, 202 TFs were expressed (43 highly differentially expressed), whereas in knl2 flower buds 330 TFs (137 highly differentially expressed) were detected. The classification of TFs is visualized in the heat map (Figure 5) and is given by the supporting information dataset (Dataset S2, Supplementary Materials). While most of the TF families showed a heterogeneous profile for the single TFs, some of the TF families behaved homogeneously (Figure 5). For example, the TF families B3 and MIKC_MADS and M-type_MADS mostly included downregulated genes in the knl2 mutant, whereas the TF families ERF, NAC, and WRKY were enriched for upregulated genes. TF families in which many genes were highly differentially expressed were bHLH, C2H2, ERF, HD-ZIP, LBD, MIKC_MADS and M-type_MADS, NAC, and WRKY. The highest expression among TFs was observed in the NAC family. AT5G14490 encoding for NAC domain-containing protein 85 was highly upregulated in flower buds (log2 FC of 3.45, adjusted p-value 2.69 × 10−12).

Figure 5.
Differentially expressed genes encoding transcription factors in the knl2 mutant line. Expression in knl2 flower buds and knl2 seedlings is depicted as F and S, respectively. Red and blue represent up- and downregulated differentially expressed genes, respectively. All values are log2 transformed. The gradient color illustrates the expression value.
TFs with an altered expression in the knl2 mutant are involved in different processes and provide insight into Arabidopsis development changes due to the inactivation of the kinetochore gene KNL2. There was a striking coordinated downregulation of genes representing the MADS-box family, which are the key regulators of seed and flower development (detailed description is presented below in 3.7). The upregulated TFs can be exemplified by the genes of the WRKY family. The TFs of this family play an important role in plant development and responses to environmental stress stimuli [41,42,43]. From 74 members of the WRKY TF family, 13 were differentially expressed in seedlings and 26 in the flower buds. The differentially expressed genes WRKY33 (AT2G38470) and WRKY46 (AT2G46400), highly upregulated in seedlings and flower buds, are known to play a role in heat stress responses. The expression of these genes was elevated in MBF1c-overexpressing plants, which showed enhanced tolerance to heat compared with wild-type plants [44]. WRKY46 additionally regulates responses to other stresses in Arabidopsis [45,46,47].
Another TF family with overrepresented genes is ERF. The most highly activated genes in flower buds include DREB19 (AT2G38340; log2 FC of 1.54, adjusted p-value 0.001), DREB 26 (AT1G21910; log2 FC of 2.43, adjusted p-value 6.77 × 10−7), and RAP2.6L (AT5G13330; log2 FC of 1.75, adjusted p-value 3.54 × 10−10). A previous study of the DREB19, DREB26, and RAP2.6L effect in Arabidopsis demonstrated the participation of the genes in plant developmental processes as well as biotic and/or abiotic stress signaling [48].
The heat map also revealed genes that are differentially expressed in flower buds but not in seedlings and vice versa. We observed that 94 highly differentially expressed genes were present only in flower buds and 18 highly regulated genes were seedling-specific (Dataset S2). The transcriptional repressor of the RAV family TEMPRANILLO 1 (TEM1), known to postpone floral induction [49], is another example of the highly upregulated gene present in the flower buds (log2 FC of 1.55).
2.7. Late Flowering of the knl2 Plants is Determined by the Altered Expression of Flowering Genes
We observed that flowering of the knl2 plants delays for 10–14 days compared to wild-type (Figure 6; Figure S2, Supplementary Materials).

Figure 6.
Effect of KNL2 depletion on the flowering time. (A) At early growth stages, no obvious phenotypical differences between wild-type (WT) and the knl2 mutant were observed. (B) The flowering time of the knl2 mutant delayed by 10–14 days compared to Arabidopsis wild-type. Seeds of the knl2 mutant and wild-type were germinated under short-day conditions, 8 h light/20° C and 16 h dark/18 °C, for two weeks and then plants were transferred to the cultivation room with long-day conditions, 16 h light/20 °C and 8 h dark/18 °C.
From this observation, we could identify a number of enriched GO terms related to the floral development in our RNA-seq data from knl2 flower buds. These represent such GO categories as “reproduction”, “regulation of pollen tube growth”, “pollen germination”, “pollination”, and “pollen development” (Table 3).
Table 3.
Assignment of genes differentially expressed in knl2 flower buds to development-related functional categories based on GO::TermFinder. Genes downregulated in the knl2 flower buds were included into the GO enrichment analysis (cutoff of 5% FDR).
Genes representing these and related categories in Table 3 were downregulated in the knl2 flower buds. Some of the genes belong to the MYB transcription factors and regulate the plant microgamete development. For instance, AtMYB103 (AT1G63910), important for pollen development [50], is one of the highly suppressed flower bud genes in knl2. The expression of other prominent transcription factors like AGAMOUS-LIKE 66 (AGL66; AT1G77980) and AGL104 (AT1G22130) involved in pollen maturation and tube growth was significantly reduced. Adamczyk and Fernandez [51] demonstrated that double mutant plants agl66 agl104 could produce pollen but had severely reduced fertility and aberrant pollen tube growth. Along with AGL66 and AGL104, AGL94 (AT1G69540) belongs to the MIKC * factors which are highly active in pollen as major regulators of pollen maturation programs [51,52]. These three genes were significantly downregulated in the knl2 flower buds (adjusted p-value < 0.001). This finding might explain the reduced fertility of knl2 plants in addition to the role of KNL2 in mitosis and meiosis [13].
The RNA-seq data analysis and observation of the delayed flowering prompted us to inspect the RNA-seq data of the knl2 seedlings. We asked the questions of whether and, if so, how many flowering-related genes based on the FLOR-ID overlap with the differentially expressed genes in the knl2 seedlings. From 306 genes, 36 genes could be identified (Dataset S3, Supplementary Materials). There was a striking coordinated downregulation of several prominent flowering-related genes. For example, the floral integrator Flowering Locus T (FT; AT1G65480), known to be expressed in leaves, was significantly downregulated in knl2 seedlings. Its reduced expression is in correspondence with the downregulation of SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1; AT2G45660) in the knl2 seedlings (log2 FC of −2.24, adjusted p-value 7.72 × 10−61). It is known that flowering activator SOC1 acts in a positive-feedback loop with AGL24, which is the important MIKCc-type transcription factor positively regulating flowering in Arabidopsis (reviewed in [53]). In our RNA-seq study, the AGL24 showed reduced expression in both knl2 seedlings and flower buds.
GIGANTEA (GI; AT1G22770), an important gene in regulating photoperiodic flowering, had a reduced expression in the knl2 mutant line compared to wild-type. Another regulator of transition to flowering FKF1 (ADO3; AT1G68050) was highly downregulated in the knl2 mutant line (log2 FC of −1.70, adjusted p-value 2.79 × 10−17). A member of the MADS-box family XAANTAL2 (XAL2/AGL14; AT4G11880), which is a necessary and sufficient agent to induce flowering [54], showed decreased expression level in the knl2 seedlings (log2 FC of −1.05, adjusted p-value 0.002).
The reduced activity of flower inductive pathway genes observed in the knl2 seedlings might explain the delay in flower initiation, at least under long-day conditions.
2.8. Knockout of KNL2 Results in Altered Expression of Genes Controlling Seed Development
The loss-of-function knl2 mutant showed reduced seed production [13]. We used the SeedGenes Project database including 481 genes in order to examine the overlap between these genes and the 4750 genes that we found to be significantly differentially expressed in response to KNL2 inactivation. From these genes, 42 were differentially expressed in the knl2 flower bud samples (Table S5, Supplementary Materials). Among downregulated genes, there are those whose disruption causes embryo abortion, such as CYL 1 (AT5G13690), EMB 1674 (AT1G58210), EMB 2184 (AT1G75350), EMB 3003 (AT1G34430), EMB 3004 (AT3G06350), and IMPL 2 (AT4G39120). Highly upregulated genes that include HSP 17.4 (AT3G46230), RLP37 (AT3G23110), and NCED3 (AT3G14440) are known for their expression in seeds and response to abiotic stresses or defense.
2.9. Real-time Quantitative PCR Confirms RNA-seq Analysis
To validate the quality of the RNA-seq data by qRT-PCR, we selected eight genes differentially expressed in seedlings and 11 genes differentially expressed in flower buds (Figure 7; Figure S3, Supplementary Materials). In the case of seedlings, four genes involved in the regulation of root growth (AT4G11880, AT4G28720, AT3G50060, AT2G43140) and two genes involved in the regulation of flowering time (AT2G45660, AT1G68050) were selected for the analysis. In the case of flower buds, an expression of five genes involved in the regulation of flowering time (AT5G48560, AT2G40080, AT5G37770, AT1G10120, AT1G02580) and four genes involved in the regulation of seed development (AT2G01860, AT3G23110, AT5G13690, AT5G23940) was validated. KNL2 (At5g02520) and cenH3 (At1g01370) genes were included for the analysis in both types of tissues. Our qRT-PCR confirmed the extreme downregulation of KNL2 gene expression in the mutant line. All RNA-seq-based determined up- and downregulated genes showed the same qualitative response in both seedlings and flower buds based on qRT-PCR (correlation coefficients between qRT-PCR and RNA-seq data of 0.996 and 0.974, respectively).
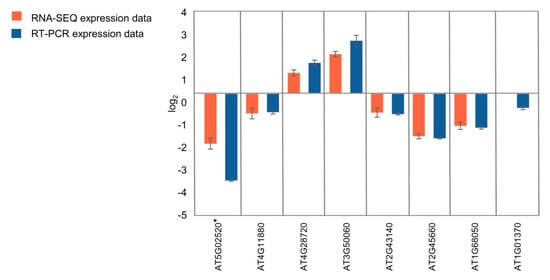
Figure 7.
qRT–PCR validation of RNA-seq results. Eight genes were selected from the genes differentially expressed in seedlings according to the RNA-seq data analysis. Detailed annotation of the selected genes is presented in Table S2 (Supplementary Materials). *At5g02520 corresponds to KNL2. Error bars indicate the standard error of three biological replicates in qRT-PCR.
2.10. Conclusions
KNL2 of Arabidopsis is involved in regulating the centromeric loading of cenH3, and its knockout results in mitotic and meiotic defects and reduced DNA methylation, followed by reduced growth rate and fertility compared to wild-type [13]. Our comparative RNA-seq analysis revealed that these abnormalities were associated with an altered expression of genes regulating corresponding processes. For instance, the reduced growth rate of roots and late flowering of the knl2 mutant correlate with the altered expression of genes regulating root growth and flowering, respectively. We propose that KNL2 has an essential regulatory function in plant growth and development that is much broader than the regulation of centromeric localization of cenH3 only.
3. Materials and Methods
3.1. Plant Materials and Growth Conditions
The A. thaliana knl2 mutant (SALK_039482) in Col-0 background was described previously [13]. Seeds of A. thaliana wild-type and knl2 were germinated in Petri dishes on half strength Murashige and Skoog (MS) medium (Murashige and Skoog, 1962) for eight days. For harvesting of flower buds, populations of wild-type and knl2 plants (30 plants per each population) were grown in soil until flowering. In both cases, plants were cultivated with a 16 h photoperiod (21 °C day/18 °C night), 70% relative humidity. Light irradiance at plant level was 130 µmol m−2 s−1.
3.2. DNA Damage Sensitivity Assays
For the pilot root length assay experiment, plants were germinated and grown for seven days on ½ strength Murashige and Skoog medium containing 0.01% DMSO and different DNA damage inducers—mitomycin C (MMC) (Duchefa, Haarlem, The Netherlands), bleomycin (Calbiochem, San Diego, CA, USA), and camptothecin (Sigma-Aldrich, Saint Louis, MO, USA) with concentrations specified in the text. For the subsequent analysis, plants were germinated and grown for 14 days on media containing different concentrations of MMC (2.5, 5, 7.5, 10, and 15 µM from Sigma-Aldrich). For PI staining assay, plants were grown on solid ½ Murashige and Skoog medium for four days, then transferred to liquid ½ Murashige and Skoog medium containing 0, 10, and 20 µM MMC and grown for 24 h. Subsequently, the plants were stained with 10 μg/mL propidium iodide solution (Sigma-Aldrich) for 3 min, rinsed with tap water and analyzed using an AxioImager Z2 (Zeiss, Jena, Germany) microscope equipped with the DSD2 confocal module (Andor Technology, Belfast, Great Britain). Plants for all procedures were grown in a Percival growth chamber under long-day (16 h light) conditions and 21 °C.
3.3. RNA Isolation and Illumina Sequencing
Total RNA was extracted from the eight-day-old seedlings of A. thaliana. At least 60 seedlings (100 mg) were pooled to produce a biological replicate. For the RNA-seq analysis of flower buds, inflorescences were harvested from three individual plants for each sample (30 mg). Flower buds older than stage 12 and flowers [55] were removed. All tests were performed on three biological replicates per condition and genotype. Total RNA was isolated from seedlings and flower bud samples using the RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s manual. The RNA preparations were checked for quality using a NanoDrop spectrophotometer and a 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). In total, 12 RNA samples (1 µg each) were provided to the IPK-Sequencing-Service (IPK, Gatersleben, Germany) for construction of cDNA libraries with the TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA). The libraries were sequenced in a HiSeq2500 rapid 100-bp single-read run system After sequencing, the adapter sequences and the barcodes were removed.
3.4. RNA-seq Data Processing
Sequencing quality of the reads was examined by using FastQC Read Quality reports, Galaxy Version 0 0.72 [56]. At least 94% of the bases of each read in all samples possessed Illumina Quality >30 and no sequence flagged as poor quality was detected.
Sequences were aligned against the Arabidopsis TAIR 10 genome assembly using HISAT2 Galaxy tool version 2.0.3. [57] with default settings. Read counts for each gene were quantified based on the BAM files produced with the HISAT2 by using the tool feature Counts, Galaxy Version 1.4.6.p5 [58]. The advanced setting parameters were strand specificity = “no”, GFF feature type filter = “exon”, and GFF gene identifier = “Parent”. Differentially expressed features were determined based on the feature Counts tables by applying the tool DESeq2, Galaxy Version 2.11.38 [21] with the setting parameter fit type = “parametric”. DESeq2 tested for differential expression based on a model using the negative binomial distribution. Differentially expressed genes were identified by comparison of two groups, namely, (1) a mutant line and the wild-type control, condition seedlings and (2) a mutant line and the wild-type control, condition flower buds.
The results of all statistical tests were adjusted for the multiple testing false discovery rate (FDR) with the Benjamini and Hochberg procedure [59]. A cutoff value of adjusted p-values equal to 0.05 was chosen as a threshold to identify significant differentially expressed genes.
3.5. Analysis of Differentially Expressed Genes (DEGs)
Functional characterization of the DEGs showing significant expression changes in response to KNL2 depletion was done based on the TAIR10 annotation (https://www.arabidopsis.org/). Gene Ontologies were analyzed for term enrichment using the Generic Gene Ontology GO::TermFinder tool ([22]. The analysis was carried out using the Benjamini–Hochberg FDR with a filter p-value of < 0.05.
Information about flowering-related genes was extracted from the Flowering Interactive Database [60]. The appearance of the transcription factors in the analyzed gene sets was confirmed with the Plant Transcription Factor Database v.4.0 [40]. Information about the genes essential for the Arabidopsis development from the SeedGenes Project [61] was used to find the corresponding genes among differentially expressed genes.
3.6. Gene Expression Validation by Reverse Transcription Quantitative PCR (RT-qPCR)
Total RNA extraction was performed as described above. The RNA was treated with DNase I (Ambion, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol to eliminate any residual genomic DNA. Reverse transcription was performed using a first-strand cDNA synthesis kit, oligo dT (18-mer) primer (both Fermentas, Thermo Fisher Scientific, Waltham, MA, USA), and 2 μg of total RNA as a starting material.
The gene-specific primers (Table S6, Supplementary Materials) were designed using the fully automated QuantPrime tool [62]. The amplification of the UBQ10 (AT4G05320) reference gene [63] was used as an internal control to normalize the data.
Quantitative real-time measurements were performed using POWER SYBR Green Master Mix reagent in a QuantStudio 6 Flex system (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. The cDNA equivalent to 40 ng of total RNA was used in a 10 µL PCR reaction.
The cycling conditions comprised 10 min polymerase activation at 95 °C and 40 cycles at 95 °C for 3 s and 60 °C for 30 s. Three biological replicates per genotype (wild-type and knl2 mutant line) in both conditions (seedlings and flower buds) were tested. Each biological replicate was represented with three technical replicates, which were analyzed during the same run. Relative gene expression was calculated using the comparative method 2−∆∆CT [64].
3.7. Data Availability
Raw reads are available under the accession number PRJEB32230 at http://www.ebi.ac.uk/ena/data/view/PRJEB32230.
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/20/22/5726/s1. Figure S1. knl2 sensitivity to DNA damaging agents. The plants were grown for seven days on media containing specified drugs. Figure S2. Effect of KNL2 depletion on the flowering time.The flowering time of the knl2 mutant delayed by 10–14 days compared to Arabidopsis wild-type. Seeds of the knl2 mutant and wild-type were germinated under short-day conditions, 8 h light/20° C and 16 h dark/18 °C, for two weeks and then plants were transferred to the cultivation room with long-day conditions, 16 h light/20° C and 8 h dark/18 °C. To demonstrate differences in flowering time between the knl2 mutant and wild-type, images of plant populations were taken with different time intervals. At early growth stages (14-day-old plants), no obvious phenotypical differences between the Col wild-type and the knl2 mutant were observed. Figure S3. qRT–PCR validation of RNA-seq results. Eleven genes were selected from the genes differentially expressed in flower buds according to the RNA-seq data analysis. Detailed annotation of the selected genes is presented in Table S2. *At5g02520 corresponds to KNL2. Error bars indicate the standard error of three biological replicates in qRT-PCR. Table S1. Results of the RNA-seq study based on the DeSeq2 in Col and the knl2 mutant line. Conditions: seedlings and flower buds. Table S2. Differentially expressed genes in knl2 seedlings and their annotation. Table S3. Differentially expressed genes in knl2 flower buds and their annotation. Table S4. Identification of root-related genes in knl2 seedlings. Table S5. Overlap between seed-related genes from the SeedGenes Project DB and differentially expressed genes in knl2 flower buds. Table S6. Selected genes and their primers used for qRT-PCR. Dataset S1. GO terms overrepresented in the knl2 mutant line. Conditions: seedlings and flower buds. Dataset S2. Overlap between TF from The Plant TF database v.4 and differentially expressed genes in the knl2 mutant line. Dataset S3. Overlap between flowering-related genes from the database FLOR-ID and differentially expressed genes in the knl2 mutant line.
Author Contributions
A.B., A.P., and I.L. designed the research. A.B., K.P., and I.L. performed experiments. A.B., A.H., A.F., K.P., A.P., and I.L. analyzed the data. A.B. and I.L. wrote the manuscript. All authors read and approved the final manuscript.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (LE2299/1–2) and the European Regional Development Fund—Project “MSCAfellow@MUNI” (No. CZ.02.2.69/0.0/0.0/17_050/0008496). A.B. was supported by the Deutsche Forschungsgemeinschaft (HO 1779/30–1). A.H. was supported by the German Federal Ministry of Education and Research (Plant 2030, Project 031B0192NN, HaploTools). A.P. and K.P. were supported by a grant from the Czech grant agency (19–13848S). A.P. was further supported by the ERDF project "Plants as a tool for sustainable global development" (No. CZ.02.1.01/0.0/0.0/16_019/0000827).
Acknowledgments
The authors wish to thank Oda Weiss and Heike Kuhlmann for technical assistance. We are grateful to Axel Himmelbach for the RNA-sequencing performed at the IPK Sequencing laboratory (Gatersleben).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Talbert, P.B.; Henikoff, S. Phylogeny as the basis for naming histones. Trends Genet. 2013, 29, 499–500. [Google Scholar] [CrossRef]
- Silva, M.C.; Jansen, L.E. At the right place at the right time: Novel CENP-A binding proteins shed light on centromere assembly. Chromosoma 2009, 118, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Transcribing centromeres: Noncoding RNAs and kinetochore assembly. Trends Genet. 2018, 34, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Perea-Resa, C.; Blower, M.D. Centromere biology: Transcription goes on stage. Mol. Cell Biol. 2018, 38, e00263-18. [Google Scholar] [CrossRef]
- Bobkov, G.O.M.; Gilbert, N.; Heun, P. Centromere transcription allows CENP-A to transit from chromatin association to stable incorporation. J. Cell Biol. 2018, 217, 1957–1972. [Google Scholar] [CrossRef]
- Bergmann, J.H.; Rodriguez, M.G.; Martins, N.M.; Kimura, H.; Kelly, D.A.; Masumoto, H.; Larionov, V.; Jansen, L.E.; Earnshaw, W.C. Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J. 2011, 30, 328–340. [Google Scholar] [CrossRef]
- Kim, I.S.; Lee, M.; Park, K.C.; Jeon, Y.; Park, J.H.; Hwang, E.J.; Jeon, T.I.; Ko, S.; Lee, H.; Baek, S.H.; et al. Roles of Mis18alpha in epigenetic regulation of centromeric chromatin and CENP-A loading. Mol. Cell 2012, 46, 260–273. [Google Scholar] [CrossRef]
- Nardi, I.K.; Zasadzinska, E.; Stellfox, M.E.; Knippler, C.M.; Foltz, D.R. Licensing of centromeric chromatin assembly through the Mis18alpha-Mis18beta heterotetramer. Mol. Cell 2016, 61, 774–787. [Google Scholar] [CrossRef]
- Fujita, Y.; Hayashi, T.; Kiyomitsu, T.; Toyoda, Y.; Kokubu, A.; Obuse, C.; Yanagida, M. Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev. Cell 2007, 12, 17–30. [Google Scholar] [CrossRef]
- Maddox, P.S.; Hyndman, F.; Monen, J.; Oegema, K.; Desai, A. Functional genomics identifies a Myb domain-containing protein family required for assembly of CENP-A chromatin. J. Cell Biol. 2007, 176, 757–763. [Google Scholar] [CrossRef]
- Foltz, D.R.; Jansen, L.E.; Bailey, A.O.; Yates, J.R., 3rd; Bassett, E.A.; Wood, S.; Black, B.E.; Cleveland, D.W. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 2009, 137, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.C.; Kuich, P.H.; Stellfox, M.E.; Ward, J.A.; Bassett, E.A.; Black, B.E.; Foltz, D.R. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J. Cell Biol. 2011, 194, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Lermontova, I.; Kuhlmann, M.; Friedel, S.; Rutten, T.; Heckmann, S.; Sandmann, M.; Demidov, D.; Schubert, V.; Schubert, I. Arabidopsis KINETOCHORE NULL2 is an upstream component for centromeric histone H3 variant cenH3 deposition at centromeres. Plant Cell 2013, 25, 3389–3404. [Google Scholar] [CrossRef]
- Sandmann, M.; Talbert, P.; Demidov, D.; Kuhlmann, M.; Rutten, T.; Conrad, U.; Lermontova, I. Targeting of arabidopsis KNL2 to centromeres depends on the conserved CENPC-k motif in Its C terminus. Plant Cell 2017, 29, 144–155. [Google Scholar] [CrossRef]
- Zhang, D.; Martyniuk, C.J.; Trudeau, V.L. SANTA domain: A novel conserved protein module in Eukaryota with potential involvement in chromatin regulation. Bioinformatics 2006, 22, 2459–2462. [Google Scholar] [CrossRef]
- Kral, L. Possible identification of CENP-C in fish and the presence of the CENP-C motif in M18BP1 of vertebrates. F1000Research 2015, 4, 474. [Google Scholar] [CrossRef]
- French, B.T.; Westhorpe, F.G.; Limouse, C.; Straight, A.F. Xenopus laevis M18BP1 directly binds existing CENP-A nucleosomes to promote centromeric chromatin assembly. Dev. Cell 2017, 42, 190e10–199e10. [Google Scholar] [CrossRef]
- Hori, T.; Shang, W.H.; Hara, M.; Ariyoshi, M.; Arimura, Y.; Fujita, R.; Kurumizaka, H.; Fukagawa, T. Association of M18BP1/KNL2 with CENP-A nucleosome is essential for centromere formation in non-mammalian vertebrates. Dev. Cell 2017, 42, 181e3–189e3. [Google Scholar] [CrossRef]
- Liebelt, F.; Jansen, N.S.; Kumar, S.; Gracheva, E.; Claessens, L.A.; Verlaan-de Vries, M.; Willemstein, E.; Vertegaal, A.C.O. The poly-SUMO2/3 protease SENP6 enables assembly of the constitutive centromere-associated network by group deSUMOylation. Nat. Commun. 2019, 10, 3987. [Google Scholar] [CrossRef]
- Silva, M.C.; Bodor, D.L.; Stellfox, M.E.; Martins, N.M.; Hochegger, H.; Foltz, D.R.; Jansen, L.E. Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev. Cell 2012, 22, 52–63. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO: TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, H.; Lloyd, C.W. Arabidopsis mutants and the network of microtubule-associated functions. Mol. Plant 2008, 1, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Wang, L.; Zhang, L.; Ren, D.; Ren, R.; Copenhaver, G.P.; Ma, H.; Wang, Y. Arabidopsis cell division cycle 20.1 is required for normal meiotic spindle assembly and chromosome segregation. Plant Cell 2015, 27, 3367–3382. [Google Scholar] [CrossRef] [PubMed]
- Kevei, Z.; Baloban, M.; Da Ines, O.; Tiricz, H.; Kroll, A.; Regulski, K.; Mergaert, P.; Kondorosi, E. Conserved CDC20 cell cycle functions are carried out by two of the five isoforms in Arabidopsis thaliana. PLoS ONE 2011, 6, e20618. [Google Scholar] [CrossRef]
- Bucher, E.; Reinders, J.; Mirouze, M. Epigenetic control of transposon transcription and mobility in Arabidopsis. Curr. Opin. Plant Biol. 2012, 15, 503–510. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef]
- Deniz, O.; Frost, J.M.; Branco, M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef]
- Rangwala, S.H.; Richards, E.J. Differential epigenetic regulation within an Arabidopsis retroposon family. Genetics 2007, 176, 151–160. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, C.; Lin, Q.; Liu, A.; Wang, T.; Feng, X.; Liu, J.; Han, H.; Ma, Y.; Bonea, D.; et al. A tetratricopeptide repeat domain-containing protein SSR1 located in mitochondria is involved in root development and auxin polar transport in Arabidopsis. Plant J. Cell Mol. Biol. 2015, 83, 582–599. [Google Scholar] [CrossRef]
- Munoz-Bertomeu, J.; Cascales-Minana, B.; Mulet, J.M.; Baroja-Fernandez, E.; Pozueta-Romero, J.; Kuhn, J.M.; Segura, J.; Ros, R. Plastidial glyceraldehyde-3-phosphate dehydrogenase deficiency leads to altered root development and affects the sugar and amino acid balance in Arabidopsis. Plant Physiol. 2009, 151, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Garay-Arroyo, A.; Ortiz-Moreno, E.; de la Paz Sanchez, M.; Murphy, A.S.; Garcia-Ponce, B.; Marsch-Martinez, N.; de Folter, S.; Corvera-Poire, A.; Jaimes-Miranda, F.; Pacheco-Escobedo, M.A.; et al. The MADS transcription factor XAL2/AGL14 modulates auxin transport during Arabidopsis root development by regulating PIN expression. EMBO J. 2013, 32, 2884–2895. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Lopez, R.; Garcia-Ponce, B.; Dubrovsky, J.G.; Garay-Arroyo, A.; Perez-Ruiz, R.V.; Kim, S.H.; Acevedo, F.; Pelaz, S.; Alvarez-Buylla, E.R. An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol. 2008, 146, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, K.; Nagata, N.; Yee, K.M.; Braybrook, S.A.; Pelletier, J.; Fujioka, S.; Yoshida, S.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. TANMEI/EMB2757 encodes a WD repeat protein required for embryo development in Arabidopsis. Plant Physiol. 2005, 139, 163–173. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Srivastava, R.; Liu, J.X.; Guo, H.; Yin, Y.; Howell, S.H. Regulation and processing of a plant peptide hormone, AtRALF23, in Arabidopsis. Plant J. Cell Mol. Biol. 2009, 59, 930–939. [Google Scholar] [CrossRef]
- Manova, V.; Gruszka, D. DNA damage and repair in plants—From models to crops. Front. Plant Sci. 2015, 6, 885. [Google Scholar] [CrossRef]
- Li, W.; Chen, C.; Markmann-Mulisch, U.; Timofejeva, L.; Schmelzer, E.; Ma, H.; Reiss, B. The Arabidopsis AtRAD51 gene is dispensable for vegetative development but required for meiosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10596–10601. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Xu, Y.; Brinkman, K.L.; Ishiyama, H.; Wong, S.T.; Xu, B. The kinetochore protein Bub1 participates in the DNA damage response. DNA Repair 2012, 11, 185–191. [Google Scholar] [CrossRef]
- Zeitlin, S.G.; Baker, N.M.; Chapados, B.R.; Soutoglou, E.; Wang, J.Y.; Berns, M.W.; Cleveland, D.W. Double-strand DNA breaks recruit the centromeric histone CENP-A. Proc. Natl. Acad. Sci. USA 2009, 106, 15762–15767. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Finatto, T.; Viana, V.E.; Woyann, L.G.; Busanello, C.; da Maia, L.C.; de Oliveira, A.C. Can WRKY transcription factors help plants to overcome environmental challenges? Genet. Mol. Biol. 2018, 41, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Rizhsky, L.; Liang, H.; Shuman, J.; Shulaev, V.; Mittler, R. Enhanced tolerance to environmental stress in transgenic plants expressing the transcriptional coactivator multiprotein bridging factor 1c. Plant Physiol. 2005, 139, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Nolan, T.M.; Ye, H.; Zhang, M.; Tong, H.; Xin, P.; Chu, J.; Chu, C.; Li, Z.; Yin, Y. Arabidopsis WRKY46, WRKY54, and WRKY70 transcription factors are involved in brassinosteroid-regulated plant growth and drought responses. Plant Cell 2017, 29, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.J.; Yan, J.Y.; Xu, X.Y.; Yu, D.Q.; Li, G.X.; Zhang, S.Q.; Zheng, S.J. Transcription factor WRKY46 regulates osmotic stress responses and stomatal movement independently in Arabidopsis. Plant J. Cell Mol. Biol. 2014, 79, 13–27. [Google Scholar] [CrossRef]
- Sheikh, A.H.; Eschen-Lippold, L.; Pecher, P.; Hoehenwarter, W.; Sinha, A.K.; Scheel, D.; Lee, J. Regulation of WRKY46 transcription factor function by mitogen-activated protein kinases in arabidopsis THALIANA. Front. Plant Sci. 2016, 7, 61. [Google Scholar] [CrossRef]
- Krishnaswamy, S.; Verma, S.; Rahman, M.H.; Kav, N.N. Functional characterization of four APETALA2-family genes (RAP2.6, RAP2.6L, DREB19 and DREB26) in Arabidopsis. Plant Mol. Biol. 2011, 75, 107–127. [Google Scholar] [CrossRef]
- Matias-Hernandez, L.; Aguilar-Jaramillo, A.E.; Marin-Gonzalez, E.; Suarez-Lopez, P.; Pelaz, S. RAV genes: Regulation of floral induction and beyond. Ann. Bot. 2014, 114, 1459–1470. [Google Scholar] [CrossRef]
- Higginson, T.; Li, S.F.; Parish, R.W. AtMYB103 regulates tapetum and trichome development in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2003, 35, 177–192. [Google Scholar] [CrossRef]
- Adamczyk, B.J.; Fernandez, D.E. MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 2009, 149, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Verelst, W.; Twell, D.; de Folter, S.; Immink, R.; Saedler, H.; Munster, T. MADS-complexes regulate transcriptome dynamics during pollen maturation. Genome Biol. 2007, 8, R249. [Google Scholar] [CrossRef] [PubMed]
- Smaczniak, C.; Immink, R.G.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ruiz, R.V.; Garcia-Ponce, B.; Marsch-Martinez, N.; Ugartechea-Chirino, Y.; Villajuana-Bonequi, M.; de Folter, S.; Azpeitia, E.; Davila-Velderrain, J.; Cruz-Sanchez, D.; Garay-Arroyo, A.; et al. XAANTAL2 (AGL14) Is an important component of the complex gene regulatory network that underlies Arabidopsis shoot apical meristem transitions. Mol. Plant 2015, 8, 796–813. [Google Scholar] [CrossRef] [PubMed]
- Smyth, D.R.; Bowman, J.L.; Meyerowitz, E.M. Early flower development in Arabidopsis. Plant Cell 1990, 2, 755–767. [Google Scholar]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 24 November 2010).
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bouché, F.; Lobet, G.; Tocquin, P.; Périlleux, C. FLOR-ID: An interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 2016, 44, D1167–D1171. [Google Scholar] [CrossRef]
- Meinke, D.; Muralla, R.; Sweeney, C.; Dickerman, A. Identifying essential genes in Arabidopsis thaliana. Trends Plant Sci. 2008, 13, 483–491. [Google Scholar] [CrossRef]
- Arvidsson, S.; Kwasniewski, M.; Riano-Pachon, D.M.; Mueller-Roeber, B. QuantPrime—A flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinform. 2008, 9, 465. [Google Scholar] [CrossRef] [PubMed]
- Czechowski, T.; Stitt, M.; Altmann, T.; Udvardi, M.K.; Scheible, W.R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).