Transplacental Gene Delivery (TPGD) as a Noninvasive Tool for Fetal Gene Manipulation in Mice



Abstract
1. Introduction
2. TPGD
2.1. Past Achievements
2.2. Optimal Timing of TPGD
2.3. Fetal Immune Responses by TPGD
2.4. Gene Delivery Cargo and TPGD
3. Mechanism of TPGD
4. Present Status of TPGD
4.1. CRISPR/Cas9 System
4.2. TPGD-GEF
5. Application of TPGD-GEF to Manipulations of Fetal Cells
5.1. Fetal Gene Therapy
5.2. Improvements of TPGD Efficiency
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| BBB | Blood-brain barrier |
| BPB | Blood-placental barrier |
| B6C3F1 | Mouse hybrid between C57BL/6 and C3H/H |
| Cas9 | Clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein-9 nuclease |
| CAT | Chloramphenicol acetyltransferase |
| CMV | Cytomegalovirus |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DSBs | Double-stranded breaks |
| DsRed | Discosoma sp. red fluorescent protein |
| EGFP | Enhanced green fluorescent protein |
| GFP | Green fluorescent protein |
| GOI | Gene of interest |
| gRNA | Guide RNA |
| HBs | Hepatitis B surface antigen |
| HBV | Hepatitis B virus |
| HCM | Hypertrophic cardiomyopathy |
| HGD | Hydrodynamics-based gene delivery |
| HIV | Human immunodeficiency virus |
| lacZ | β-Galactosidase |
| MHC | Myosin heavy chain |
| MVBs | Multivesicular bodies |
| NHEJ | Nonhomologous end-joining |
| NLS | Nuclear location signal |
| PAM | Protospacer adjacent motif |
| rAAV | Recombinant adeno-associated virus |
| RNAi | RNA interference |
| RNP | Ribonucleoprotein |
| sFlt-1 | Soluble fms-like tyrosine kinase-1 |
| shRNA | Short hairpin RNA |
| Sry | Sex-determining region Y |
| ST | Syncytiotrophoblast |
| TALEN | Transcription activator-like effector nucleases |
| Tg mouse | Transgenic mouse |
| THL | Trojan horse liposome |
| TPFE | Tetra (piperazino) fullerene epoxide |
| TPGD | Transplacental gene delivery |
| TPGD-GEF | Transplacental gene delivery (TPGD) for acquiring genome-edited fetuses |
| VE | Visceral endoderm |
| VS | Villous stroma |
| ZFN | Zinc-finger nuclease |
References
- Ho, B.X.; Loh, S.J.H.; Chan, W.K.; Soh, B.S. In vivo genome editing as a therapeutic approach. Int. J. Mol. Sci. 2018, 19, 2721. [Google Scholar] [CrossRef]
- Wang, L.; Li, F.; Dang, L.; Liang, C.; Wang, C.; He, B.; Liu, J.; Li, D.; Wu, X.; Xu, X.; et al. In vivo delivery systems for therapeutic genome editing. Int. J. Mol. Sci. 2016, 17, 626. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef]
- Burnett, J.C.; Zaia, J.A.; Rossi, J.J. Creating genetic resistance to HIV. Curr. Opin. Immunol. 2012, 24, 625–632. [Google Scholar] [CrossRef]
- Qasim, W.; Amrolia, P.J.; Samarasinghe, S.; Ghorashian, S.; Zhan, H.; Stafford, S.; Butler, K.; Ahsan, G.; Gilmour, K.; Adams, S.; et al. First clinical application of TALEN engineered universal CAR19 T cells in B-ALL. Blood 2015, 126, 2046. [Google Scholar] [CrossRef]
- Chira, S.; Gulei, D.; Hajitou, A.; Zimta, A.A.; Cordelier, P.; Berindan-Neagoe, I. CRISPR/Cas9: Transcending the reality of genome editing. Mol. Ther. Nucleic Acids 2017, 7, 211–222. [Google Scholar] [CrossRef]
- Li, L.; Hu, S.; Chen, X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials 2018, 171, 207–218. [Google Scholar] [CrossRef]
- Larson, J.E.; Cohen, J.C. In utero gene therapy. Ochsner J. 2000, 2, 107–110. [Google Scholar]
- Okuda, K.; Xin, K.Q.; Haruki, A.; Kawamoto, S.; Kojima, Y.; Hirahara, F.; Okada, H.; Klinman, D.; Hamajima, K. Transplacental genetic immunization after intravenous delivery of plasmid DNA to pregnant mice. J. Immunol. 2001, 167, 5478–5484. [Google Scholar] [CrossRef]
- O’Shea, K.S.; De Boer, L.S.; Slawny, N.A.; Gratsch, T.E. Transplacental RNAi: Deciphering gene function in the postimplantation-staged embryo. J. Biomed. Biotechnol. 2006, 2006, 18657. [Google Scholar] [CrossRef]
- Baldwin, H.S.; Mickanin, C.; Buck, C. Adenovirus-mediated gene transfer during initial organogenesis in the mammalian embryo is promoter-dependent and tissue-specific. Gene Ther. 1997, 4, 1142–1149. [Google Scholar] [CrossRef]
- Larson, J.E.; Morrow, S.L.; Happel, L.; Sharp, J.F.; Cohen, J.C. Reversal of cystic fibrosis phenotype in mice by gene therapy in utero. Lancet 1997, 349, 619–620. [Google Scholar] [CrossRef]
- Gaensler, K.M.; Tu, G.; Bruch, S.; Liggitt, D.; Lipshutz, G.S.; Metkus, A.; Harrison, M.; Heath, T.D.; Debs, R.J. Fetal gene transfer by transuterine injection of cationic liposome-DNA complexes. Nat. Biotechnol. 1999, 17, 1188–1192. [Google Scholar] [CrossRef]
- Papaioannou, V.E. In utero manipulation. In Postimplantation Mammalian Embryos; IRL Press (at Oxford University Press): Oxford, UK, 1990; pp. 61–80. [Google Scholar]
- Turkay, A.; Saunders, T.; Kurachi, K. Intrauterine gene transfer: Gestational stage-specific gene delivery in mice. Gene Ther. 1999, 6, 1685–1694. [Google Scholar] [CrossRef]
- Douar, A.M.; Adebakin, S.; Themis, M.; Pavirani, A.; Cook, T.; Coutelle, C. Foetal gene delivery in mice by intra-amniotic administration of retroviral producer cells and adenovirus. Gene Ther. 1997, 4, 883–890. [Google Scholar] [CrossRef]
- Schachtner, S.; Buck, C.; Bergelson, J.; Baldwin, H. Temporally regulated expression patterns following in utero adenovirus-mediated gene transfer. Gene Ther. 1999, 6, 1249–1257. [Google Scholar] [CrossRef]
- Tsukamoto, M.; Ochiya, T.; Yoshida, S.; Sugimura, T.; Terada, M. Gene transfer and expression in progeny after intravenous DNA injection into pregnant mice. Nat. Genet. 1995, 9, 243–248. [Google Scholar] [CrossRef]
- Nakamura, S.; Ishihara, M.; Ando, N.; Watanabe, S.; Sakurai, T.; Sato, M. Transplacental delivery of genome editing components causes mutations in embryonic cardiomyocytes of mid-gestational murine fetuses. IUBMB Life 2019, 71, 835–844. [Google Scholar] [CrossRef]
- Ochiya, T.; Takahama, Y.; Baba-Toriyama, H.; Tsukamoto, M.; Yasuda, Y.; Kikuchi, H.; Terada, M. Evaluation of cationic liposome suitable for gene transfer into pregnant animals. Biochem. Biophys. Res. Commun. 1999, 258, 358–365. [Google Scholar] [CrossRef]
- Kikuchi, N.; Nakamura, S.; Ohtsuka, M.; Kimura, M.; Sato, M. Possible mechanism of gene transfer into early to mid-gestational mouse fetuses by tail vein injection. Gene Ther. 2002, 9, 1529–1541. [Google Scholar] [CrossRef]
- Srivastava, A.S.; Chauhan, D.P.; Carrier, E. In utero detection of T7 phage after systemic administration to pregnant mice. Biotechniques 2004, 37, 81–83. [Google Scholar] [CrossRef]
- Maeda-Mamiya, R.; Noiri, E.; Isobe, H.; Nakanishi, W.; Okamoto, K.; Doi, K.; Sugaya, T.; Izumi, T.; Homma, T.; Nakamura, E. In vivo gene delivery by cationic tetraamino fullerene. Proc. Natl. Acad. Sci. USA 2010, 107, 5339–5344. [Google Scholar] [CrossRef]
- Efremov, A.M.; Buglaeva, A.O.; Orlov, S.V.; Burov, S.V.; Ignatovich, I.A.; Dizhe, E.B.; Shavva, V.S.; Perevozchikov, A.P. Transfer of genetic constructions through the transplacental barrier into mice embryos. Russ. J. Dev. Biol. 2010, 41, 71–76. [Google Scholar] [CrossRef]
- Wu, N.; Yu, A.B.; Zhu, H.B.; Lin, X.K. Effective silencing of Sry gene with RNA interference in developing mouse embryos resulted in feminization of XY gonad. J. Biomed. Biotechnol. 2012, 2012, 343891. [Google Scholar] [CrossRef]
- Picconi, J.L.; Muff-Luett, M.A.; Wu, D.; Bunchman, E.; Schaefer, F.; Brophy, P.D. Kidney-specific expression of GFP by in-utero delivery of pseudotyped adeno-associated virus 9. Mol. Ther. Methods Clin. Dev. 2014, 1, 14014. [Google Scholar] [CrossRef]
- Carver, A.R.; Andrikopoulou, M.; Lei, J.; Tamayo, E.; Gamble, P.; Hou, Z.; Zhang, J.; Mori, S.; Saade, G.R.; Costantine, M.M.; et al. Maternal pravastatin prevents altered fetal brain development in a preeclamptic CD-1 mouse model. PLoS ONE 2014, 9, e100873. [Google Scholar] [CrossRef]
- Cornford, E.M.; Hyman, S.; Cornford, M.E.; Chytrova, G.; Rhee, J.; Suzuki, T.; Yamagata, T.; Yamakawa, K.; Penichet, M.L.; Pardridge, W.M. Non-invasive gene targeting to the fetal brain after intravenous administration and transplacental transfer of plasmid DNA using PEGylated immunoliposomes. J. Drug Target. 2016, 24, 58–67. [Google Scholar] [CrossRef]
- Rugh, R. The Mouse: Its Reproduction and Development; Oxford Science Publications: Oxford, UK, 1990; pp. 116–146. [Google Scholar]
- Mor, G.; Singla, M.; Steinberg, A.D.; Hoffman, S.L.; Okuda, K.; Klinman, D.M. Do DNA vaccines induce autoimmune disease? Hum. Gene Ther. 1997, 8, 293–300. [Google Scholar] [CrossRef]
- Ichino, M.; Mor, G.; Conover, J.; Weiss, W.R.; Takeno, M.; Ishii, K.J.; Klinman, D.M. Factors associated with the development of neonatal tolerance after the administration of a plasmid DNA vaccine. J. Immunol. 1999, 162, 3814–3818. [Google Scholar]
- Zimmerman, R.K.; Ruben, F.L.; Ahwesh, E.R. Hepatitis B virus infection, hepatitis B vaccine, and hepatitis B immune globulin. J. Fam. Pract. 1997, 45, 295–315. [Google Scholar]
- Rinaldi, M.; Signori, E.; Rosati, P.; Cannelli, G.; Parrella, P.; Iannace, E.; Monego, G.; Ciafre, S.A.; Farace, M.G.; Iurescia, S.; et al. Feasibilty of in utero DNA vaccination following naked gene transfer into pig fetal muscle: Transgene expression, immunity and safety. Vaccine 2006, 24, 4586–4591. [Google Scholar] [CrossRef]
- Liu, J.; Chang, J.; Jiang, Y.; Meng, X.; Sun, T.; Mao, L.; Xu, Q.; Wang, M. Fast and efficient CRISPR/Cas9 genome editing in vivo enabled by bioreducible lipid and messenger RNA nanoparticles. Adv. Mater. 2019, 31, e1902575. [Google Scholar] [CrossRef]
- Timin, A.S.; Muslimov, A.R.; Lepik, K.V.; Epifanovskaya, O.S.; Shakirova, A.I.; Mock, U.; Riecken, K.; Okilova, M.V.; Sergeev, V.S.; Afanasyev, B.V.; et al. Efficient gene editing via non-viral delivery of CRISPR-Cas9 system using polymeric and hybrid microcarriers. Nanomedicine 2018, 14, 97–108. [Google Scholar] [CrossRef]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef]
- Hosseini, E.S.; Nikkhah, M.; Hosseinkhani, S. Cholesterol-rich lipid-mediated nanoparticles boost of transfection efficiency, utilized for gene editing by CRISPR-Cas9. Int. J. Nanomed. 2019, 14, 4353–4366. [Google Scholar] [CrossRef]
- Zhang, H.; Bahamondez-Canas, T.F.; Zhang, Y.; Leal, J.; Smyth, H.D.C. PEGylated chitosan for nonviral aerosol and mucosal delivery of the CRISPR/Cas9 system in vitro. Mol. Pharm. 2018, 15, 4814–4826. [Google Scholar] [CrossRef]
- Ryu, N.; Kim, M.A.; Park, D.; Lee, B.; Kim, Y.R.; Kim, K.H.; Baek, J.I.; Kim, W.J.; Lee, K.Y.; Kim, U.K. Effective PEI-mediated delivery of CRISPR-Cas9 complex for targeted gene therapy. Nanomedicine 2018, 14, 2095–2102. [Google Scholar] [CrossRef]
- Cheng, W.J.; Chen, L.C.; Ho, H.O.; Lin, H.L.; Sheu, M.T. Stearyl polyethylenimine complexed with plasmids as the core of human serum albumin nanoparticles noncovalently bound to CRISPR/Cas9 plasmids or siRNA for disrupting or silencing PD-L1 expression for immunotherapy. Int. J. Nanomed. 2018, 13, 7079–7094. [Google Scholar] [CrossRef]
- Zhang, Z.; Wan, T.; Chen, Y.; Chen, Y.; Sun, H.; Cao, T.; Songyang, Z.; Tang, G.; Wu, C.; Ping, Y.; et al. Cationic polymer-mediated CRISPR/Cas9 plasmid delivery for genome editing. Macromol. Rapid Commun. 2019, 40, e1800068. [Google Scholar] [CrossRef]
- Kretzmann, J.A.; Ho, D.; Evans, C.W.; Plani-Lam, J.H.C.; Garcia-Bloj, B.; Mohamed, A.E.; O’Mara, M.L.; Ford, E.; Tan, D.E.K.; Lister, R.; et al. Synthetically controlling dendrimer flexibility improves delivery of large plasmid DNA. Chem. Sci. 2017, 8, 2923–2930. [Google Scholar] [CrossRef]
- Gulei, D.; Berindan-Neagoe, I. Activation of necroptosis by engineered self tumor-derived exosomes loaded with CRISPR/Cas9. Mol. Ther. Nucleic Acids 2019, 17, 448–451. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, J.; Gu, W.; Huang, Y.; Tong, Z.; Huang, L.; Tan, J. Exosome-liposome hybrid nanoparticles deliver CRISPR/Cas9 system in MSCs. Adv. Sci. 2018, 5, 1700611. [Google Scholar] [CrossRef]
- Ibraheim, R.; Song, C.Q.; Mir, A.; Amrani, N.; Xue, W.; Sontheimer, E.J. All-in-one adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome Biol. 2018, 19, 137. [Google Scholar] [CrossRef]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Wang, D.; Mou, H.; Li, S.; Li, Y.; Hough, S.; Tran, K.; Li, J.; Yin, H.; Anderson, D.G.; Sontheimer, E.J.; et al. Adenovirus-mediated somatic genome editing of PTEN by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum. Gene Ther. 2015, 26, 432–442. [Google Scholar] [CrossRef]
- Cheng, R.; Peng, J.; Yan, Y.; Cao, P.; Wang, J.; Qiu, C.; Tang, L.; Liu, D.; Tang, L.; Jin, J.; et al. Efficient gene editing in adult mouse livers via adenoviral delivery of CRISPR/Cas9. FEBS Lett. 2014, 588, 3954–3958. [Google Scholar] [CrossRef]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Kay, M.A. State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 2011, 12, 316–328. [Google Scholar] [CrossRef]
- Gaspar, H.B.; Parsley, K.L.; Howe, S.; King, D.; Gilmour, K.C.; Sinclair, J.; Brouns, G.; Schmidt, M.; Von Kalle, C.; Barington, T.; et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet 2004, 364, 2181–2187. [Google Scholar] [CrossRef]
- Portman, O.W.; Behrman, R.E.; Soltys, P. Transfer of free fatty acids across the primate placenta. Am. J. Physiol. 1969, 216, 143–147. [Google Scholar] [CrossRef]
- Srivastava, A.S.; Kaido, T.; Carrier, E. Immunological factors that affect the in vivo fate of T7 phage in the mouse. J. Virol. Methods 2004, 115, 99–104. [Google Scholar] [CrossRef]
- Xu, X.; Wan, T.; Xin, H.; Li, D.; Pan, H.; Wu, J.; Ping, Y. Delivery of CRISPR/Cas9 for therapeutic genome editing. J. Gene Med. 2019, 21, e3107. [Google Scholar] [CrossRef]
- Lang, J.F.; Toulmin, S.A.; Brida, K.L.; Eisenlohr, L.C.; Davidson, B.L. Standard screening methods underreport AAV-mediated transduction and gene editing. Nat. Commun. 2019, 10, 3415. [Google Scholar] [CrossRef]
- Marshall, E. Gene therapy death prompts review of adenovirus vector. Science 1999, 286, 2244–2245. [Google Scholar] [CrossRef]
- Marshall, E. Clinical research. Gene therapy a suspect in leukemia-like disease. Science 2002, 298, 34–35. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, H.; Colosi, P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Nelson, C.E.; Gersbach, C.A. Engineering delivery vehicles for genome editing. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 637–662. [Google Scholar] [CrossRef]
- Tan, Y.; Chu, A.H.Y.; Bao, S.; Hoang, D.A.; Kebede, F.T.; Xiong, W.; Ji, M.; Shi, J.; Zheng, Z. Rationally engineered Staphylococcus aureus Cas9 nucleases with high genome-wide specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 20969–20976. [Google Scholar] [CrossRef]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef]
- Gude, N.M.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef]
- Beckman, D.A.; Lloyd, J.B.; Brent, R.L. Investigations into mechanisms of amino acid supply to the rat embryo using whole-embryo culture. Int. J. Dev. Biol. 1997, 41, 315–318. [Google Scholar]
- Terasawa, Y.; Cases, S.J.; Wong, J.S.; Jamil, H.; Jothi, S.; Traber, M.G.; Packer, L.; Gordon, D.A.; Hamilton, R.L.; Farese, R.V., Jr. Apolipoprotein B-related gene expression and ultrastructural characteristics of lipoprotein secretion in mouse yolk sac during embryonic development. J. Lipid Res. 1999, 40, 1967–1977. [Google Scholar]
- Guschanski, K.; Warnefors, M.; Kaessmann, H. The evolution of duplicate gene expression in mammalian organs. Genome Res. 2017, 27, 1461–1474. [Google Scholar] [CrossRef]
- Zhang, B.; Liang, R.; Zheng, M.; Cai, L.; Fan, X. Surface-functionalized nanoparticles as efficient tools in targeted therapy of pregnancy complications. Int. J. Mol. Sci. 2019, 20, 3642. [Google Scholar] [CrossRef]
- Menjoge, A.R.; Rinderknecht, A.L.; Navath, R.S.; Faridnia, M.; Kim, C.J.; Romero, R.; Miller, R.K.; Kannan, R.M. Transfer of PAMAM dendrimers across human placenta: Prospects of its use as drug carrier during pregnancy. J. Control. Release 2011, 150, 326–338. [Google Scholar] [CrossRef]
- Schneider, H.; Miller, R.K. Receptor-mediated uptake and transport of macromolecules in the human placenta. Int. J. Dev. Biol. 2010, 54, 367–375. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Peng, R.; Lin, G.; Li, J. Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. 2016, 283, 1218–1231. [Google Scholar] [CrossRef]
- Wu, W.Y.; Lebbink, J.H.G.; Kanaar, R.; Geijsen, N.; van der Oost, J. Genome editing by natural and engineered CRISPR-associated nucleases. Nat. Chem. Biol. 2018, 14, 642–651. [Google Scholar] [CrossRef]
- Xu, X.; Qi, L.S. A CRISPR-dCas toolbox for genetic engineering and synthetic biology. J. Mol. Biol. 2019, 431, 34–47. [Google Scholar] [CrossRef]
- Sato, M.; Ohtsuka, M.; Nakamura, S.; Sakurai, T.; Watanabe, S.; Gurumurthy, C.B. In vivo genome editing targeted towards the female reproductive system. Arch. Pharm. Res. 2018, 41, 898–910. [Google Scholar] [CrossRef]
- Watanabe, S.; Sakurai, T.; Nakamura, S.; Miyoshi, K.; Sato, M. The combinational use of CRISPR/Cas9 and targeted toxin technology enables efficient isolation of bi-allelic knockout non-human mammalian clones. Int. J. Mol. Sci. 2018, 19, 1075. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Karvelis, T.; Gasiunas, G.; Siksnys, V. Methods for decoding Cas9 protospacer adjacent motif (PAM) sequences: A brief overview. Methods 2017, 121-122, 3–8. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Wagner, A.M.; Schoeberlein, A.; Surbek, D. Fetal gene therapy: Opportunities and risks. Adv. Drug Deliv. Rev. 2009, 61, 813–821. [Google Scholar] [CrossRef]
- Rossidis, A.C.; Stratigis, J.D.; Chadwick, A.C.; Hartman, H.A.; Ahn, N.J.; Li, H.; Singh, K.; Coons, B.E.; Li, L.; Lv, W.; et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med. 2018, 24, 1513–1518. [Google Scholar] [CrossRef]
- Alapati, D.; Zacharias, W.J.; Hartman, H.A.; Rossidis, A.C.; Stratigis, J.D.; Ahn, N.J.; Coons, B.; Zhou, S.; Li, H.; Singh, K.; et al. In utero gene editing for monogenic lung disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Jiang, J.; Wakimoto, H.; Seidman, J.G.; Seidman, C.E. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 2013, 342, 111–114. [Google Scholar] [CrossRef]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef]
- Nakamura, S.; Maehara, T.; Watanabe, S.; Ishihara, M.; Sato, M. Improvement of hydrodynamics-based gene transfer of nonviral DNA targeted to murine hepatocytes. Biomed. Res. Int. 2013, 2013, 928790. [Google Scholar] [CrossRef]
- Nakamura, S.; Maehara, T.; Watanabe, S.; Ishihara, M.; Sato, M. Liver lobe and strain difference in gene expression after hydrodynamics-based gene delivery in mice. Anim. Biotechnol. 2015, 26, 51–57. [Google Scholar] [CrossRef]
- Nakamura, S.; Ishihara, M.; Watanabe, S.; Ando, N.; Ohtsuka, M.; Sato, M. Intravenous delivery of piggybac transposons as a useful tool for liver-specific gene-switching. Int. J. Mol. Sci. 2018, 19, 3452. [Google Scholar] [CrossRef]
- Kertschanska, S.; Stulcova, B.; Kaufmann, P.; Stulc, J. Distensible transtrophoblastic channels in the rat placenta. Placenta 2000, 21, 670–677. [Google Scholar] [CrossRef]
- Suda, T.; Liu, D. Hydrodynamic delivery. Adv. Genet. 2015, 89, 89–111. [Google Scholar]
- Dragovic, R.A.; Gardiner, C.; Brooks, A.S.; Tannetta, D.S.; Ferguson, D.J.P.; Hole, P.; Carr, B.; Redman, C.W.G.; Harris, A.L.; Dobson, P.J.; et al. Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomedicine 2011, 7, 780–788. [Google Scholar] [CrossRef]
- Thebaud, B.; Stewart, D.J. Exosomes: Cell garbage can, therapeutic carrier, or trojan horse? Circulation 2012, 126, 2553–2555. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Sato, S.; Yoshida, K.; Kawauchi, S.; Hosoe, K.; Akutsu, Y.; Fujimoto, N.; Nawashiro, H.; Terakawa, M. Highly site-selective transvascular drug delivery by the use of nanosecond pulsed laser-induced photomechanical waves. J. Control. Release 2014, 192, 228–235. [Google Scholar] [CrossRef]
- Sakurai, T.; Kamiyoshi, A.; Kawate, H.; Mori, C.; Watanabe, S.; Tanaka, M.; Uetake, R.; Sato, M.; Shindo, T. A non-inheritable maternal Cas9-based multiple-gene editing system in mice. Sci. Rep. 2016, 6, 20011. [Google Scholar] [CrossRef]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef]
- Carroll, K.J.; Makarewich, C.A.; McAnally, J.; Anderson, D.M.; Zentilin, L.; Liu, N.; Giacca, M.; Bassel-Duby, R.; Olson, E.N. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc. Natl. Acad. Sci. USA 2016, 113, 338–343. [Google Scholar] [CrossRef]
- Doetschman, T.; Georgieva, T. Gene editing with CRISPR/Cas9 RNA-directed nuclease. Circ. Res. 2017, 120, 876–894. [Google Scholar] [CrossRef]
- Lattanzi, A.; Meneghini, V.; Pavani, G.; Amor, F.; Ramadier, S.; Felix, T.; Antoniani, C.; Masson, C.; Alibeu, O.; Lee, C.; et al. Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Mol. Ther. 2019, 27, 137–150. [Google Scholar] [CrossRef]
- Sun, W.; Ji, W.; Hall, J.M.; Hu, Q.; Wang, C.; Beisel, C.L.; Gu, Z. Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew. Chem. Int. Ed. Engl. 2015, 54, 12029–12033. [Google Scholar] [CrossRef]
- D’Astolfo, D.S.; Pagliero, R.J.; Pras, A.; Karthaus, W.R.; Clevers, H.; Prasad, V.; Lebbink, R.J.; Rehmann, H.; Geijsen, N. Efficient intracellular delivery of native proteins. Cell 2015, 161, 674–690. [Google Scholar] [CrossRef]
- Guan, X.; Luo, Z.; Sun, W. A peptide delivery system sneaks CRISPR into cells. J. Biol. Chem. 2018, 293, 17306–17307. [Google Scholar] [CrossRef]
- Gaj, T.; Staahl, B.T.; Rodrigues, G.M.C.; Limsirichai, P.; Ekman, F.K.; Doudna, J.A.; Schaffer, D.V. Targeted gene knock-in by homology-directed genome editing using Cas9 ribonucleoprotein and AAV donor delivery. Nucleic Acids Res. 2017, 45, e98. [Google Scholar] [CrossRef]
- Senut, M.C.; Suhr, S.T.; Gage, F.H. Gene transfer to the rodent placenta in situ. A new strategy for delivering gene products to the fetus. J. Clin. Investig. 1998, 101, 1565–1571. [Google Scholar] [CrossRef][Green Version]
- Molas, M.; Gomez-Valades, A.G.; Vidal-Alabro, A.; Miguel-Turu, M.; Bermudez, J.; Bartrons, R.; Perales, J.C. Receptor-mediated gene transfer vectors: Progress towards genetic pharmaceuticals. Curr. Gene Ther. 2003, 3, 468–485. [Google Scholar] [CrossRef]





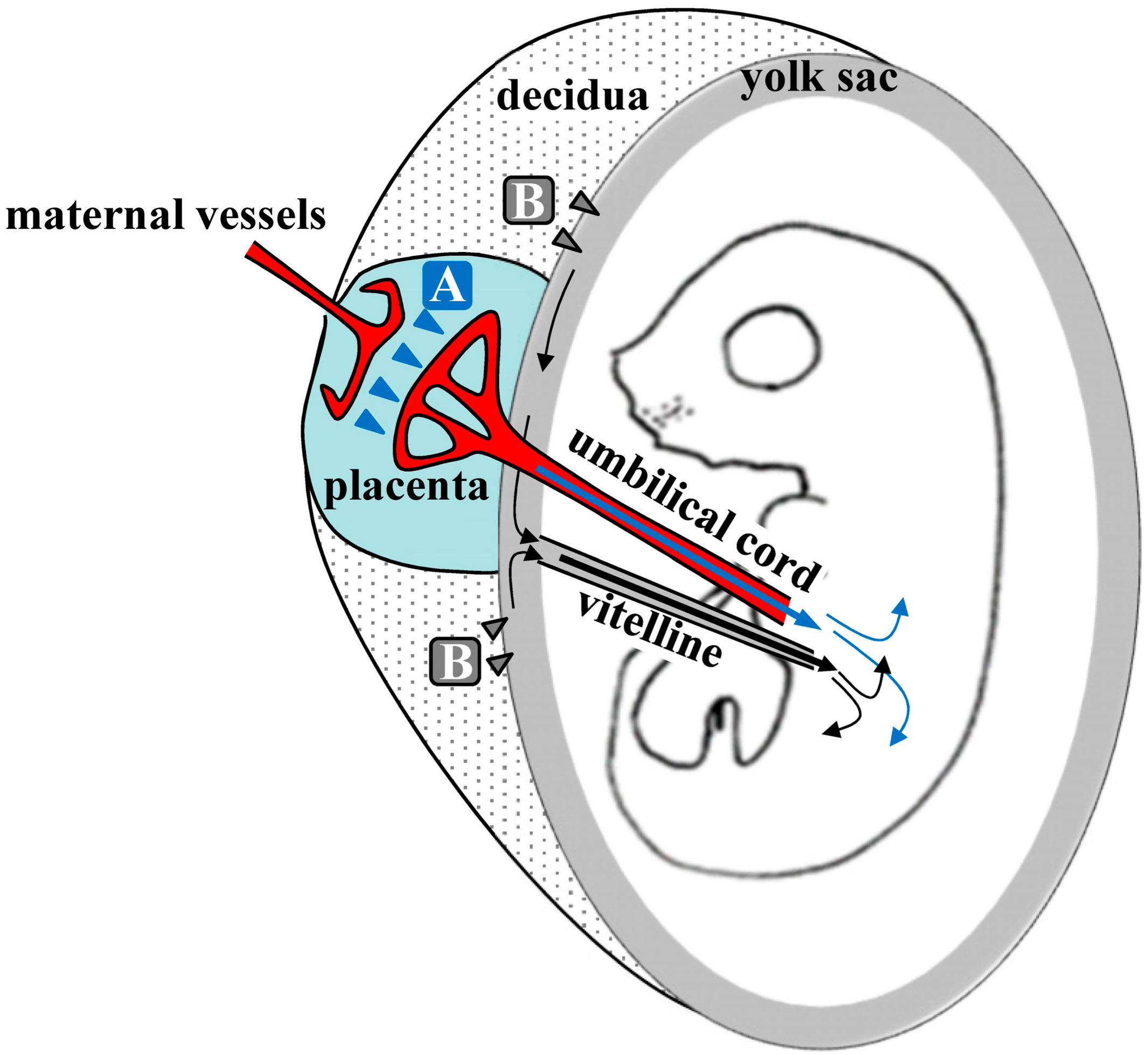{kind=link}
{kind=link}
{kind=link}
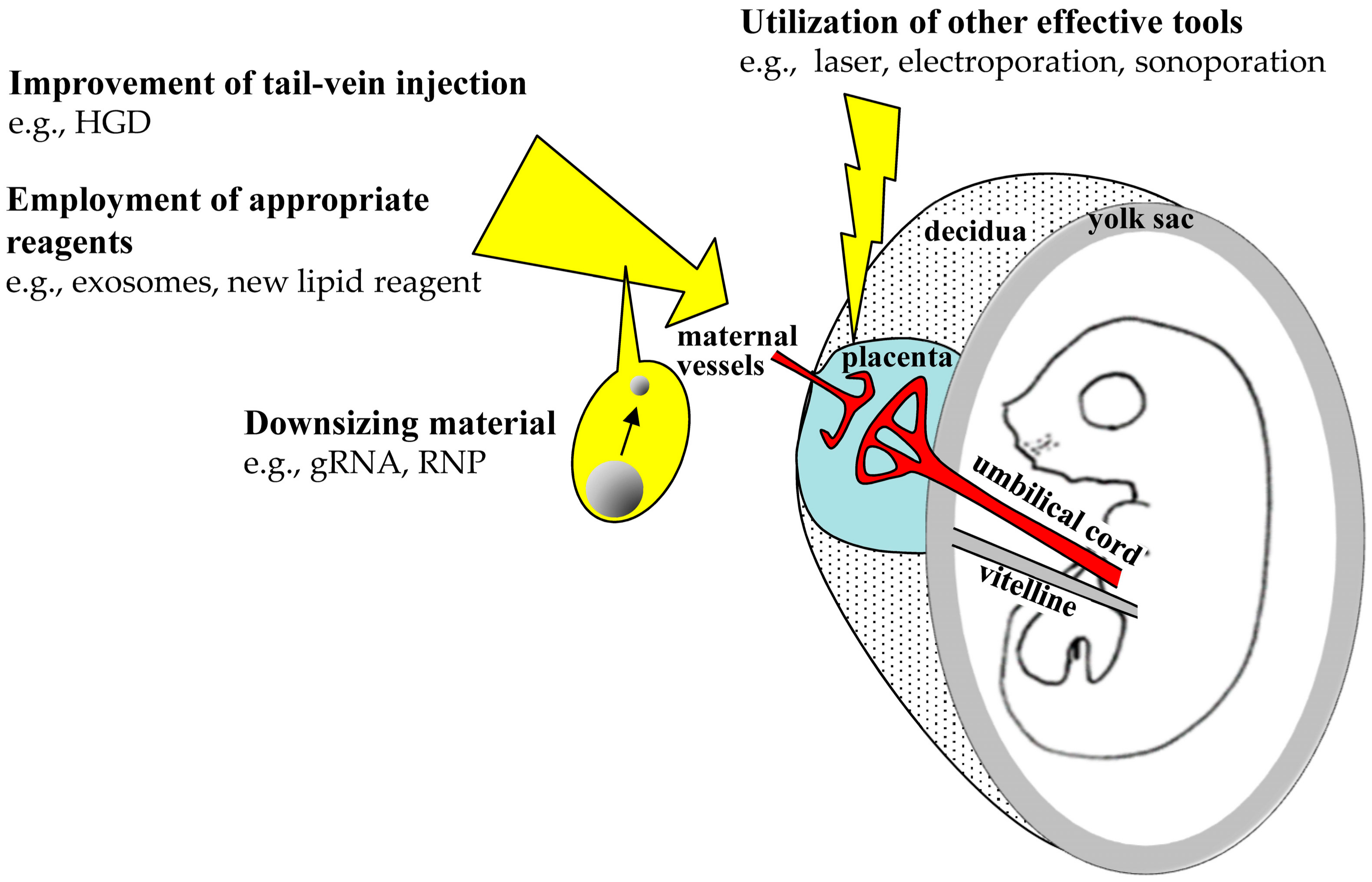{kind=link}
| Pregnant Mice | Injected Time (E) 1 | Injected Material | GOI 2 | Reagents Used for Gene Delivery | Note | Year, Reference |
|---|---|---|---|---|---|---|
| ICR | 3.0–15.0 | Plasmid DNA | Carrying CAT or lacZ gene | Commercially available lipopolyamine reagent (Transfectam) | This is the first report concerning TPGD. E9.5 is the day allowing to achieve most efficient TPGD efficiency. | 1995 [18] |
| ICR | 11.5 | Plasmid DNA | Carrying lacZ gene | Commercially available lipopolyamine reagent (DMRIE-C) | Although the transferred efficiency of DNA into embryos were low, expression of the reporter gene was observed. | 1999 [20] |
| BALB/c | 5.5, 9.5, 14.5 | Plasmid DNA | Carrying gene encoding antigen from HIV-1 or influenza virus | Cationic liposome prepared in-house | DNA-vaccinated mothers confer the antigen-specific immunity to their progeny. | 2001 [9] |
| B6C3F1 3 | 4.5–13.5 | Plasmid DNA | Carrying Cre gene | Commercially available lipid reagents (FuGENE6/Lipofectin/DOSPER) | This is the first report that the TPGD can mediate Cre/loxP-based recombination even in a fetus. | 2002 [21] |
| BALB/c | 14 | T7 phage particles | none | none | T7 Phage were detected in various fetal tissues. | 2004 [22] |
| Multiple strains of mice | 6.5 | Plasmid DNA | Carrying DsRed cDNA and shRNA for geminin gene | none | This is the first report that the TPGD is useful for RNAi-based gene silencing in a fetus. | 2006 [10] |
| C57BL/6 | 8 | Plasmid DNA | Carrying GFP cDNA | Tetra (piperazino) fullerene epoxide (TPFE) | Injected plasmid DNA was detected in the fetus, but the transfection efficiency was very low. | 2010 [23] |
| C57BL/6 | 17–19 | Plasmid DNA | Carrying luciferase gene | Nuclear location signal (NLS)-alarelin peptide | This is the first report that the TPGD coupled with hydrodynamics-based gene delivery (HGD) is useful for efficient transfection of a fetus. | 2010 [24] |
| ICR | 5.5–10.5 | Plasmid DNA | Carrying GFP cDNA and shRNA for Sry gene | Polyethylenimines | This report employs HGD and shows that the transfection efficiency is associated with the injection-time, -speed, and -volume. | 2012 [25] |
| C57BL/6 | 12.5 | Recombinant adeno-associated virus | Carrying GFP cDNA | None | Kidney-specific GOI expression was observed in a fetus, although the expression was also found in the dam. | 2014 [26] |
| CD-1 | 8 | Adenovirus | Carrying sFlt-1 gene | None | The authors created disease animal model by TPGD to evaluate the role of drugs in preventing the disease. | 2014 [27] |
| C57BL/6 | 17 | Plasmid DNA | Carrying luciferase gene or lac Z gene | PEGylated immunoliposomes within immunoliposomes bearing 8D3 monoclonal antibodies | Receptor-mediated transport of GOI via placental barrier is possible. | 2016 [28] |
| B6C3F1 4 | 12.5 | Plasmid DNA | Carrying humanized Cas9 gene and gRNA to eGFP | Commercially available lipid reagent (FuGENE6) | This is the first report that the TPGD is useful for inducing genome editing in fetal cardiac cells. | 2019 [19] |
| Delivery System | Representative Advantage | Representative Disadvantage | e.g., | Application Examples in TPGD | Application Examples in Genome Editing Systems | |
|---|---|---|---|---|---|---|
| Non-viral method | Cationic lipid | Low cost; great stability; simple and easy handling | Low efficiency; delayed onset | commercially available reagent for gene delivery (FuGENE6, etc.) | 4 cases reported [18,19,20,21] | Many cases reported [7,34,35,36] |
| Immunoliposome | 1 case reported [9] | none | ||||
| PEGylation | 1 case reported [28] | Some cases reported [37,38] | ||||
| Chemical reagent | Easy to produce; large packaging capacity | Low targeting efficiency; toxic | Carbon nanotube | 1 case reported [23] | none | |
| Polyethylenimines | 1 case reported [25] | Some cases reported [39,40] | ||||
| Polymers | easy to optimize | Cannot be applied to deliver the native form of Cas9 protein | Peptide | 1 case reported [24] | Many cases reported [7,35,41,42] | |
| Secretion | High efficiency; tissue-specificity | There are many unexplained parts | Exosome | None | Some cases reported [43,44] | |
| Viral method | Virus | Generally considered a safe and effective delivery vehicle | Low packaging capacity (less than 4.7 kb); difficulty in production of high-affinity virus targeted to liver | Adeno-associated virus | 1 case reported [26] | Many cases reported [45,46,47,48] |
| High efficiency; high packaging capacity | High immunogenicity | Adenovirus | 1 case reported [27] | Some cases reported [49,50] | ||
| High efficiency | Does not efficiently infect human cells | Bacteriophage | 1 case reported [22] | Some cases reported [51,52] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, S.; Watanabe, S.; Ando, N.; Ishihara, M.; Sato, M. Transplacental Gene Delivery (TPGD) as a Noninvasive Tool for Fetal Gene Manipulation in Mice. Int. J. Mol. Sci. 2019, 20, 5926. https://doi.org/10.3390/ijms20235926
Nakamura S, Watanabe S, Ando N, Ishihara M, Sato M. Transplacental Gene Delivery (TPGD) as a Noninvasive Tool for Fetal Gene Manipulation in Mice. International Journal of Molecular Sciences. 2019; 20(23):5926. https://doi.org/10.3390/ijms20235926
Chicago/Turabian StyleNakamura, Shingo, Satoshi Watanabe, Naoko Ando, Masayuki Ishihara, and Masahiro Sato. 2019. "Transplacental Gene Delivery (TPGD) as a Noninvasive Tool for Fetal Gene Manipulation in Mice" International Journal of Molecular Sciences 20, no. 23: 5926. https://doi.org/10.3390/ijms20235926
APA StyleNakamura, S., Watanabe, S., Ando, N., Ishihara, M., & Sato, M. (2019). Transplacental Gene Delivery (TPGD) as a Noninvasive Tool for Fetal Gene Manipulation in Mice. International Journal of Molecular Sciences, 20(23), 5926. https://doi.org/10.3390/ijms20235926