Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors

 ,
,  and
and 
Abstract
:1. Introduction
2. The Introduction of TP53 and Tumor Suppressive Role of p53
2.1. The Finding of TP53
2.2. The Functional Role of Wild Type p53
3. TP53 Mutation in CRC and Other Solid Tumors
3.1. TP53 Mutational Spectrum in CRC
3.2. The Mechanisms of p53 Mutants with Gain of Fucntion (GOF)
3.3. Oncogenic Roles of p53 Mutants with GOF
3.3.1. Enhancing Cell Proliferation and Colony Formation
3.3.2. Promoting Migration, Invasion and Metastasis
3.3.3. Inducing Angiogenesis
3.3.4. Inducing Chromatin Remodeling
4. Targeting p53 Mutants in Tumorigenicity
4.1. Restoring the Function of Wild Type p53
4.1.1. Cysteine-Binding Compounds
4.1.2. Zn2+-Chelating Compounds
4.1.3. Peptides
4.1.4. Other Types of Compounds
4.2. Depleting Mutated p53 Proteins
4.3. Inducing Synthetic Lethality to p53 Mutants
4.4. Targeting the Oncogenic Downstreams of p53 Mutants
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bhandari, A.; Woodhouse, M.; Gupta, S. Colorectal cancer is a leading cause of cancer incidence and mortality among adults younger than 50 years in the USA: A SEER-based analysis with comparison to other young-onset cancers. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2017, 65, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Otori, K.; Konishi, M.; Sugiyama, K.; Hasebe, T.; Shimoda, T.; Kikuchi-Yanoshita, R.; Mukai, K.; Fukushima, S.; Miyaki, M.; Esumi, H. Infrequent somatic mutation of the adenomatous polyposis coli gene in aberrant crypt foci of human colon tissue. Cancer 1998, 83, 896–900. [Google Scholar] [CrossRef]
- Miyaki, M.; Konishi, M.; Kikuchi-Yanoshita, R.; Enomoto, M.; Igari, T.; Tanaka, K.; Muraoka, M.; Takahashi, H.; Amada, Y.; Fukayama, M.; et al. Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res. 1994, 54, 3011–3020. [Google Scholar] [PubMed]
- Cottrell, S.; Bicknell, D.; Kaklamanis, L.; Bodmer, W.F. Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet 1992, 340, 626–630. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- DeLeo, A.B.; Jay, G.; Appella, E.; Dubois, G.C.; Law, L.W.; Old, L.J. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc. Natl. Acad. Sci. USA 1979, 76, 2420–2424. [Google Scholar] [CrossRef]
- Rotter, V. p53, a transformation-related cellular-encoded protein, can be used as a biochemical marker for the detection of primary mouse tumor cells. Proc. Natl. Acad. Sci. USA 1983, 80, 2613–2617. [Google Scholar] [CrossRef]
- Eliyahu, D.; Raz, A.; Gruss, P.; Givol, D.; Oren, M. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 1984, 312, 646–649. [Google Scholar] [CrossRef]
- Weiss, B.; Nitschko, H.; Ghattas, I.; Wright, R.; Schlesinger, S. Evidence for specificity in the encapsidation of Sindbis virus RNAs. J. Virol. 1989, 63, 5310–5318. [Google Scholar] [PubMed]
- Hinds, P.W.; Finlay, C.A.; Quartin, R.S.; Baker, S.J.; Fearon, E.R.; Vogelstein, B.; Levine, A.J. Mutant p53 DNA clones from human colon carcinomas cooperate with ras in transforming primary rat cells: A comparison of the "hot spot" mutant phenotypes. Cell Growth Differ. 1990, 1, 571–580. [Google Scholar] [PubMed]
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093. [Google Scholar] [CrossRef]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Takahashi, T.; Nau, M.M.; Chiba, I.; Birrer, M.J.; Rosenberg, R.K.; Vinocour, M.; Levitt, M.; Pass, H.; Gazdar, A.F.; Minna, J.D. p53: A frequent target for genetic abnormalities in lung cancer. Science 1989, 246, 491–494. [Google Scholar] [CrossRef]
- Unger, T.; Nau, M.M.; Segal, S.; Minna, J.D. p53: A transdominant regulator of transcription whose function is ablated by mutations occurring in human cancer. EMBO J. 1992, 11, 1383–1390. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. Embo J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef]
- Romer, L.; Klein, C.; Dehner, A.; Kessler, H.; Buchner, J. p53—A natural cancer killer: Structural insights and therapeutic concepts. Angew. Chem. Int. Ed. Engl. 2006, 45, 6440–6460. [Google Scholar] [CrossRef]
- Li, Q.; Lozano, G. Molecular pathways: Targeting Mdm2 and Mdm4 in cancer therapy. Clin. Cancer Res. 2013, 19, 34–41. [Google Scholar] [CrossRef]
- Li, X.L.; Zhou, J.; Chen, Z.R.; Chng, W.J. P53 mutations in colorectal cancer-molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Dysregulation of apoptosis in cancer. J. Clin. Oncol. 1999, 17, 2941–2953. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef]
- Lin, Y.; Ma, W.; Benchimol, S. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat. Genet. 2000, 26, 122–127. [Google Scholar] [CrossRef]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [PubMed]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. p53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Ferraiuolo, M.; Verduci, L.; Blandino, G.; Strano, S. Mutant p53 Protein and the Hippo Transducers YAP and TAZ: A Critical Oncogenic Node in Human Cancers. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dameron, K.M.; Volpert, O.V.; Tainsky, M.A.; Bouck, N. The p53 tumor suppressor gene inhibits angiogenesis by stimulating the production of thrombospondin. Cold Spring Harb. Symp. Quant. Biol. 1994, 59, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.G.; Parker, A.E.; Zhu, X.; Green, M.R. p53-mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science 2006, 313, 968–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, W.; Zhou, L. MiR-590-3p regulates osteogenic differentiation of human mesenchymal stem cells by regulating APC gene. Biochem. Biophys. Res. Commun. 2016, 478, 1582–1587. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.C.; Ji, W.L.; Yue, N.; Huang, Y.C.; Ma, X.M. The relationship between the PD-1/PD-L1 pathway and DNA mismatch repair in cervical cancer and its clinical significance. Cancer Manag. Res. 2018, 10, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef]
- Marin, M.C.; Jost, C.A.; Brooks, L.A.; Irwin, M.S.; O’Nions, J.; Tidy, J.A.; James, N.; McGregor, J.M.; Harwood, C.A.; Yulug, I.G.; et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat. Genet. 2000, 25, 47–54. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, Y.; Tang, Y.; Butler, N.; Kim, J.; Guessous, F.; Schiff, D.; Mandell, J.; Abounader, R. A novel PTEN/mutant p53/c-Myc/Bcl-XL axis mediates context-dependent oncogenic effects of PTEN with implications for cancer prognosis and therapy. Neoplasia 2013, 15, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Xu, J.; Liu, J.; Amjad, A.; Zhang, K.; Liu, Q.; Zhou, L.; Xiao, J.; Li, X. Mutant p53 promotes tumor cell malignancy by both positive and negative regulation of the transforming growth factor beta (TGF-beta) pathway. J. Biol. Chem. 2015, 290, 11729–11740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Shah, A.S.; Ahmad, A. Gain-of-function of mutant p53: Mutant p53 enhances cancer progression by inhibiting KLF17 expression in invasive breast carcinoma cells. Cancer Lett. 2014, 354, 87–96. [Google Scholar] [CrossRef]
- Xu, J.; Wang, J.; Hu, Y.; Qian, J.; Xu, B.; Chen, H.; Zou, W.; Fang, J.Y. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis. 2014, 5, e1108. [Google Scholar] [CrossRef] [Green Version]
- Karsy, M.; Arslan, E.; Moy, F. Current Progress on Understanding MicroRNAs in Glioblastoma Multiforme. Genes Cancer 2012, 3, 3–15. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Kollareddy, M.; Dimitrova, E.; Vallabhaneni, K.C.; Chan, A.; Le, T.; Chauhan, K.M.; Carrero, Z.I.; Ramakrishnan, G.; Watabe, K.; Haupt, Y.; et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat. Commun. 2015, 6, 7389. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Bao, W.; Jiang, F.; Che, Q.; Chen, Z.; Wang, F.; Tong, H.; Dai, C.; He, X.; Liao, Y.; et al. Mutant p53 (p53-R248Q) functions as an oncogene in promoting endometrial cancer by up-regulating REGgamma. Cancer Lett. 2015, 360, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Wang, Z.; Fu, J.; Ji, L.; Liu, J.; Li, L.; Wang, H.; Chen, J.; Caulin, C.; Myers, J.N.; et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-beta signalling and mutant p53 in cancer cells. Nat. Commun. 2013, 4, 2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Ling, S.; Lin, W.C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell Biol. 2011, 31, 4464–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.H.; Lang, B.J.; Chai, R.C.; Vieusseux, J.L.; Kouspou, M.M.; Price, J.T. Heat-shock factor 1 both positively and negatively affects cellular clonogenic growth depending on p53 status. Biochem. J. 2013, 452, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Chen, X. Identification of GRO1 as a critical determinant for mutant p53 gain of function. J. Biol. Chem. 2009, 284, 12178–12187. [Google Scholar] [CrossRef] [Green Version]
- Yeudall, W.A.; Vaughan, C.A.; Miyazaki, H.; Ramamoorthy, M.; Choi, M.Y.; Chapman, C.G.; Wang, H.; Black, E.; Bulysheva, A.A.; Deb, S.P.; et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 2012, 33, 442–451. [Google Scholar] [CrossRef]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, M.; Francis, P.; Bilke, S.; Li, X.L.; Hara, T.; Lu, X.; Jones, M.F.; Walker, R.L.; Zhu, Y.; Pineda, M.; et al. A mutant p53/let-7i-axis-regulated gene network drives cell migration, invasion and metastasis. Oncogene 2015, 34, 1094–1104. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef]
- Muller, P.A.; Trinidad, A.G.; Caswell, P.T.; Norman, J.C.; Vousden, K.H. Mutant p53 regulates Dicer through p63-dependent and -independent mechanisms to promote an invasive phenotype. J. Biol. Chem. 2014, 289, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Trinidad, A.G.; Timpson, P.; Morton, J.P.; Zanivan, S.; van den Berghe, P.V.; Nixon, C.; Karim, S.A.; Caswell, P.T.; Noll, J.E.; et al. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene 2013, 32, 1252–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.t.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arjonen, A.; Kaukonen, R.; Mattila, E.; Rouhi, P.; Hognas, G.; Sihto, H.; Miller, B.W.; Morton, J.P.; Bucher, E.; Taimen, P.; et al. Mutant p53-associated myosin-X upregulation promotes breast cancer invasion and metastasis. J. Clin. Invest. 2014, 124, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Sakai, E.; Echizen, K.; Yamada, Y.; Oshima, H.; Han, T.S.; Ohki, R.; Fujii, S.; Ochiai, A.; Robine, S.; et al. Intestinal cancer progression by mutant p53 through the acquisition of invasiveness associated with complex glandular formation. Oncogene 2017, 36, 5885–5896. [Google Scholar] [CrossRef] [Green Version]
- Khromova, N.V.; Kopnin, P.B.; Stepanova, E.V.; Agapova, L.S.; Kopnin, B.P. p53 hot-spot mutants increase tumor vascularization via ROS-mediated activation of the HIF1/VEGF-A pathway. Cancer Lett. 2009, 276, 143–151. [Google Scholar] [CrossRef]
- Joshi, H.; Bhanot, G.; Borresen-Dale, A.L.; Kristensen, V. Potential tumorigenic programs associated with TP53 mutation status reveal role of VEGF pathway. Br. J. Cancer 2012, 107, 1722–1728. [Google Scholar] [CrossRef]
- Montero, E.; Abreu, C.; Tonino, P. Relationship between VEGF and p53 expression and tumor cell proliferation in human gastrointestinal carcinomas. J. Cancer Res. Clin. Oncol. 2008, 134, 193–201. [Google Scholar] [CrossRef]
- Fontemaggi, G.; Dell’Orso, S.; Trisciuoglio, D.; Shay, T.; Melucci, E.; Fazi, F.; Terrenato, I.; Mottolese, M.; Muti, P.; Domany, E.; et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol. 2009, 16, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Pfister, N.T.; Fomin, V.; Regunath, K.; Zhou, J.Y.; Zhou, W.; Silwal-Pandit, L.; Freed-Pastor, W.A.; Laptenko, O.; Neo, S.P.; Bargonetti, J.; et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 2015, 29, 1298–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahnamoun, H.; Lu, H.; Duttke, S.H.; Benner, C.; Glass, C.K.; Lauberth, S.M. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat. Commun. 2017, 8, 754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Su, Z.Y.; Wang, L.; Shu, L.; Yang, Y.; Guo, Y.; Pung, D.; Bountra, C.; Kong, A.N. Epigenetic blockade of neoplastic transformation by bromodomain and extra-terminal (BET) domain protein inhibitor JQ-1. Biochem. Pharm. 2016, 117, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Tong, Y.; Zhang, X.; Pan, M.; Chen, S. Arsenic sulfide combined with JQ1, chemotherapy agents, or celecoxib inhibit gastric and colon cancer cell growth. Drug Des. Devel. 2015, 9, 5851–5862. [Google Scholar]
- Zhang, Y.; Tian, S.; Xiong, J.; Zhou, Y.; Song, H.; Liu, C. JQ-1 Inhibits Colon Cancer Proliferation via Suppressing Wnt/beta-Catenin Signaling and miR-21. Chem. Res. Toxicol. 2018, 31, 302–307. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, C.A.; Frum, R.; Pearsall, I.; Singh, S.; Windle, B.; Yeudall, A.; Deb, S.P.; Deb, S. Allele specific gain-of-function activity of p53 mutants in lung cancer cells. Biochem. Biophys. Res. Commun. 2012, 428, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Gurtner, A.; Starace, G.; Norelli, G.; Piaggio, G.; Sacchi, A.; Bossi, G. Mutant p53-induced up-regulation of mitogen-activated protein kinase kinase 3 contributes to gain of function. J. Biol. Chem. 2010, 285, 14160–14169. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Liang, Y.; Zhu, H.; Zhang, J.; Zhong, X. R280T mutation of p53 gene promotes proliferation of human glioma cells through GSK-3beta/PTEN pathway. Neurosci. Lett. 2012, 529, 60–65. [Google Scholar] [CrossRef]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Wischhusen, J.; Naumann, U.; Ohgaki, H.; Rastinejad, F.; Weller, M. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene 2003, 22, 8233–8245. [Google Scholar] [CrossRef] [Green Version]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [Green Version]
- Rippin, T.M.; Bykov, V.J.; Freund, S.M.; Selivanova, G.; Wiman, K.G.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zhu, Y.; Han, L.; Kim, A.L.; Kopelovich, L.; Bickers, D.R.; Athar, M. CP-31398 restores mutant p53 tumor suppressor function and inhibits UVB-induced skin carcinogenesis in mice. J. Clin. Invest. 2007, 117, 3753–3764. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; Synnott, N.C.; McGowan, P.M.; Crown, J.; O’Connor, D.; Gallagher, W.M. p53 as a target for the treatment of cancer. Cancer Treat. Rev. 2014, 40, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- p53 Suppressor Activation in Recurrent High Grade Serous Ovarian Cancer, a Phase Ib/II Study of Systemic Carboplatin Combination Chemotherapy with or without APR-246. 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02098343 (accessed on 27 November 2019).
- A Study of APR-246 in Oesophageal Cancer (APROC). 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02999893 (accessed on 27 November 2019).
- Phase 1b/2 Safety and Efficacy of APR-246 w/Azacitidine for tx of TP53 Mutant Myeloid Neoplasms. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03072043 (accessed on 27 November 2019).
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [Green Version]
- Zache, N.; Lambert, J.M.R.; Rokaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J.N. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2017, 11, 595. [Google Scholar] [CrossRef] [Green Version]
- Punganuru, S.R.; Madala, H.R.; Venugopal, S.N.; Samala, R.; Mikelis, C.; Srivenugopal, K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016, 107, 233–244. [Google Scholar] [CrossRef]
- Bauer, M.R.; Joerger, A.C.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280. [Google Scholar] [CrossRef] [Green Version]
- Kaar, J.L.; Basse, N.; Joerger, A.C.; Stephens, E.; Rutherford, T.J.; Fersht, A.R. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. A Publ. Protein Soc. 2010, 19, 2267–2278. [Google Scholar] [CrossRef] [Green Version]
- Puca, R.; Nardinocchi, L.; Porru, M.; Simon, A.J.; Rechavi, G.; Leonetti, C.; Givol, D.; D’Orazi, G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle 2011, 10, 1679–1689. [Google Scholar] [CrossRef]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Study of COTI-2 as Monotherapy or Combination Therapy for the Treatment of Malignancies (COTI2-101). 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02433626 (accessed on 27 November 2019).
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef] [Green Version]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; Dasmahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008, 15, 718–729. [Google Scholar] [CrossRef]
- Demma, M.; Maxwell, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, a small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef] [Green Version]
- Hiraki, M.; Hwang, S.Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [Green Version]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Zhang, Y.; Zhang, J.; Liu, S.; Cho, S.J.; Chen, X. Mutant p53 protein is targeted by arsenic for degradation and plays a role in arsenic-mediated growth suppression. J. Biol. Chem. 2011, 286, 17478–17486. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Jung, Y.S.; Zhang, Y.; Chen, X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS ONE 2014, 9, e103497. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, Q.; Qi, Q.; Gu, H.Y.; Rong, J.J.; Mu, R.; Zou, M.J.; Tao, L.; You, Q.D.; Guo, Q.L. Gambogic acid-induced degradation of mutant p53 is mediated by proteasome and related to CHIP. J. Cell. Biochem. 2011, 112, 509–519. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [Green Version]
- Rifai, K.; Judes, G.; Idrissou, M.; Daures, M.; Bignon, Y.J.; Penault-Llorca, F.; Bernard-Gallon, D. Dual SIRT1 expression patterns strongly suggests its bivalent role in human breast cancer. Oncotarget 2017, 8, 110922–110930. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhou, L.; Hong, B.; van den Heuvel, A.P.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015, 75, 3842–3852. [Google Scholar] [CrossRef] [Green Version]
- Paranjpe, A.; Srivenugopal, K.S. Degradation of NF-kappaB, p53 and other regulatory redox-sensitive proteins by thiol-conjugating and -nitrosylating drugs in human tumor cells. Carcinogenesis 2013, 34, 990–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Fan, S.; Eastman, A.; Worland, P.J.; Sausville, E.A.; O’Connor, P.M. UCN-01: A potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J. Natl. Cancer Inst. 1996, 88, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Pagliarini, R.; Bunz, F.; Rago, C.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc. Natl. Acad. Sci. USA 2009, 106, 3964–3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, J.; Booher, R.N.; Kraker, A.; Lawrence, T.; Leopold, W.R.; Sun, Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001, 61, 8211–8217. [Google Scholar]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]

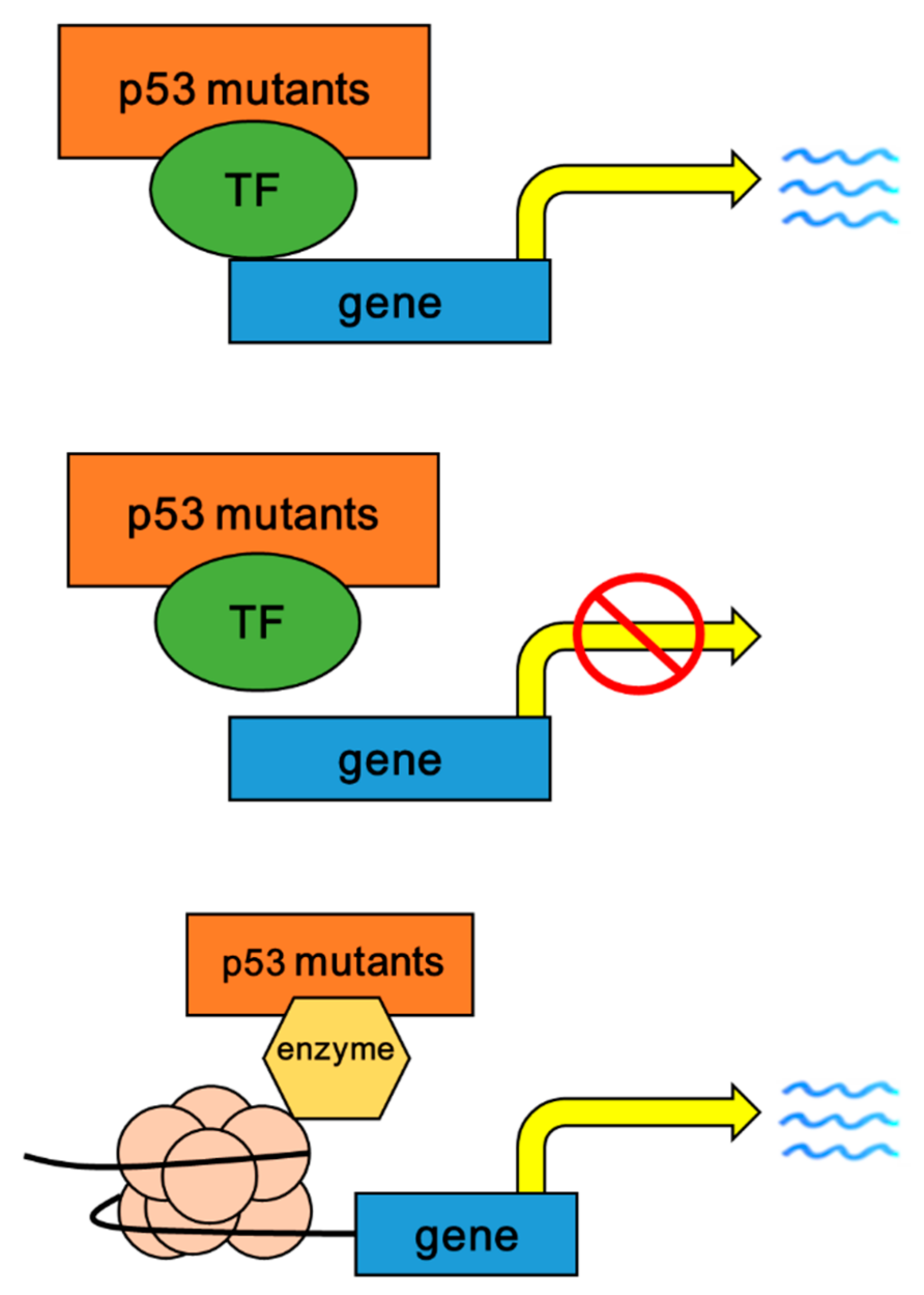

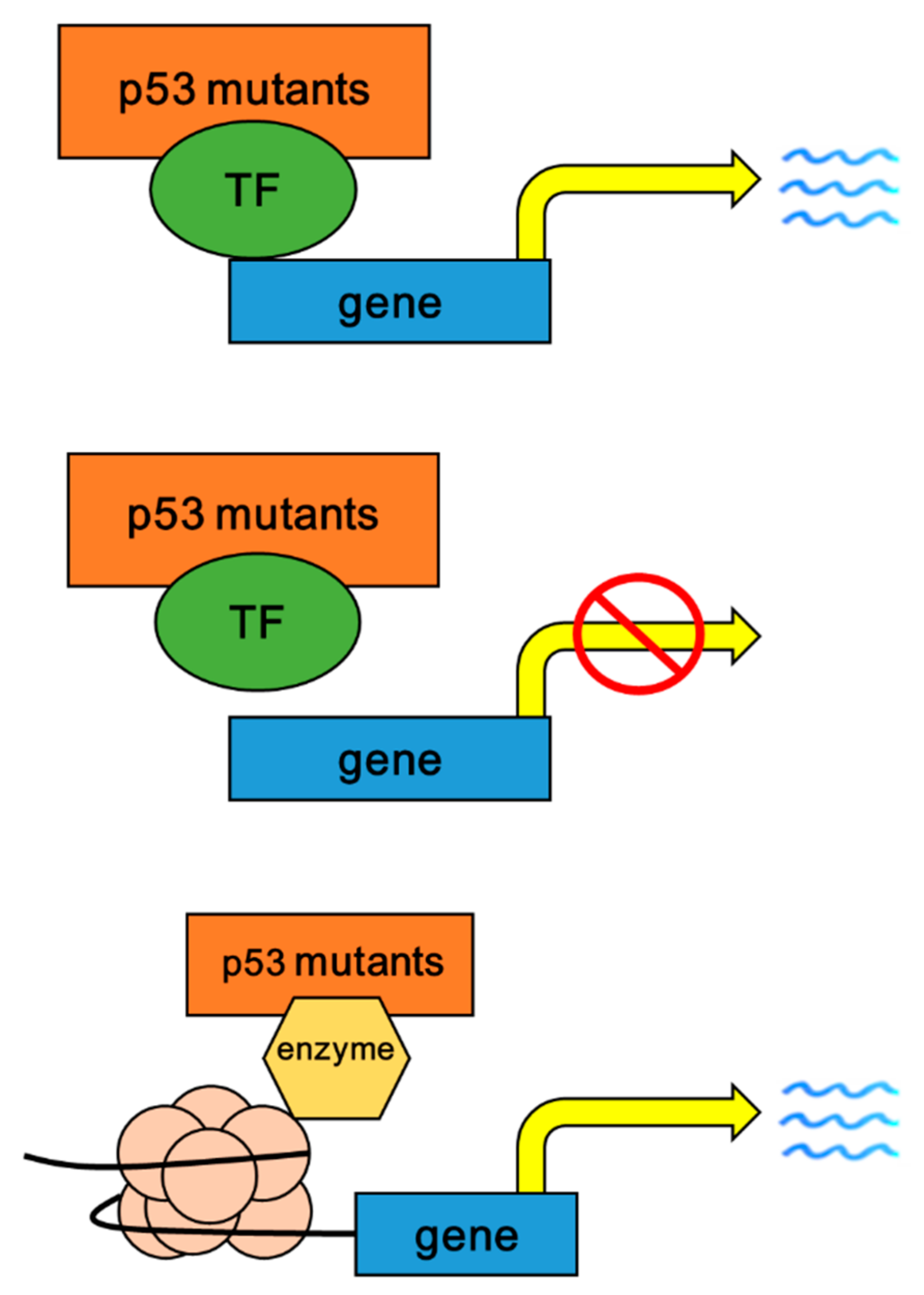
{kind=link}
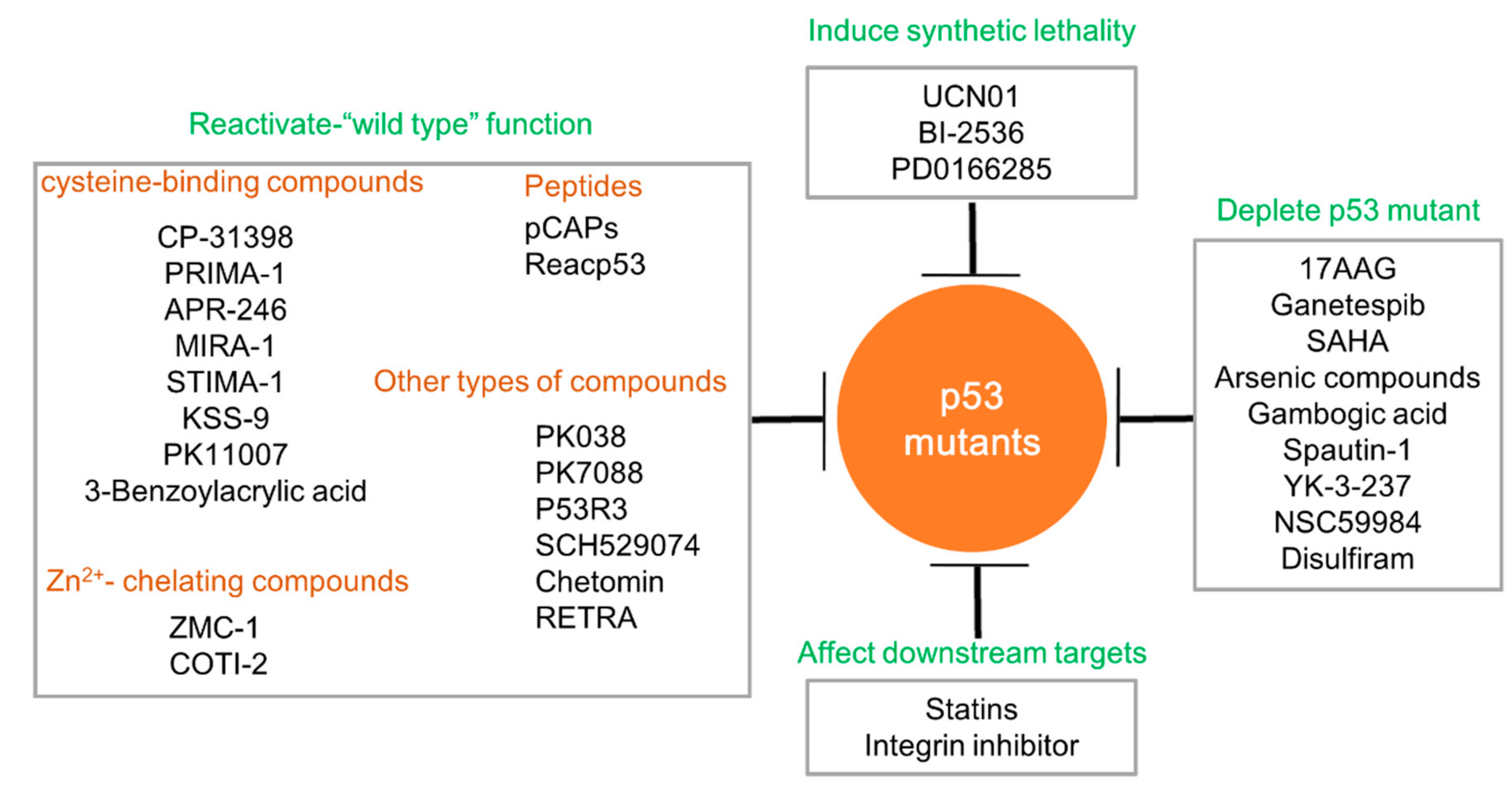
{kind=link}
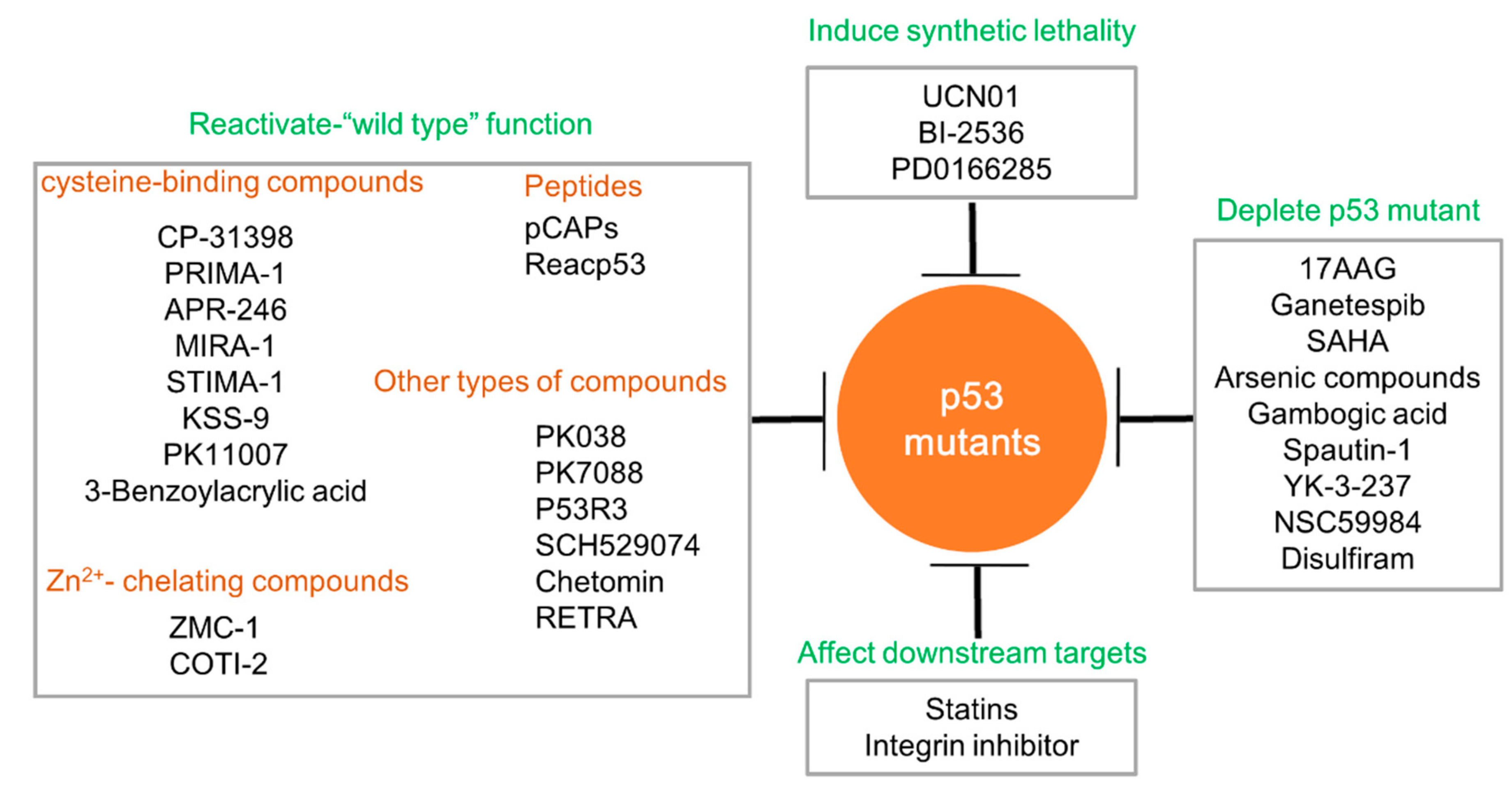{kind=link}
| Mutation Type | Cell Lines | Downstream Effectors | Refs |
|---|---|---|---|
| Promoting cell proliferation | |||
| R273H | U373/SNB19 | c-Myc/Bcl-XL | [40] |
| R280K/R282W | MDA-MB-231/MDA-MB-1386 | KLF17 | [46] |
| R273H | H1299 | miR-27a/EGFR | [47] |
| R273H/R175H/D281G | H1299 | Axl | [48] |
| R273C/R267P | H1048/H1437 | ||
| R238Q/R172H | - | HSP90/HDAC6 | [49] |
| R249S/R273L/R280K | BT549/HCC38/MDAMB231 | EST2/NMGs | [50] |
| R249S/R175H | MCF10a/H1299 | ||
| R248Q | HEC-1B | REG-γ | [51] |
| R175H | H1299/UMSCC-1 | REG-γ | [52] |
| R280K/R282W/R273H | MDA-MB-231/MDA-MB-1386 | ||
| P278S/R267P | ABC1/H1437 | Axl | [78] |
| R175H/R273H | H1299 | TopBP1 | [54] |
| R273C/R248Q/R175H | C33A/OVCAR-3/SKBr3 | ||
| R175H/R273H/R280K | SKBR3/HT29/MDA-MB468/MDA-MB231 | MAP2K3 | [79] |
| R280T | SWO-38 | GSK-3β/PTEN | [80] |
| R280K/R273H | MDA-MB-231/MDA-468 | SREBPs | [64] |
| Increasing colony formation ability | |||
| R273H | U373/SNB19 | c-Myc/Bcl-XL | [40] |
| R273H | H1299 | NRF2 | [53] |
| R175H/R273H | H1299 | TopBP1 | [54] |
| R273C/R248Q/R175H | C33A/OVCAR-3/SKBr3 | ||
| R273H | MCF10a | HSF1 | [55] |
| R273H | H1299 | miR-27a/EGFR | [47] |
| R175H | HCT116-/- | GRO1 | [56] |
| R273H/P309S | SW480 | ||
| R248W | MIA-PaCa-2 | ||
| Increasing cell invasion and migration | |||
| R273H | U373/SNB19 | c-Myc/Bcl-XL | [40] |
| R280K/R282W | MDA-MB-231/MDA-MB-1386 | KLF17 | [46] |
| R249S/R273L/R280K | BT549/HCC38/MDAMB231 | EST2/NMGs | [50] |
| R249S/R175H | MCF10a/H1299 | ||
| R248Q | HEC-1B | REG-γ | [51] |
| R175H/R273H | MCF10a/H1299 | RCP/integrin/EGFR | [60] |
| R273H | H1299 | TAp63/Dicer | [61] |
| R280K/R273H | MDA-MB-231/HT29/A431 | ||
| R175H/R273H | H1299 | TAp63/Met | [62] |
| R175H | H1299 | TAp63/Sharp 1/Cyclin G2 | [41] |
| R175H | H1299 | Smad3 | [42] |
| R175H | H1299 | let-7i/E2F5/LIN28B/MYC | [59] |
| R248W/R220C/H242R/H155P | Miapaca2/BXPC3/CFPAC/A2.1 | p73/NF-Y/PDGFR-beta | [63] |
| R273H/R280K | SW620/H1975/MDA-MB-231 | ||
| R280K/R273H | MDA-MB-231/MDA-468 | SREBPs | [64] |
| R280K | MDA-MB-231 | Myo10 | [65] |
| R175H/R273H/C135Y | HEC-50 | miR-130b/ZEB1 | [58] |
| R273H | U373/SNB19 | c-Myc/Bcl-XL | [40] |
| R280K | MDA-MB-231 | KLF17 | [46] |
| R282W | MDA-MB-1386 | ||
| R273H/R175H/D281G | H1299 | Axl | [48] |
| R273C/R267P | H1048/H1437 | ||
| R175H | H1299 | TAp63/Sharp 1/Cyclin G2 | [41] |
| R175H | H1299 | let-7i/E2F5/LIN28B/MYC | [59] |
| R175H/R273H/D281G | H1299 | CXCL5/CXCL8/CXCL12 | [57] |
| R280K | MDA-MB-231 | Pin1 | [81] |
| Inducing angiogenesis | |||
| R175H/R273H/R248W | HCT116-/- | HIF1/VEGF-A | [68] |
| R175H/R273H | H1299 | ID4/IL8/GRO-a | [71] |
| R280K | MDA-MB-231 | ||
| Chromatin remodeling | |||
| R248Q | HCC70 | MLL1/MLL2/MOZ | [73] |
| R249S | BT-549 | ||
| R273H | MDA-MB-468 | ||
| R273H | SW480 | MMP9/CCL2/CYP24A1/CPA4 | [74] |
| R273H | MDA-468 | VEGFR2/SWI/SNF | [72] |
| Reactivating the Wild Type p53 Function | |||
|---|---|---|---|
| Compounds (Small Molecules) | Mechanisms | Clinical Trial (Cancers) | Refs. |
| Cysteine-binding compounds | |||
| CP-31398 | binds to the cysteine residues | [85,86,87,88] | |
| PRIMA-1 | converts to methylene quinuclidinone | [89,90,91,92] | |
| APR-246 | converts to methylene quinuclidinone | phase Ib/II (lymphoma, ovarian, esophageal) | [90,91,92,93,94] |
| MIRA-1 | prevents unfolding of wild-type and mutant p53 | [98] | |
| STIMA-1 | prevents unfolding of wild-type and mutant p53 | [99] | |
| KSS-9 | prevents unfolding of wild-type and mutant p53 | [100] | |
| PK11007 | binds p53 by nucleophilic aromatic substitution | [101] | |
| 3-Benzoylacrylic acid | binds p53 by Michael addition | [102] | |
| Zn2+-chelating compounds | |||
| ZMC-1 | Zn2+ chelator | [104] | |
| COTI-2 | Zn2+ chelator | phase I (gynecological, head and neck cancer) | [105] |
| Peptides | |||
| pCAPs | promote refolding | [107] | |
| Reacp53 | blocks aggregation | [108] | |
| Other types of compounds | |||
| PK083 | restores wild-type conformation | [109] | |
| PK7088 | restores wild-type conformation | [105] | |
| P53R3 | restores DNA-binding ability | [106] | |
| SCH529074 | restores DNA-binding ability | [112] | |
| Chetomin | promotes refolding | [113] | |
| RETRA | disrupts mutant p53–p73 complexes | [114] | |
| Depleting the GOF of p53 mutants | |||
| 17AAG | Hsp90 inhibitors | [116] | |
| Ganetespib | Hsp90 inhibitors | phase III (lung cancer) | [49] |
| SAHA | HDAC inhibitors | [117] | |
| Arsenic compounds | increases transcripts of Pirh2,and | [118] | |
| induces degradation of mutant p53 | [119] | ||
| Gambogic acid | inhibits the mutant p53-Hsp90 complex | [121] | |
| Spautin-1 | induces mutant p53 degradation | [122] | |
| YK-3-237 | activates SIRT1 and deacetylate lysine 382 | [123] | |
| NSC59984 | induces MDM2-mediated mutant p53 degradation | [124] | |
| Disulfiram | induces p53 degradation | [125] | |
| Inducing synthetic lethality | |||
| UCN01 | protein kinase C inhibitor | [126] | |
| BI-2536 | polo-like kinase 1 inhibitor | [127] | |
| PD0166285 | Wee1 kinase inhibitor | [128] | |
| Blocking the oncogenic downstreams of p53 mutants | |||
| Statins | HMG-CoA reductase inhibitor | [64] | |
| inhibits YAP/TAZ activation | [129] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Zhang, J.; Tong, J.H.M.; Chan, A.W.H.; Yu, J.; Kang, W.; To, K.F. Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors. Int. J. Mol. Sci. 2019, 20, 5999. https://doi.org/10.3390/ijms20235999
Li H, Zhang J, Tong JHM, Chan AWH, Yu J, Kang W, To KF. Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors. International Journal of Molecular Sciences. 2019; 20(23):5999. https://doi.org/10.3390/ijms20235999
Chicago/Turabian StyleLi, Hui, Jinglin Zhang, Joanna Hung Man Tong, Anthony Wing Hung Chan, Jun Yu, Wei Kang, and Ka Fai To. 2019. "Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors" International Journal of Molecular Sciences 20, no. 23: 5999. https://doi.org/10.3390/ijms20235999
APA StyleLi, H., Zhang, J., Tong, J. H. M., Chan, A. W. H., Yu, J., Kang, W., & To, K. F. (2019). Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors. International Journal of Molecular Sciences, 20(23), 5999. https://doi.org/10.3390/ijms20235999