Calcium Signaling in ß-cell Physiology and Pathology: A Revisit

 , ,
, ,  and
and 
Abstract
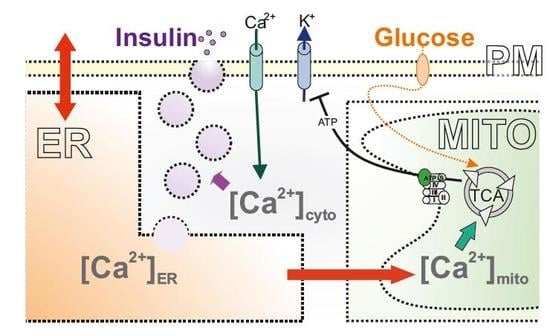
:
{kind=link}
{kind=link}
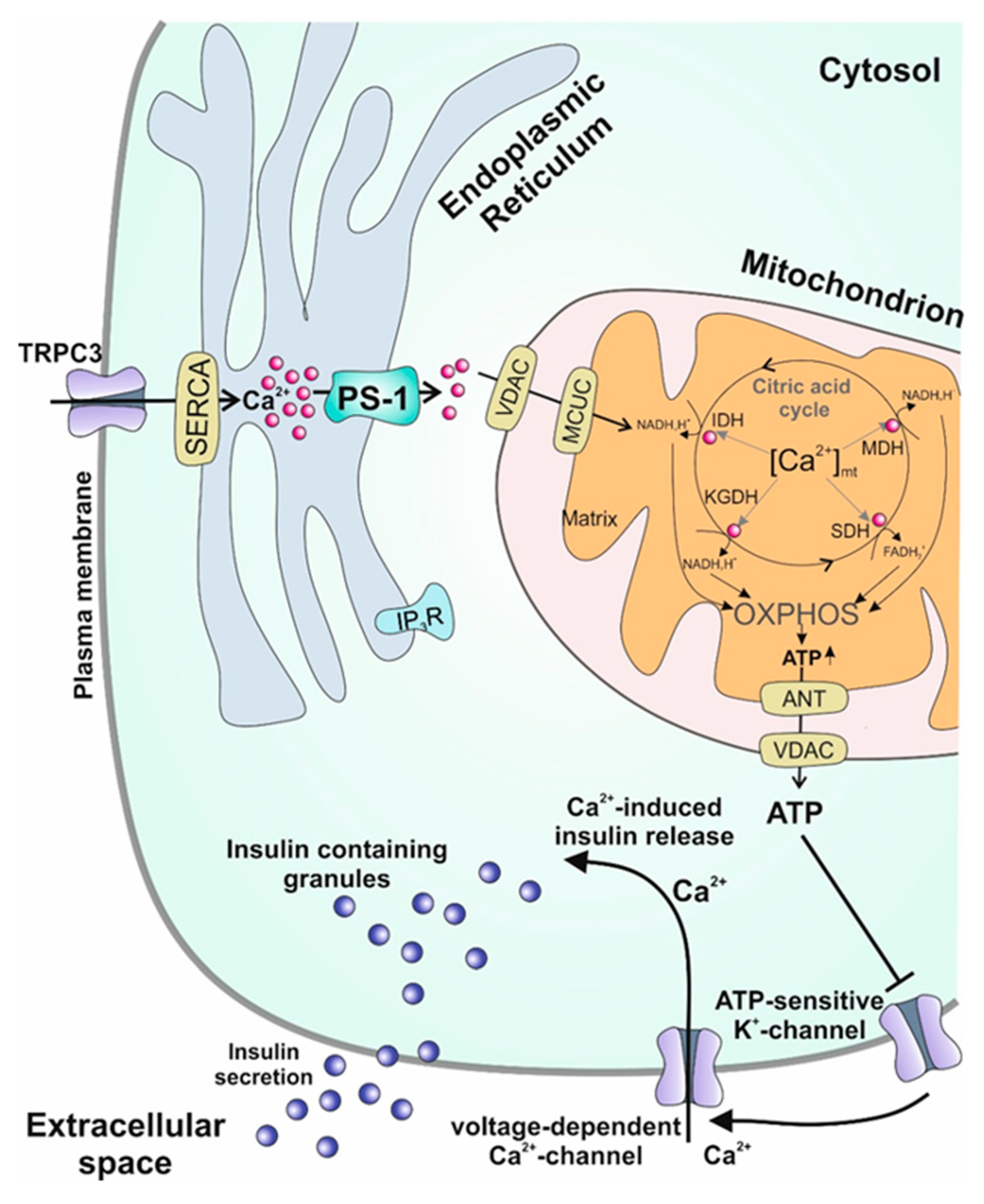{kind=link}
1. Introduction
2. The Role of Ca2+ Signaling in Pancreatic β-cells: The Basics
3. The Role of Ca2+ Signaling in Pancreatic β-Cells: A Closer Look
3.1. Ca2+ in β-Cell Proliferation
3.2. Ca2+ in β-cell Survival
3.3. Ca2+ Oscillations for Proper β-Cell Function
3.4. Ca2+ in Biphasic Insulin Secretion
4. Ca2+ on Subcellular Level
4.1. Plasma Membrane
4.2. Mitochondria
4.3. Endoplasmic Reticulum
4.4. The Golgi-Apparatus
5. Ca2+ in the Development of T2DM
6. Presenilin-1, the Missing Link between Diabetes and Alzheimer’s Disease?: Excursus
7. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| ATP | Adenosine triphosphate |
| CaMK | Ca2+/calmodulin dependent kinase |
| CICR | Calcium induced calcium release |
| DM | Diabetes mellitus |
| ER | Endoplasmic reticulum |
| GLUT | Glucose transporter |
| GSIS | Glucose stimulated insulin secretion |
| GSK3β | Glycogen synthase kinase 3 β |
| IP3 | Inositol trisphosphate |
| IP3R | IP3 receptor |
| IR | Insulin receptor |
| IRS | Insulin receptor substrate |
| MCU | Mitochondrial calcium uniporter |
| MICU | Mitochondrial calcium uptake 1 |
| NCX | Na+/Ca2+ exchanger |
| NCLX | Mitochondrial Na+/Ca2+ exchanger |
| NFAT | Nuclear factor of activated T-cells |
| PM | Plasma membrane |
| PMCA | Plamsa membrane Ca2+-ATPase |
| PTP | Permeability transition pore |
| RyR | Ryanodine receptor |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| T1/2DM | Type 1 or type 2 diabetes mellitus |
| VDCC | Voltage dependent calcium channels |
References
- Mayer, J. Glucostatic mechanism of regulation of food intake. N. Engl. J. Med. 1953, 249, 13–16. [Google Scholar] [CrossRef]
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [Green Version]
- Steiner, D.J.; Kim, A.; Miller, K.; Hara, M. Pancreatic islet plasticity: Interspecies comparison of islet architecture and composition. Islets 2010, 2, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Diaz, R.; Molano, R.D.; Weitz, J.R.; Abdulreda, M.H.; Berman, D.M.; Leibiger, B.; Leibiger, I.B.; Kenyon, N.S.; Ricordi, C.; Pileggi, A.; et al. Paracrine Interactions within the Pancreatic Islet Determine the Glycemic Set Point. Cell Metab. 2018, 27, 549–558. [Google Scholar] [CrossRef]
- Mari, A.; Tura, A.; Natali, A.; Laville, M.; Laakso, M.; Gabriel, R.; Beck-Nielsen, H.; Ferrannini, E.; Investigators, R. Impaired beta cell glucose sensitivity rather than inadequate compensation for insulin resistance is the dominant defect in glucose intolerance. Diabetologia 2010, 53, 749–756. [Google Scholar] [CrossRef] [Green Version]
- German, M.S. Glucose sensing in pancreatic islet beta cells: The key role of glucokinase and the glycolytic intermediates. Proc. Natl. Acad. Sci. USA 1993, 90, 1781–1785. [Google Scholar] [CrossRef] [Green Version]
- Bosboom, R.S.; Zweens, J.; Bouman, P.R. Effects of feeding and fasting on the insulin secretory response to glucose and sulfonylureas in intact rats and isolated perfused rat pancreas. Diabetologia 1973, 9, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; Asterholm, I.W.; Benrick, A.; Briant, L.J.B.; Chibalina, M.V.; et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 139. [Google Scholar] [CrossRef]
- IDF. DF Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017; Available online: http://www.diabetesatlas.org (accessed on 23 October 2019).
- Gepts, W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 1965, 14, 619–633. [Google Scholar] [CrossRef]
- Kahn, S.E.; Zraika, S.; Utzschneider, K.M.; Hull, R.L. The beta cell lesion in type 2 diabetes: There has to be a primary functional abnormality. Diabetologia 2009, 52, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R. Insulin resistance and type 2 diabetes. Diabetes 2012, 61, 778–779. [Google Scholar] [CrossRef] [Green Version]
- Renstrom, E.; Ding, W.G.; Bokvist, K.; Rorsman, P. Neurotransmitter-induced inhibition of exocytosis in insulin-secreting beta cells by activation of calcineurin. Neuron 1996, 17, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Alfa, R.W.; Park, S.; Skelly, K.R.; Poffenberger, G.; Jain, N.; Gu, X.; Kockel, L.; Wang, J.; Liu, Y.; Powers, A.C.; et al. Suppression of insulin production and secretion by a decretin hormone. Cell Metab. 2015, 21, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Henquin, J.C.; Nenquin, M.; Ravier, M.A.; Szollosi, A. Shortcomings of current models of glucose-induced insulin secretion. Diabetes Obes. Metab. 2009, 11 (Suppl. 4), 168–179. [Google Scholar] [CrossRef]
- Alam, M.R.; Groschner, L.N.; Parichatikanond, W.; Kuo, L.; Bondarenko, A.I.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J. Biol. Chem. 2012, 287, 34445–34454. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A.; Tsuboi, T.; Ravier, M.A. Ca2+ microdomains and the control of insulin secretion. Cell Calcium 2006, 40, 539–551. [Google Scholar] [CrossRef]
- Sherman, A.; Rinzel, J. Model for synchronization of pancreatic beta-cells by gap junction coupling. Biophys. J. 1991, 59, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef]
- Philipson, K.D.; Nicoll, D.A. Sodium-calcium exchange: A molecular perspective. Annu. Rev. Physiol. 2000, 62, 111–133. [Google Scholar] [CrossRef]
- Strehler, E.E.; Treiman, M. Calcium pumps of plasma membrane and cell interior. Curr. Mol. Med. 2004, 4, 323–335. [Google Scholar] [CrossRef]
- Prasad, V.; Okunade, G.W.; Miller, M.L.; Shull, G.E. Phenotypes of SERCA and PMCA knockout mice. Biochem. Biophys. Res. Commun. 2004, 322, 1192–1203. [Google Scholar] [CrossRef]
- Carafoli, E. Biogenesis: Plasma membrane calcium ATPase: 15 years of work on the purified enzyme. FASEB J. 1994, 8, 993–1002. [Google Scholar] [CrossRef]
- Noble, D.; Herchuelz, A. Role of Na/Ca exchange and the plasma membrane Ca2+-ATPase in cell function. Conference on Na/Ca exchange. EMBO Rep. 2007, 8, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Bagur, R.; Hajnoczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Ravier, M.A.; Daro, D.; Roma, L.P.; Jonas, J.C.; Cheng-Xue, R.; Schuit, F.C.; Gilon, P. Mechanisms of control of the free Ca2+ concentration in the endoplasmic reticulum of mouse pancreatic beta-cells: Interplay with cell metabolism and [Ca2+]c and role of SERCA2b and SERCA3. Diabetes 2011, 60, 2533–2545. [Google Scholar] [CrossRef] [Green Version]
- Cassel, R.; Ducreux, S.; Alam, M.R.; Dingreville, F.; Berle, C.; Burda-Jacob, K.; Chauvin, M.A.; Chikh, K.; Paita, L.; Al-Mawla, R.; et al. Protection of Human Pancreatic Islets from Lipotoxicity by Modulation of the Translocon. PLoS ONE 2016, 11, e0148686. [Google Scholar] [CrossRef]
- Klec, C.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Sachdev, V.; Duta-Mare, M.; Gottschalk, B.; Depaoli, M.R.; Rost, R.; Hay, J.; Waldeck-Weiermair, M.; et al. Glycogen Synthase Kinase 3 Beta Controls Presenilin-1-Mediated Endoplasmic Reticulum Ca2+ Leak Directed to Mitochondria in Pancreatic Islets and beta-Cells. Cell. Physiol. Biochem. 2019, 52, 57–75. [Google Scholar] [CrossRef] [Green Version]
- Luciani, D.S.; Gwiazda, K.S.; Yang, T.L.; Kalynyak, T.B.; Bychkivska, Y.; Frey, M.H.; Jeffrey, K.D.; Sampaio, A.V.; Underhill, T.M.; Johnson, J.D. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes 2009, 58, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Bootman, M.D.; Cheek, T.R.; Moreton, R.B.; Bennett, D.L.; Berridge, M.J. Smoothly graded Ca2+ release from inositol 1,4,5-trisphosphate-sensitive Ca2+ stores. J. Biol. Chem. 1994, 269, 24783–24791. [Google Scholar]
- Isenberg, G.; Han, S. Gradation of Ca2+-induced Ca2+ release by voltage-clamp pulse duration in potentiated guinea-pig ventricular myocytes. J. Physiol. 1994, 480 Pt 3, 423–438. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Jonas, J.C.; Weir, G.C.; Laychock, S.G. Glucose regulates expression of inositol 1,4,5-trisphosphate receptor isoforms in isolated rat pancreatic islets. Endocrinology 1999, 140, 2173–2182. [Google Scholar] [CrossRef]
- Johnson, J.D.; Kuang, S.; Misler, S.; Polonsky, K.S. Ryanodine receptors in human pancreatic beta cells: Localization and effects on insulin secretion. FASEB J. 2004, 18, 878–880. [Google Scholar] [CrossRef]
- Sabatini, P.V.; Speckmann, T.; Lynn, F.C. Friend and foe: Beta-cell Ca2+ signaling and the development of diabetes. Mol. Metab. 2019, 21, 1–12. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Yeromin, A.V.; Zhang, S.L.; Jiang, W.; Yu, Y.; Safrina, O.; Cahalan, M.D. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 2006, 443, 226–229. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Soboloff, J.; Spassova, M.A.; Hewavitharana, T.; He, L.P.; Xu, W.; Johnstone, L.S.; Dziadek, M.A.; Gill, D.L. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr. Biol. 2006, 16, 1465–1470. [Google Scholar] [CrossRef] [Green Version]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [Green Version]
- Inayama, M.; Suzuki, Y.; Yamada, S.; Kurita, T.; Yamamura, H.; Ohya, S.; Giles, W.R.; Imaizumi, Y. Orai1-Orai2 complex is involved in store-operated calcium entry in chondrocyte cell lines. Cell Calcium 2015, 57, 337–347. [Google Scholar] [CrossRef]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Karoly Jani, P.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Erxleben, C.; Abramowitz, J.; Flockerzi, V.; Zhu, M.X.; Armstrong, D.L.; Birnbaumer, L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc. Natl. Acad. Sci. USA 2008, 105, 2895–2900. [Google Scholar] [CrossRef] [Green Version]
- Sabourin, J.; Le Gal, L.; Saurwein, L.; Haefliger, J.A.; Raddatz, E.; Allagnat, F. Store-operated Ca2+ Entry Mediated by Orai1 and TRPC1 Participates to Insulin Secretion in Rat beta-Cells. J. Biol. Chem. 2015, 290, 30530–30539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamarina, N.A.; Kuznetsov, A.; Rhodes, C.J.; Bindokas, V.P.; Philipson, L.H. Inositol (1,4,5)-trisphosphate dynamics and intracellular calcium oscillations in pancreatic beta-cells. Diabetes 2005, 54, 3073–3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harootunian, A.T.; Kao, J.P.; Paranjape, S.; Adams, S.R.; Potter, B.V.; Tsien, R.Y. Cytosolic Ca2+ oscillations in REF52 fibroblasts: Ca(2+)-stimulated IP3 production or voltage-dependent Ca2+ channels as key positive feedback elements. Cell Calcium 1991, 12, 153–164. [Google Scholar] [CrossRef]
- Pasti, L.; Volterra, A.; Pozzan, T.; Carmignoto, G. Intracellular calcium oscillations in astrocytes: A highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J. Neurosci. 1997, 17, 7817–7830. [Google Scholar] [CrossRef] [Green Version]
- Nunemaker, C.S.; Bertram, R.; Sherman, A.; Tsaneva-Atanasova, K.; Daniel, C.R.; Satin, L.S. Glucose modulates [Ca2+]i oscillations in pancreatic islets via ionic and glycolytic mechanisms. Biophys. J. 2006, 91, 2082–2096. [Google Scholar] [CrossRef] [Green Version]
- Dolmetsch, R.E.; Xu, K.; Lewis, R.S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 1998, 392, 933–936. [Google Scholar] [CrossRef]
- German, M.S.; Moss, L.G.; Rutter, W.J. Regulation of insulin gene expression by glucose and calcium in transfected primary islet cultures. J. Biol. Chem. 1990, 265, 22063–22066. [Google Scholar]
- Luzi, L.; Pozza, G. Glibenclamide: An old drug with a novel mechanism of action? Acta Diabetol. 1997, 34, 239–244. [Google Scholar] [CrossRef]
- Wang, Q.; Heimberg, H.; Pipeleers, D.; Ling, Z. Glibenclamide activates translation in rat pancreatic beta cells through calcium-dependent mTOR, PKA and MEK signalling pathways. Diabetologia 2008, 51, 1202–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamateris, R.E.; Sharma, R.B.; Kong, Y.; Ebrahimpour, P.; Panday, D.; Ranganath, P.; Zou, B.; Levitt, H.; Parambil, N.A.; O’Donnell, C.P.; et al. Glucose Induces Mouse beta-Cell Proliferation via IRS2, MTOR, and Cyclin D2 but Not the Insulin Receptor. Diabetes 2016, 65, 981–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, L.C.; Yokoe, T.; Zhang, P.; Scott, D.K.; Kim, S.K.; O’Donnell, C.P.; Garcia-Ocana, A. Glucose infusion in mice: A new model to induce beta-cell replication. Diabetes 2007, 56, 1792–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitt, H.E.; Cyphert, T.J.; Pascoe, J.L.; Hollern, D.A.; Abraham, N.; Lundell, R.J.; Rosa, T.; Romano, L.C.; Zou, B.; O’Donnell, C.P.; et al. Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia 2011, 54, 572–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porat, S.; Weinberg-Corem, N.; Tornovsky-Babaey, S.; Schyr-Ben-Haroush, R.; Hija, A.; Stolovich-Rain, M.; Dadon, D.; Granot, Z.; Ben-Hur, V.; White, P.; et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab. 2011, 13, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nat. Commun. 2015, 6, 8372. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Walker, J.R.; Wang, X.; Tremblay, M.S.; Lee, J.W.; Wu, X.; Schultz, P.G. Identification of small-molecule inducers of pancreatic beta-cell expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 1427–1432. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Murao, K.; Sayo, Y.; Imachi, H.; Cao, W.M.; Ohtsuka, S.; Niimi, M.; Tokumitsu, H.; Inuzuka, H.; Wong, N.C.; et al. The role of calcium/calmodulin-dependent protein kinase cascade in glucose upregulation of insulin gene expression. Diabetes 2004, 53, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Barbosa-Sampaio, H.; Jones, P.M.; Persaud, S.J.; Muller, D.S. The CaMK4/CREB/IRS-2 cascade stimulates proliferation and inhibits apoptosis of beta-cells. PLoS ONE 2012, 7, e45711. [Google Scholar] [CrossRef]
- Heit, J.J.; Apelqvist, A.A.; Gu, X.; Winslow, M.M.; Neilson, J.R.; Crabtree, G.R.; Kim, S.K. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 2006, 443, 345–349. [Google Scholar] [CrossRef]
- Dai, C.; Hang, Y.; Shostak, A.; Poffenberger, G.; Hart, N.; Prasad, N.; Phillips, N.; Levy, S.E.; Greiner, D.L.; Shultz, L.D.; et al. Age-dependent human beta cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J. Clin. Investig. 2017, 127, 3835–3844. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Butler, A.E.; Saisho, Y.; Monchamp, T.; Galasso, R.; Bhushan, A.; Rizza, R.A.; Butler, P.C. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008, 57, 1584–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEvoy, R.C. Changes in the volumes of the A-, B-, and D-cell populations in the pancreatic islets during the postnatal development of the rat. Diabetes 1981, 30, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Finegood, D.T.; Scaglia, L.; Bonner-Weir, S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 1995, 44, 249–256. [Google Scholar] [CrossRef]
- Nielsen, J.H.; Brunstedt, J.; Andersson, A.; Frimodt-Moller, C. Preservation of beta cell function in adult human pancreatic islets for several months in vitro. Diabetologia 1979, 16, 97–100. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.; Distelhorst, C.W. Bcl-2 protein family members: Versatile regulators of calcium signaling in cell survival and apoptosis. Annu. Rev. Physiol. 2008, 70, 73–91. [Google Scholar] [CrossRef]
- Srinivasan, S.; Bernal-Mizrachi, E.; Ohsugi, M.; Permutt, M.A. Glucose promotes pancreatic islet beta-cell survival through a PI 3-kinase/Akt-signaling pathway. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E784–E793. [Google Scholar] [CrossRef] [Green Version]
- Bonner-Weir, S. Perspective: Postnatal pancreatic beta cell growth. Endocrinology 2000, 141, 1926–1929. [Google Scholar] [CrossRef]
- Soleimanpour, S.A.; Crutchlow, M.F.; Ferrari, A.M.; Raum, J.C.; Groff, D.N.; Rankin, M.M.; Liu, C.; De Leon, D.D.; Naji, A.; Kushner, J.A.; et al. Calcineurin signaling regulates human islet {beta}-cell survival. J. Biol. Chem. 2010, 285, 40050–40059. [Google Scholar] [CrossRef] [Green Version]
- Nir, T.; Melton, D.A.; Dor, Y. Recovery from diabetes in mice by beta cell regeneration. J. Clin. Investig. 2007, 117, 2553–2561. [Google Scholar] [CrossRef] [Green Version]
- del Castillo, J.M.B.; Arias-Diaz, J.; Garcia Martin, M.C.; Vives-Pi, M.; Garcia Perez, J.C.; Cantero Cid, R.; Vara Ameigeiras, E.; Balibrea Cantero, J.L. Cytoprotective effect of low-dose tacrolimus on islets of Langerhans in cultures subjected to stimulation by acute rejection cytokines. Cirugía Española 2010, 87, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Ranta, F.; Avram, D.; Berchtold, S.; Dufer, M.; Drews, G.; Lang, F.; Ullrich, S. Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes 2006, 55, 1380–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Mahadevan, J.; Kanekura, K.; Hara, M.; Lu, S.; Urano, F. Calcium efflux from the endoplasmic reticulum leads to beta-cell death. Endocrinology 2014, 155, 758–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyadomari, S.; Takeda, K.; Takiguchi, M.; Gotoh, T.; Matsumoto, M.; Wada, I.; Akira, S.; Araki, E.; Mori, M. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 10845–10850. [Google Scholar] [CrossRef] [Green Version]
- Gwiazda, K.S.; Yang, T.L.; Lin, Y.; Johnson, J.D. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E690–E701. [Google Scholar] [CrossRef]
- Moore, C.E.; Omikorede, O.; Gomez, E.; Willars, G.B.; Herbert, T.P. PERK activation at low glucose concentration is mediated by SERCA pump inhibition and confers preemptive cytoprotection to pancreatic beta-cells. Mol. Endocrinol. 2011, 25, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, P.J.; Paolisso, G.; Scheen, A.J.; Henquin, J.C. Pulsatility of insulin and glucagon release: Physiological significance and pharmacological implications. Diabetologia 1987, 30, 443–452. [Google Scholar] [CrossRef]
- Goodner, C.J.; Walike, B.C.; Koerker, D.J.; Ensinck, J.W.; Brown, A.C.; Chideckel, E.W.; Palmer, J.; Kalnasy, L. Insulin, glucagon, and glucose exhibit synchronous, sustained oscillations in fasting monkeys. Science 1977, 195, 177–179. [Google Scholar] [CrossRef]
- Kennedy, R.T.; Kauri, L.M.; Dahlgren, G.M.; Jung, S.K. Metabolic oscillations in beta-cells. Diabetes 2002, 51 (Suppl. 1), S152–S161. [Google Scholar] [CrossRef] [Green Version]
- Lang, D.A.; Matthews, D.R.; Peto, J.; Turner, R.C. Cyclic oscillations of basal plasma glucose and insulin concentrations in human beings. N. Engl. J. Med. 1979, 301, 1023–1027. [Google Scholar] [CrossRef]
- Bingley, P.J.; Matthews, D.R.; Williams, A.J.; Bottazzo, G.F.; Gale, E.A. Loss of regular oscillatory insulin secretion in islet cell antibody positive non-diabetic subjects. Diabetologia 1992, 35, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rahilly, S.; Turner, R.C.; Matthews, D.R. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N. Engl. J. Med. 1988, 318, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Grapengiesser, E.; Gylfe, E.; Hellman, B. Glucose-induced oscillations of cytoplasmic Ca2+ in the pancreatic beta-cell. Biochem. Biophys. Res. Commun. 1988, 151, 1299–1304. [Google Scholar] [CrossRef]
- Hellman, B.; Gylfe, E.; Grapengiesser, E.; Lund, P.E.; Berts, A. Cytoplasmic Ca2+ oscillations in pancreatic beta-cells. Biochim. Biophys. Acta 1992, 1113, 295–305. [Google Scholar] [CrossRef]
- Santos, R.M.; Rosario, L.M.; Nadal, A.; Garcia-Sancho, J.; Soria, B.; Valdeolmillos, M. Widespread synchronous [Ca2+]i oscillations due to bursting electrical activity in single pancreatic islets. Pflug. Arch. 1991, 418, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.L.; Kellard, J.A.; Hara, M.; Johnson, J.D.; Rodriguez, B.; Briant, L.J.B. Beta-cell hubs maintain Ca2+ oscillations in human and mouse islet simulations. Islets 2018, 10, 151–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdeolmillos, M.; Nadal, A.; Soria, B.; Garcia-Sancho, J. Fluorescence digital image analysis of glucose-induced [Ca2+]i oscillations in mouse pancreatic islets of Langerhans. Diabetes 1993, 42, 1210–1214. [Google Scholar] [CrossRef]
- Fridlyand, L.E.; Tamarina, N.; Philipson, L.H. Bursting and calcium oscillations in pancreatic beta-cells: Specific pacemakers for specific mechanisms. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E517–E532. [Google Scholar] [CrossRef] [Green Version]
- Hellman, B.; Gylfe, E.; Bergsten, P.; Grapengiesser, E.; Lund, P.E.; Berts, A.; Tengholm, A.; Pipeleers, D.G.; Ling, Z. Glucose induces oscillatory Ca2+ signalling and insulin release in human pancreatic beta cells. Diabetologia 1994, 37 (Suppl. 2), S11–S20. [Google Scholar] [CrossRef] [Green Version]
- Goodner, C.J.; Sweet, I.R.; Harrison, H.C., Jr. Rapid reduction and return of surface insulin receptors after exposure to brief pulses of insulin in perifused rat hepatocytes. Diabetes 1988, 37, 1316–1323. [Google Scholar] [CrossRef]
- Jones, P.M.; Persaud, S.J.; Howell, S.L. Ca2+-induced insulin secretion from electrically permeabilized islets. Loss of the Ca2(+)-induced secretory response is accompanied by loss of Ca2+-induced protein phosphorylation. Biochem. J. 1992, 285 Pt 3, 973–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orrenius, S.; McConkey, D.J.; Bellomo, G.; Nicotera, P. Role of Ca2+ in toxic cell killing. Trends Pharmacol. Sci. 1989, 10, 281–285. [Google Scholar] [CrossRef]
- Curry, D.L.; Bennett, L.L.; Grodsky, G.M. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology 1968, 83, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Porte, D., Jr.; Pupo, A.A. Insulin responses to glucose: Evidence for a two pool system in man. J. Clin. Investig. 1969, 48, 2309–2319. [Google Scholar] [CrossRef] [Green Version]
- Henquin, J.C.; Ishiyama, N.; Nenquin, M.; Ravier, M.A.; Jonas, J.C. Signals and pools underlying biphasic insulin secretion. Diabetes 2002, 51 (Suppl. 1), S60–S67. [Google Scholar] [CrossRef] [Green Version]
- Cerasi, E.; Luft, R. The plasma insulin response to glucose infusion in healthy subjects and in diabetes mellitus. Eur. J. Endocrinol. 1967, 55, 278–304. [Google Scholar] [CrossRef]
- Fujita, Y.; Herron, A.L., Jr.; Seltzer, H.S. Confirmation of impaired early insulin response to glycemic stimulus in nonobese mild diabetics. Diabetes 1975, 24, 17–27. [Google Scholar] [CrossRef]
- Metz, S.A.; Halter, J.B.; Robertson, R.P. Paradoxical inhibition of insulin secretion by glucose in human diabetes mellitus. J. Clin. Endocrinol. Metab. 1979, 48, 827–835. [Google Scholar] [CrossRef]
- Pfeiffer, B.; Sarrazin, W.; Weitzel, G. Insulin-like effects of agmatine derivatives in vitro and in vivo (author’s transl). Hoppe-Seyler’s Z. Physiol. Chemie 1981, 362, 1331–1337. [Google Scholar] [CrossRef]
- Luzi, L.; DeFronzo, R.A. Effect of loss of first-phase insulin secretion on hepatic glucose production and tissue glucose disposal in humans. Am. J. Physiol. 1989, 257, E241–E246. [Google Scholar] [CrossRef]
- Rebrin, K.; Steil, G.M.; Mittelman, S.D.; Bergman, R.N. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J. Clin. Investig. 1996, 98, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getty, L.; Hamilton-Wessler, M.; Ader, M.; Dea, M.K.; Bergman, R.N. Biphasic insulin secretion during intravenous glucose tolerance test promotes optimal interstitial insulin profile. Diabetes 1998, 47, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Wollheim, C.B.; Kikuchi, M.; Renold, A.E.; Sharp, G.W. The roles of intracellular and extracellular Ca++ in glucose-stimulated biphasic insulin release by rat islets. J. Clin. Investig. 1978, 62, 451–458. [Google Scholar] [CrossRef] [Green Version]
- Henquin, J.C. Relative importance of extracellular and intracellular calcium for the two phases of glucose-stimulated insulin release: Studies with theophylline. Endocrinology 1978, 102, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.E., 3rd. The role of ion channels in insulin secretion. J. Cell. Biochem. 1992, 48, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Klec, C.; Madreiter-Sokolowski, C.T.; Ziomek, G.; Stryeck, S.; Sachdev, V.; Duta-Mare, M.; Gottschalk, B.; Depaoli, M.R.; Rost, R.; Hay, J.; et al. Presenilin-1 Established ER-Ca2+ Leak: A Follow Up on Its Importance for the Initial Insulin Secretion in Pancreatic Islets and beta-Cells upon Elevated Glucose. Cell. Physiol. Biochem. 2019, 53, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Hamming, K.S.; Riedel, M.J.; Soliman, D.; Matemisz, L.C.; Webster, N.J.; Searle, G.J.; MacDonald, P.E.; Light, P.E. Splice variant-dependent regulation of beta-cell sodium-calcium exchange by acyl-coenzyme As. Mol. Endocrinol. 2008, 22, 2293–2306. [Google Scholar] [CrossRef] [Green Version]
- Hamming, K.S.; Soliman, D.; Webster, N.J.; Searle, G.J.; Matemisz, L.C.; Liknes, D.A.; Dai, X.Q.; Pulinilkunnil, T.; Riedel, M.J.; Dyck, J.R.; et al. Inhibition of beta-cell sodium-calcium exchange enhances glucose-dependent elevations in cytoplasmic calcium and insulin secretion. Diabetes 2010, 59, 1686–1693. [Google Scholar] [CrossRef] [Green Version]
- Pachera, N.; Papin, J.; Zummo, F.P.; Rahier, J.; Mast, J.; Meyerovich, K.; Cardozo, A.K.; Herchuelz, A. Heterozygous inactivation of plasma membrane Ca(2+)-ATPase in mice increases glucose-induced insulin release and beta cell proliferation, mass and viability. Diabetologia 2015, 58, 2843–2850. [Google Scholar] [CrossRef] [Green Version]
- Balaban, R.S.; Kantor, H.L.; Katz, L.A.; Briggs, R.W. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science 1986, 232, 1121–1123. [Google Scholar] [CrossRef]
- Lin, A.L.; Fox, P.T.; Hardies, J.; Duong, T.Q.; Gao, J.H. Nonlinear coupling between cerebral blood flow, oxygen consumption, and ATP production in human visual cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 8446–8451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellman, B.; Idahl, L.A.; Lernmark, A.; Sehlin, J.; Taljedal, I.B. The pancreatic beta-cell recognition of insulin secretagogues. Effects of calcium and sodium on glucose metabolism and insulin release. Biochem. J. 1974, 138, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweet, I.R.; Cook, D.L.; DeJulio, E.; Wallen, A.R.; Khalil, G.; Callis, J.; Reems, J. Regulation of ATP/ADP in pancreatic islets. Diabetes 2004, 53, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederkehr, A.; Szanda, G.; Akhmedov, D.; Mataki, C.; Heizmann, C.W.; Schoonjans, K.; Pozzan, T.; Spat, A.; Wollheim, C.B. Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab. 2011, 13, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, A.; Park, K.S.; Dupont, O.; Demaurex, N.; Pozzan, T.; Cline, G.W.; Wollheim, C.B. Matrix alkalinization: A novel mitochondrial signal for sustained pancreatic beta-cell activation. EMBO J. 2009, 28, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M.; McCormack, J.G. On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett. 1980, 119, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M.; McCormack, J.G. Ca2+ transport by mammalian mitochondria and its role in hormone action. Am. J. Physiol. 1985, 249, E543–E554. [Google Scholar] [CrossRef]
- Hansford, R.G. Relation between mitochondrial calcium transport and control of energy metabolism. Rev. Physiol. Biochem. Pharmacol. 1985, 102, 1–72. [Google Scholar]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Wiederkehr, A.; Wollheim, C.B. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic beta-cell. Cell Calcium 2008, 44, 64–76. [Google Scholar] [CrossRef]
- Rutter, G.A.; McCormack, J.G.; Midgley, P.J.; Denton, R.M. The role of Ca2+ in the hormonal regulation of the activities of pyruvate dehydrogenase and oxoglutarate dehydrogenase complexes. Ann. N. Y. Acad. Sci. 1989, 573, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Maechler, P.; Wang, H.; Wollheim, C.B. Continuous monitoring of ATP levels in living insulin secreting cells expressing cytosolic firefly luciferase. FEBS Lett. 1998, 422, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-stimulated insulin secretion: A newer perspective. J. Diabetes Investig. 2013, 4, 511–516. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in beta-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmugi, A.; Parnis, J.; Chen, X.; Carmichael, L.; Hardy, J.; Mannan, N.; Marchetti, P.; Piemonti, L.; Bosco, D.; Johnson, P.; et al. Sorcin Links Pancreatic beta-Cell Lipotoxicity to ER Ca2+ Stores. Diabetes 2016, 65, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Kone, M.; Pullen, T.J.; Sun, G.; Ibberson, M.; Martinez-Sanchez, A.; Sayers, S.; Nguyen-Tu, M.S.; Kantor, C.; Swisa, A.; Dor, Y.; et al. LKB1 and AMPK differentially regulate pancreatic beta-cell identity. FASEB J. 2014, 28, 4972–4985. [Google Scholar] [CrossRef] [Green Version]
- Noordeen, N.A.; Meur, G.; Rutter, G.A.; Leclerc, I. Glucose-induced nuclear shuttling of ChREBP is mediated by sorcin and Ca2+ ions in pancreatic beta-cells. Diabetes 2012, 61, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Lokuta, A.J.; Meyers, M.B.; Sander, P.R.; Fishman, G.I.; Valdivia, H.H. Modulation of cardiac ryanodine receptors by sorcin. J. Biol. Chem. 1997, 272, 25333–25338. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Hisamatsu, Y.; Ohkusa, T.; Inoue, N.; Sato, T.; Suzuki, S.; Ikeda, Y.; Matsuzaki, M. Sorcin interacts with sarcoplasmic reticulum Ca(2+)-ATPase and modulates excitation-contraction coupling in the heart. Basic Res. Cardiol. 2005, 100, 250–262. [Google Scholar] [CrossRef]
- Cushman, S.W.; Wardzala, L.J. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J. Biol. Chem. 1980, 255, 4758–4762. [Google Scholar]
- Borge, P.D.; Moibi, J.; Greene, S.R.; Trucco, M.; Young, R.A.; Gao, Z.; Wolf, B.A. Insulin receptor signaling and sarco/endoplasmic reticulum calcium ATPase in beta-cells. Diabetes 2002, 51 (Suppl. 3), S427–S433. [Google Scholar] [CrossRef] [Green Version]
- Varadi, A.; Lebel, L.; Hashim, Y.; Mehta, Z.; Ashcroft, S.J.; Turner, R. Sequence variants of the sarco(endo)plasmic reticulum Ca(2+)-transport ATPase 3 gene (SERCA3) in Caucasian type II diabetic patients (UK Prospective Diabetes Study 48). Diabetologia 1999, 42, 1240–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micaroni, M.; Mironov, A.A. Roles of Ca and secretory pathway Ca-ATPase pump type 1 (SPCA1) in intra-Golgi transport. Commun. Integr. Biol. 2010, 3, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, P.; Pozzan, T.; Rizzuto, R. The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. 1998, 17, 5298–5308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lissandron, V.; Podini, P.; Pizzo, P.; Pozzan, T. Unique characteristics of Ca2+ homeostasis of the trans-Golgi compartment. Proc. Natl. Acad. Sci. USA 2010, 107, 9198–9203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunteski-Hamblin, A.M.; Clarke, D.M.; Shull, G.E. Molecular cloning and tissue distribution of alternatively spliced mRNAs encoding possible mammalian homologues of the yeast secretory pathway calcium pump. Biochemistry 1992, 31, 7600–7608. [Google Scholar] [CrossRef] [PubMed]
- Vanoevelen, J.; Dode, L.; Van Baelen, K.; Fairclough, R.J.; Missiaen, L.; Raeymaekers, L.; Wuytack, F. The secretory pathway Ca2+/Mn2+-ATPase 2 is a Golgi-localized pump with high affinity for Ca2+ ions. J. Biol. Chem. 2005, 280, 22800–22808. [Google Scholar] [CrossRef] [Green Version]
- Micaroni, M.; Perinetti, G.; Berrie, C.P.; Mironov, A.A. The SPCA1 Ca2+ pump and intracellular membrane trafficking. Traffic 2010, 11, 1315–1333. [Google Scholar] [CrossRef]
- Bone, R.N.; Kono, T.; Evans-Mollina, C. Reduced ß Cell SPCA1 Leads to Impaired Calcium Oscillations and Decreased Autophagy. Diabetes 2018, 67. [Google Scholar] [CrossRef]
- Sun, G.; Vasdev, S.; Martin, G.R.; Gadag, V.; Zhang, H. Altered calcium homeostasis is correlated with abnormalities of fasting serum glucose, insulin resistance, and beta-cell function in the Newfoundland population. Diabetes 2005, 54, 3336–3339. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Han, T.; Gao, J.; Zhang, Y.; Zhao, S.; Sun, R.; Sun, C.; Niu, Y.; Li, Y. Association of Serum Calcium and Insulin Resistance With Hypertension Risk: A Prospective Population-Based Study. J. Am. Heart Assoc. 2019, 8, e009585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, L.; Tabas, I. Calcium signalling and ER stress in insulin resistance and atherosclerosis. J. Intern. Med. 2016, 280, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.E.; Klein, R.; Moss, S.E. Prevalence of cataracts in a population-based study of persons with diabetes mellitus. Ophthalmology 1985, 92, 1191–1196. [Google Scholar] [CrossRef]
- Sowers, J.R.; Epstein, M.; Frohlich, E.D. Diabetes, hypertension, and cardiovascular disease: An update. Hypertension 2001, 37, 1053–1059. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.; Gavin, J.R., 3rd; Sowers, J.R. Diabetes mellitus: A disease of abnormal cellular calcium metabolism? Am. J. Med. 1994, 96, 260–273. [Google Scholar] [CrossRef]
- van Dam, R.M.; Hu, F.B.; Rosenberg, L.; Krishnan, S.; Palmer, J.R. Dietary calcium and magnesium, major food sources, and risk of type 2 diabetes in U.S. black women. Diabetes Care 2006, 29, 2238–2243. [Google Scholar] [CrossRef] [Green Version]
- Pittas, A.G.; Lau, J.; Hu, F.B.; Dawson-Hughes, B. The role of vitamin D and calcium in type 2 diabetes. A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2007, 92, 2017–2029. [Google Scholar] [CrossRef]
- Nikooyeh, B.; Neyestani, T.R.; Farvid, M.; Alavi-Majd, H.; Houshiarrad, A.; Kalayi, A.; Shariatzadeh, N.; Gharavi, A.; Heravifard, S.; Tayebinejad, N.; et al. Daily consumption of vitamin D- or vitamin D + calcium-fortified yogurt drink improved glycemic control in patients with type 2 diabetes: A randomized clinical trial. Am. J. Clin. Nutr. 2011, 93, 764–771. [Google Scholar] [CrossRef]
- Borissova, A.M.; Tankova, T.; Kirilov, G.; Dakovska, L.; Kovacheva, R. The effect of vitamin D3 on insulin secretion and peripheral insulin sensitivity in type 2 diabetic patients. Int. J. Clin. Pract. 2003, 57, 258–261. [Google Scholar]
- Berridge, M.J. Vitamin D deficiency and diabetes. Biochem. J. 2017, 474, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Barbachano, A.; Singh, P.K.; Campbell, M.J.; Munoz, A.; Larriba, M.J. Vitamin D has wide regulatory effects on histone demethylase genes. Cell Cycle 2012, 11, 1081–1089. [Google Scholar] [CrossRef]
- Roe, M.W.; Philipson, L.H.; Frangakis, C.J.; Kuznetsov, A.; Mertz, R.J.; Lancaster, M.E.; Spencer, B.; Worley, J.F., 3rd; Dukes, I.D. Defective glucose-dependent endoplasmic reticulum Ca2+ sequestration in diabetic mouse islets of Langerhans. J. Biol. Chem. 1994, 269, 18279–18282. [Google Scholar]
- Levy, J.; Zhu, Z.; Dunbar, J.C. The effect of glucose and calcium on Ca2+-adenosine triphosphatase in pancreatic islets isolated from a normal and a non-insulin-dependent diabetes mellitus rat model. Metabolism 1998, 47, 185–189. [Google Scholar] [CrossRef]
- Raggi, P.; Shaw, L.J.; Berman, D.S.; Callister, T.Q. Prognostic value of coronary artery calcium screening in subjects with and without diabetes. J. Am. Coll. Cardiol. 2004, 43, 1663–1669. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Ishida, H.; Tsuura, Y.; Okamoto, Y.; Tsuji, K.; Horie, M.; Okada, Y.; Seino, Y. Increased calcium-channel currents of pancreatic beta cells in neonatally streptozocin-induced diabetic rats. Metabolism 1994, 43, 1395–1400. [Google Scholar] [CrossRef]
- Rasmussen, H. The calcium messenger system (2). N. Engl. J. Med. 1986, 314, 1164–1170. [Google Scholar] [CrossRef]
- Porte, D., Jr. Banting lecture 1990. Beta-cells in type II diabetes mellitus. Diabetes 1991, 40, 166–180. [Google Scholar] [CrossRef]
- Davies, M.J.; Rayman, G.; Grenfell, A.; Gray, I.P.; Day, J.L.; Hales, C.N. Loss of the first phase insulin response to intravenous glucose in subjects with persistent impaired glucose tolerance. Diabet. Med. 1994, 11, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Pathogenesis and treatment of type 2 (noninsulin-dependent) diabetes mellitus (NIDDM). Horm. Metab. Res. 1996, 28, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, R.W.; Wahl, P.W.; Leonetti, D.L.; Fujimoto, W.Y. Association of fasting glucose levels with a delayed secretion of insulin after oral glucose in subjects with glucose intolerance. J. Clin. Endocrinol. Metab. 1990, 71, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Pratley, R.E.; Weyer, C. The role of impaired early insulin secretion in the pathogenesis of Type II diabetes mellitus. Diabetologia 2001, 44, 929–945. [Google Scholar] [CrossRef] [Green Version]
- Del Prato, S. Loss of early insulin secretion leads to postprandial hyperglycaemia. Diabetologia 2003, 46 (Suppl. 1), M2–M8. [Google Scholar] [CrossRef] [Green Version]
- Del Prato, S.; Marchetti, P.; Bonadonna, R.C. Phasic insulin release and metabolic regulation in type 2 diabetes. Diabetes 2002, 51 (Suppl. 1), S109–S116. [Google Scholar] [CrossRef] [Green Version]
- Reaven, G.M.; Thompson, L.W.; Nahum, D.; Haskins, E. Relationship between hyperglycemia and cognitive function in older NIDDM patients. Diabetes Care 1990, 13, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Perlmuter, L.C.; Hakami, M.K.; Hodgson-Harrington, C.; Ginsberg, J.; Katz, J.; Singer, D.E.; Nathan, D.M. Decreased cognitive function in aging non-insulin-dependent diabetic patients. Am. J. Med. 1984, 77, 1043–1048. [Google Scholar] [CrossRef]
- Pike, C.J.; Burdick, D.; Walencewicz, A.J.; Glabe, C.G.; Cotman, C.W. Neurodegeneration induced by beta-amyloid peptides In Vitro: The role of peptide assembly state. J. Neurosci. 1993, 13, 1676–1687. [Google Scholar] [CrossRef]
- Gschwind, M.; Huber, G. Apoptotic cell death induced by beta-amyloid 1-42 peptide is cell type dependent. J. Neurochem. 1995, 65, 292–300. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.; McGrath, G.; Zhang, J.; Ruddy, D.A.; Sym, M.; Apfeld, J.; Nicoll, M.; Maxwell, M.; Hai, B.; Ellis, M.C.; et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev. Cell 2002, 3, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Nishimura, M.; Arawaka, S.; Levitan, D.; Zhang, L.; Tandon, A.; Song, Y.Q.; Rogaeva, E.; Chen, F.; Kawarai, T.; et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 2000, 407, 48–54. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef] [Green Version]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Pimplikar, S.W.; Nixon, R.A.; Robakis, N.K.; Shen, J.; Tsai, L.H. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J. Neurosci. 2010, 30, 14946–14954. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Gutierrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Bentahir, M.; Nyabi, O.; Verhamme, J.; Tolia, A.; Horre, K.; Wiltfang, J.; Esselmann, H.; De Strooper, B. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J. Neurochem. 2006, 96, 732–742. [Google Scholar] [CrossRef]
- Shimojo, M.; Sahara, N.; Murayama, M.; Ichinose, H.; Takashima, A. Decreased Abeta secretion by cells expressing familial Alzheimer’s disease-linked mutant presenilin 1. Neurosci. Res. 2007, 57, 446–453. [Google Scholar] [CrossRef]
- Cacquevel, M.; Aeschbach, L.; Houacine, J.; Fraering, P.C. Alzheimer’s disease-linked mutations in presenilin-1 result in a drastic loss of activity in purified gamma-secretase complexes. PLoS ONE 2012, 7, e35133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilig, E.A.; Xia, W.; Shen, J.; Kelleher, R.J., 3rd. A presenilin-1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of gamma-secretase activity. J. Biol. Chem. 2010, 285, 22350–22359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A. Amyloid plaque imaging In Vivo: Current achievement and future prospects. Eur. J. Nucl. Med. Mol. Imaging 2008, 35 (Suppl. 1), S46–S50. [Google Scholar] [CrossRef] [Green Version]
- Villemagne, V.L.; Fodero-Tavoletti, M.T.; Pike, K.E.; Cappai, R.; Masters, C.L.; Rowe, C.C. The ART of loss: Abeta imaging in the evaluation of Alzheimer’s disease and other dementias. Mol. Neurobiol. 2008, 38, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Raux, G.; Gantier, R.; Thomas-Anterion, C.; Boulliat, J.; Verpillat, P.; Hannequin, D.; Brice, A.; Frebourg, T.; Campion, D. Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology 2000, 55, 1577–1578. [Google Scholar] [CrossRef]
- Tang-Wai, D.; Lewis, P.; Boeve, B.; Hutton, M.; Golde, T.; Baker, M.; Hardy, J.; Michels, V.; Ivnik, R.; Jack, C.; et al. Familial frontotemporal dementia associated with a novel presenilin-1 mutation. Dement. Geriatr. Cogn. Disord. 2002, 14, 13–21. [Google Scholar] [CrossRef]
- Dermaut, B.; Kumar-Singh, S.; Engelborghs, S.; Theuns, J.; Rademakers, R.; Saerens, J.; Pickut, B.A.; Peeters, K.; van den Broeck, M.; Vennekens, K.; et al. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann. Neurol. 2004, 55, 617–626. [Google Scholar] [CrossRef]
- Cruts, M.; Theuns, J.; Van Broeckhoven, C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012, 33, 1340–1344. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; de Strooper, B.; Bezprozvanny, I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supnet, C.; Bezprozvanny, I. Presenilins function in ER calcium leak and Alzheimer’s disease pathogenesis. Cell Calcium 2011, 50, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezprozvanny, I. Presenilins and calcium signaling-systems biology to the rescue. Sci. Signal. 2013, 6, pe24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, W.; Ostaszewski, B.L.; Kimberly, W.T.; Rahmati, T.; Moore, C.L.; Wolfe, M.S.; Selkoe, D.J. FAD mutations in presenilin-1 or amyloid precursor protein decrease the efficacy of a gamma-secretase inhibitor: Evidence for direct involvement of PS1 in the gamma-secretase cleavage complex. Neurobiol. Dis. 2000, 7, 673–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szostak, J.W.; Bartel, D.P.; Luisi, P.L. Synthesizing life. Nature 2001, 409, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ruder, W.C.; LeDuc, P.R. Artificial cells: Building bioinspired systems using small-scale biology. Trends Biotechnol. 2008, 26, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, R.; Corbo, C.; Martinez, J.O.; Taraballi, F.; Evangelopoulos, M.; Minardi, S.; Yazdi, I.K.; Zhao, P.; De Rosa, E.; Sherman, M.B.; et al. Biomimetic proteolipid vesicles for targeting inflamed tissues. Nat. Mater. 2016, 15, 1037–1046. [Google Scholar] [CrossRef]
- Hu, C.M.; Fang, R.H.; Wang, K.C.; Luk, B.T.; Thamphiwatana, S.; Dehaini, D.; Nguyen, P.; Angsantikul, P.; Wen, C.H.; Kroll, A.V.; et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature 2015, 526, 118–121. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, J.; Sun, W.; Archibong, E.; Kahkoska, A.R.; Zhang, X.; Lu, Y.; Ligler, F.S.; Buse, J.B.; Gu, Z. Synthetic beta cells for fusion-mediated dynamic insulin secretion. Nat. Chem. Biol. 2018, 14, 86–93. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klec, C.; Ziomek, G.; Pichler, M.; Malli, R.; Graier, W.F. Calcium Signaling in ß-cell Physiology and Pathology: A Revisit. Int. J. Mol. Sci. 2019, 20, 6110. https://doi.org/10.3390/ijms20246110
Klec C, Ziomek G, Pichler M, Malli R, Graier WF. Calcium Signaling in ß-cell Physiology and Pathology: A Revisit. International Journal of Molecular Sciences. 2019; 20(24):6110. https://doi.org/10.3390/ijms20246110
Chicago/Turabian StyleKlec, Christiane, Gabriela Ziomek, Martin Pichler, Roland Malli, and Wolfgang F. Graier. 2019. "Calcium Signaling in ß-cell Physiology and Pathology: A Revisit" International Journal of Molecular Sciences 20, no. 24: 6110. https://doi.org/10.3390/ijms20246110
APA StyleKlec, C., Ziomek, G., Pichler, M., Malli, R., & Graier, W. F. (2019). Calcium Signaling in ß-cell Physiology and Pathology: A Revisit. International Journal of Molecular Sciences, 20(24), 6110. https://doi.org/10.3390/ijms20246110